
Minimal Change Disease: Pathogenetic Insights from Glomerular Proteomics
 ,
,  , ,
, ,  ,
,  ,
,  , ,
, ,  , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Characteristics of the Study Participants
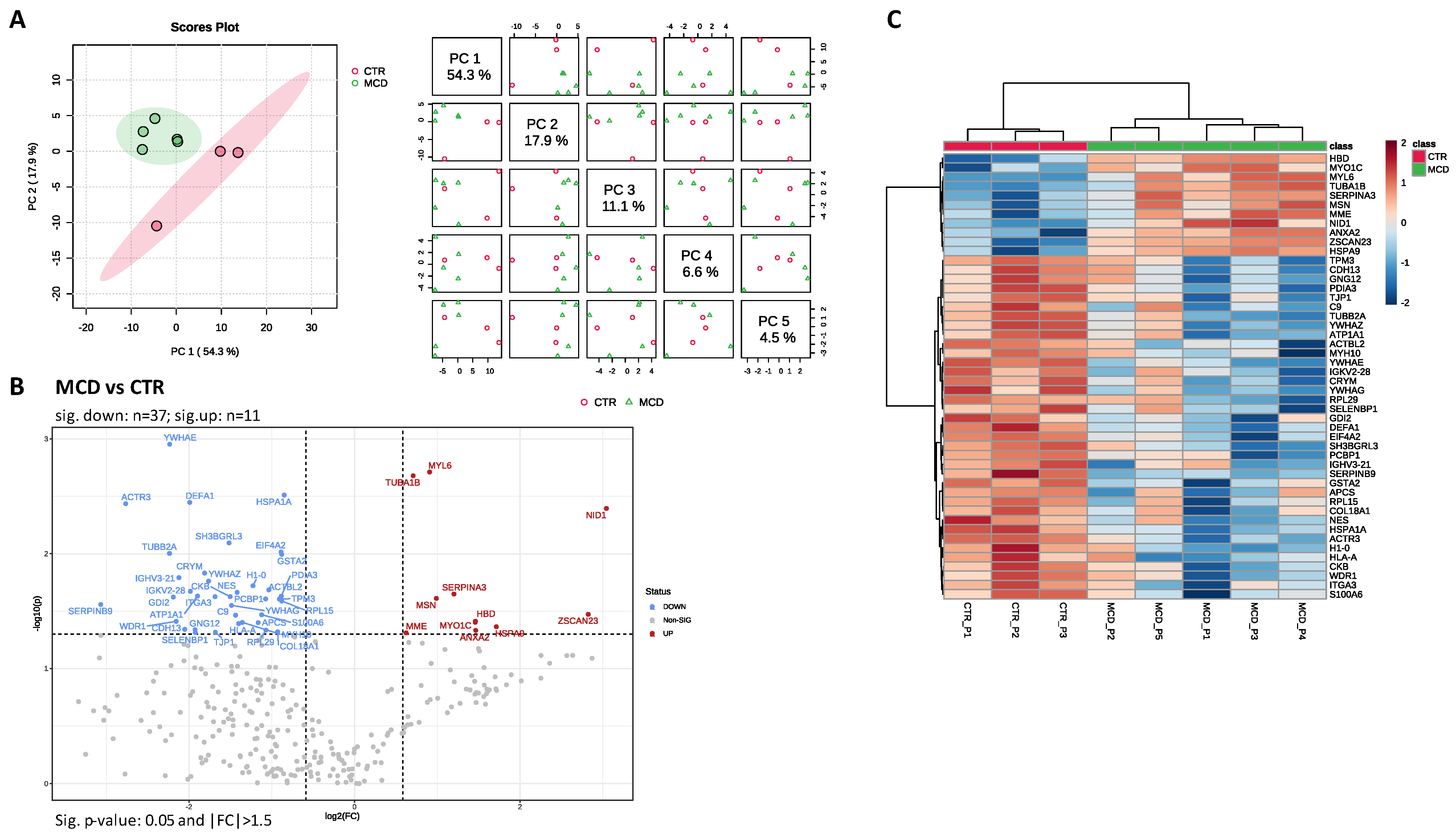
2.2. LC–MS/MS Analysis
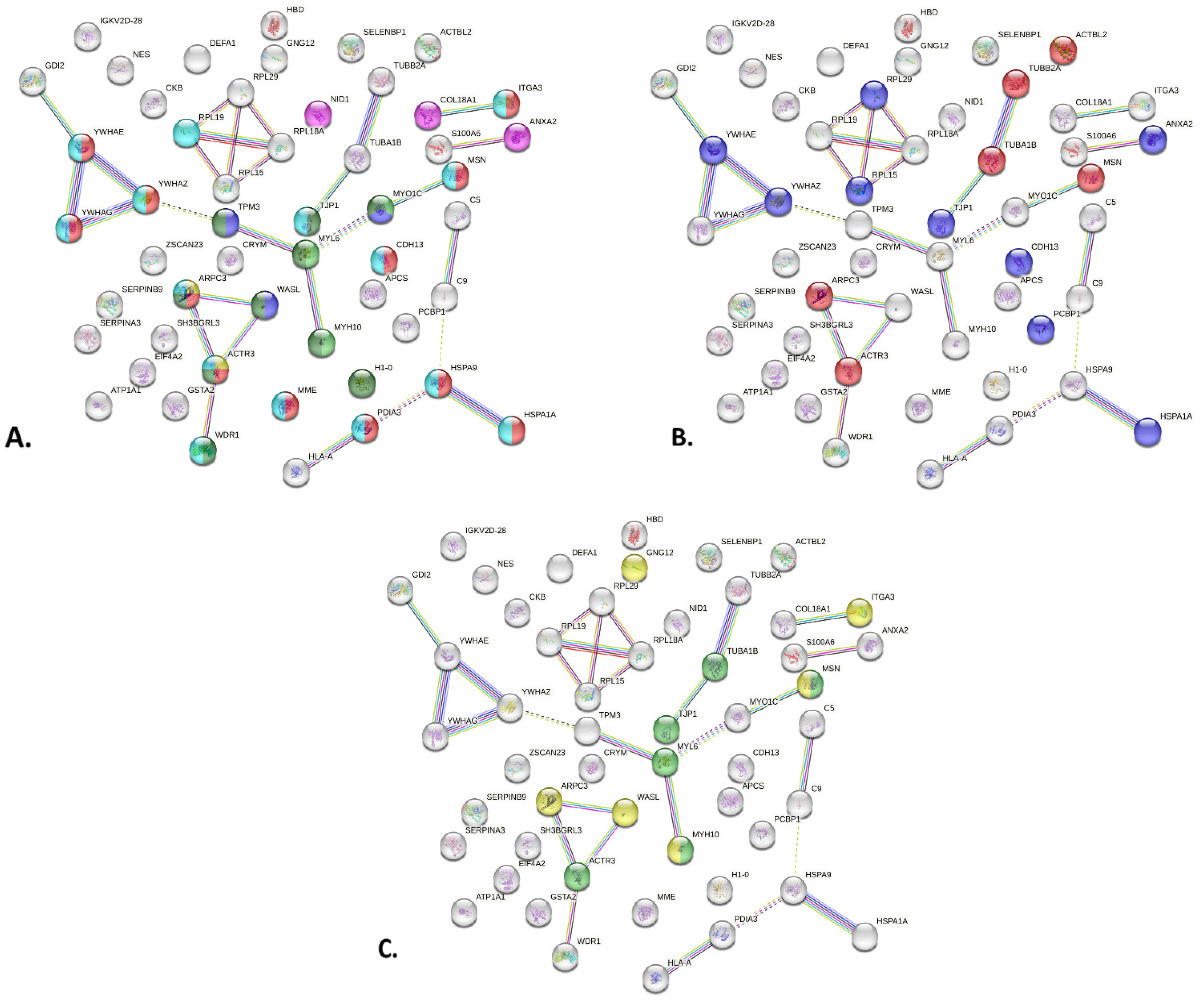
2.3. PPI Network of MCD Related Proteins
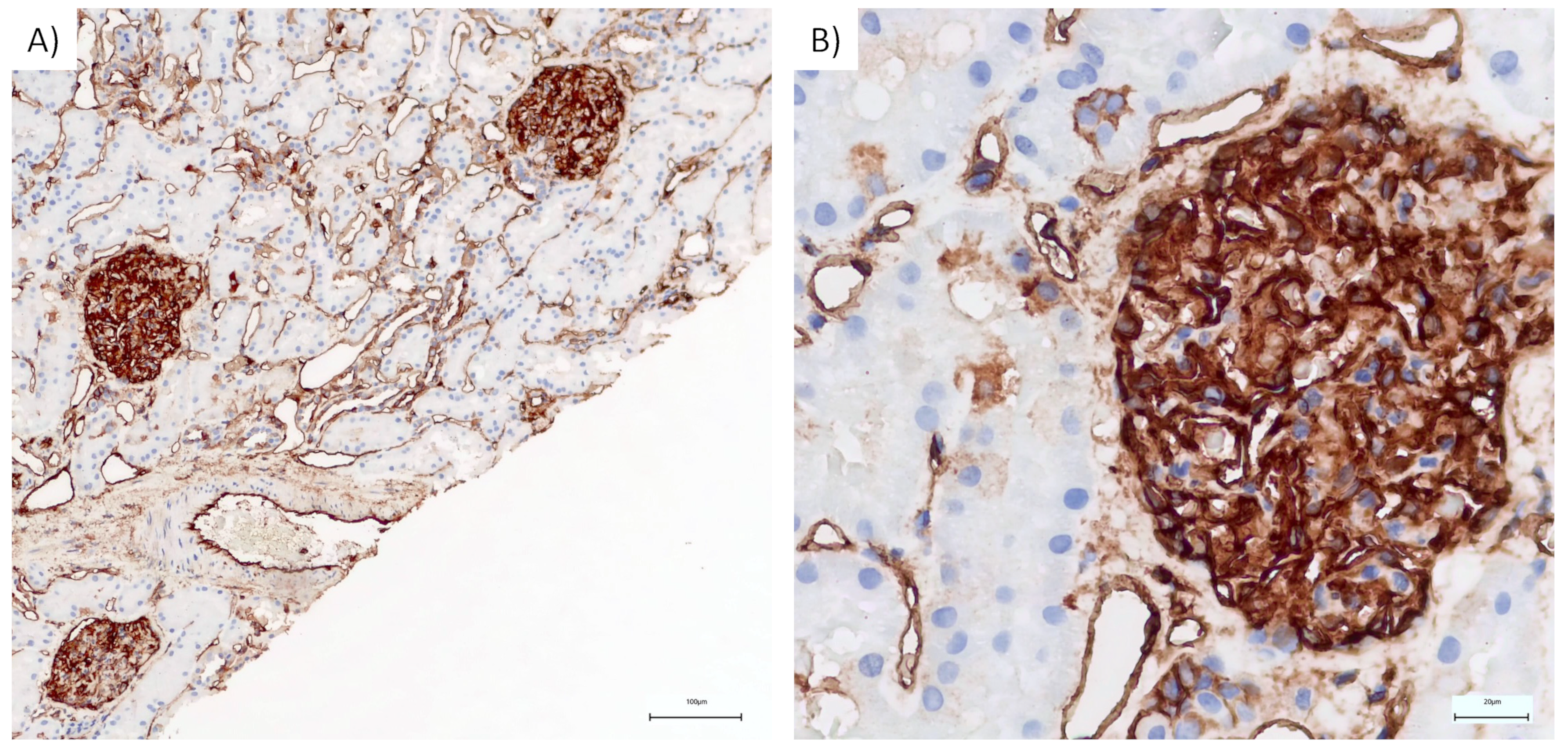
2.4. Immunohistochemistry
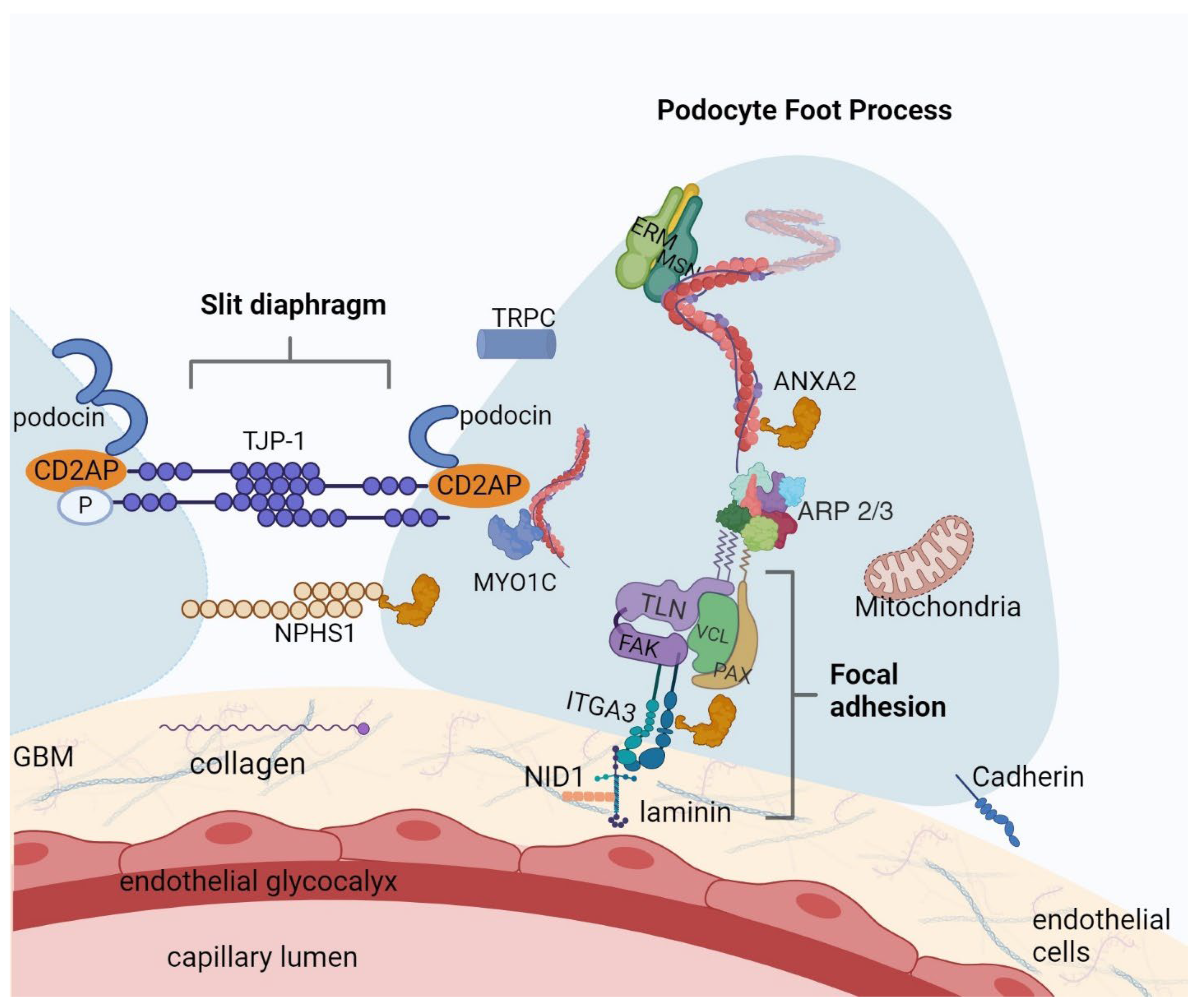
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Storage
4.2. Laser Capture Microdissection Sample Preparation
4.3. Sample Preparation for Proteome Profiling
4.4. LC−MS Analysis
4.5. Data Processing and Label-Free Quantification
4.6. Immunohistochemistry
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barisoni, L.; Schnaper, H.W.; Kopp, J.B. A Proposed Taxonomy for the Podocytopathies. Clin. J. Am. Soc. Nephrol. 2007, 2, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Purohit, S.; Piani, F.; Ordoñez, F.A.; de Lucas-Collantes, C.; Bauer, C.; Cara-Fuentes, G. Molecular Mechanisms of Proteinuria in Minimal Change Disease. Front. Med. 2021, 8, 761600. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, R.; Bonanni, A.; Caridi, G.; Canepa, A.; Ghiggeri, G.M. Molecular and Cellular Mechanisms for Proteinuria in Minimal Change Disease. Front. Med. 2018, 5, 170. [Google Scholar] [CrossRef] [PubMed]
- New, L.A.; Martin, C.E.; Scott, R.P.; Platt, M.J.; Keyvani Chahi, A.; Stringer, C.D.; Lu, P.; Samborska, B.; Eremina, V.; Takano, T.; et al. Nephrin Tyrosine Phosphorylation Is Required to Stabilize and Restore Podocyte Foot Process Architecture. J. Am. Soc. Nephrol. 2016, 27, 2422–2435. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Venkatareddy, M.; Kalinowski, A.; Li, T.; Kukla, J.; Mollin, A.; Cara-Fuentes, G.; Patel, S.R.; Garg, P. Nephrin Is Necessary for Podocyte Recovery Following Injury in an Adult Mature Glomerulus. PLoS ONE 2018, 13, e0198013. [Google Scholar] [CrossRef] [PubMed]
- Perico, L.; Conti, S.; Benigni, A.; Remuzzi, G. Podocyte–Actin Dynamics in Health and Disease. Nat. Rev. Nephrol. 2016, 12, 692–710. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, K.; Kurihara, H.; Sakai, T. Actin Filament Organization of Foot Processes in Rat Podocytes. J. Histochem. Cytochem. 2003, 51, 1589–1600. [Google Scholar] [CrossRef]
- Kriz, W.; Shirato, I.; Nagata, M.; LeHir, M.; Lemley, K.V. The Podocyte’s Response to Stress: The Enigma of Foot Process Effacement. Am. J. Physiol. Physiol. 2013, 304, F333–F347. [Google Scholar] [CrossRef]
- Kriz, W. The Pathogenesis of “classic” Focal Segmental Glomerulosclerosis--Lessons from Rat Models. Nephrol. Dial. Transplant. 2003, 18, vi39–vi44. [Google Scholar] [CrossRef]
- Trimarchi, H. Podocyturia: Potential Applications and Current Limitations. World J. Nephrol. 2017, 6, 221. [Google Scholar] [CrossRef]
- Sever, S.; Schiffer, M. Actin Dynamics at Focal Adhesions: A Common Endpoint and Putative Therapeutic Target for Proteinuric Kidney Diseases. Kidney Int. 2018, 93, 1298–1307. [Google Scholar] [CrossRef]
- Wharram, B.L.; Goyal, M.; Wiggins, J.E.; Sanden, S.K.; Hussain, S.; Filipiak, W.E.; Saunders, T.L.; Dysko, R.C.; Kohno, K.; Holzman, L.B.; et al. Podocyte Depletion Causes Glomerulosclerosis. J. Am. Soc. Nephrol. 2005, 16, 2941–2952. [Google Scholar] [CrossRef]
- Wiggins, R.-C. The Spectrum of Podocytopathies: A Unifying View of Glomerular Diseases. Kidney Int. 2007, 71, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Kestilä, M.; Lenkkeri, U.; Männikkö, M.; Lamerdin, J.; McCready, P.; Putaala, H.; Ruotsalainen, V.; Morita, T.; Nissinen, M.; Herva, R.; et al. Positionally Cloned Gene for a Novel Glomerular Protein—Nephrin—Is Mutated in Congenital Nephrotic Syndrome. Mol. Cell 1998, 1, 575–582. [Google Scholar] [CrossRef]
- Beltcheva, O.; Martin, P.; Lenkkeri, U.; Tryggvason, K. Mutation Spectrum in the Nephrin Gene (NPHS1) in Congenital Nephrotic Syndrome. Hum. Mutat. 2001, 17, 368–373. [Google Scholar] [CrossRef]
- Philippe, A.A.A.; Nevo, F.; Esquivel, E.L.; Reklaityte, D.; Gribouval, O.; Te[Combining Circumflex Accent]te, M.-J.G.A.; Loirat, C.; Dantal, J.; Fischbach, M.; Pouteil-Noble, C.; et al. Nephrin Mutations Can Cause Childhood-Onset Steroid-Resistant Nephrotic Syndrome. J. Am. Soc. Nephrol. 2008, 19, 1871–1878. [Google Scholar] [CrossRef] [PubMed]
- Shalhoub, R. Pathogenesis of lipoid nephrosis: A disorder of T-cell function. Lancet 1974, 304, 556–560. [Google Scholar] [CrossRef]
- Kopp, J.B.; Anders, H.-J.; Susztak, K.; Podestà, M.A.; Remuzzi, G.; Hildebrandt, F.; Romagnani, P. Podocytopathies. Nat. Rev. Dis. Prim. 2020, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Davin, J.-C. The Glomerular Permeability Factors in Idiopathic Nephrotic Syndrome. Pediatr. Nephrol. 2016, 31, 207–215. [Google Scholar] [CrossRef]
- Colucci, M.; Corpetti, G.; Emma, F.; Vivarelli, M. Immunology of Idiopathic Nephrotic Syndrome. Pediatr. Nephrol. 2018, 33, 573–584. [Google Scholar] [CrossRef]
- Eikrem, Ø.; Lillefosse, B.; Delaleu, N.; Strauss, P.; Osman, T.; Vikse, B.E.; Debiec, H.; Ronco, P.; Sekulic, M.; Koch, E.; et al. Network-Based Assessment of Minimal Change Disease Identifies Glomerular Response to IL-7 and IL-12 Pathways Activation as Innovative Treatment Target. Biomedicines 2023, 11, 226. [Google Scholar] [CrossRef] [PubMed]
- Mariani, L.H.; Eddy, S.; AlAkwaa, F.M.; McCown, P.J.; Harder, J.L.; Nair, V.; Eichinger, F.; Martini, S.; Ademola, A.D.; Boima, V.; et al. Precision Nephrology Identified Tumor Necrosis Factor Activation Variability in Minimal Change Disease and Focal Segmental Glomerulosclerosis. Kidney Int. 2023, 103, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, L.; Murea, M.; Freedman, B.I.; Ma, L. Bioinformatics Analysis Reveals a Shared Pathway for Common Forms of Adult Nephrotic Syndrome. Kidney360 2023, 4, e515–e524. [Google Scholar] [CrossRef] [PubMed]
- Pérez, V.; López, D.; Boixadera, E.; Ibernón, M.; Espinal, A.; Bonet, J.; Romero, R. Comparative Differential Proteomic Analysis of Minimal Change Disease and Focal Segmental Glomerulosclerosis. BMC Nephrol. 2017, 18, 49. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.W.; Kim, Y.G.; Song, M.-Y.; Moon, J.-Y.; Jeong, K.-H.; Lee, T.-W.; Ihm, C.-G.; Park, K.-S.; Lee, S.-H. Potential Urine Proteomics Biomarkers for Primary Nephrotic Syndrome. Clin. Proteom. 2017, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Suresh, C.P.; Saha, A.; Kaur, M.; Kumar, R.; Dubey, N.K.; Basak, T.; Tanwar, V.S.; Bhardwaj, G.; Sengupta, S.; Batra, V.V.; et al. Differentially Expressed Urinary Biomarkers in Children with Idiopathic Nephrotic Syndrome. Clin. Exp. Nephrol. 2016, 20, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Weng, Q.; Zhou, Q.; Tong, J.; Jin, Y.; Liu, Y.; Yu, X.; Pan, X.; Ren, H.; Wang, W.; Xie, J.; et al. New Risk Score for Predicting Steroid Resistance in Patients with Focal Segmental Glomerulosclerosis or Minimal Change Disease. Clin. Proteom. 2020, 17, 18. [Google Scholar] [CrossRef] [PubMed]
- Almaani, S.; Madhavan, S.M.; Shapiro, J.P.; Satoskar, A.A.; Ayoub, I.; Rovin, B.H.; Parikh, S.V. Comparison of Glomerular Proteomics Profiles of Healthy Human Kidney and Minimal Change Disease Identifies Distinct Targets and Pathways. In Proceedings of the Glomerular Diseases: Fibrosis and Extracellular Matrix, Virtual, 4 November 2021. [Google Scholar]
- Madhavan, S.M.; Almaani, S.; Shapiro, J.P.; Satoskar, A.A.; Ayoub, I.; Rovin, B.H. Differentiating Steroid-Sensitive Minimal Change Disease and Primary and Secondary Focal Segmental Glomerulosclerosis: A Proteomics-Based Approach. In Proceedings of the Glomerular Diseases: Antibodies, Complement, and Inflammatory Mediators, Virtual, 5 November 2021. [Google Scholar]
- Rinschen, M.M.; Schroeter, C.B.; Koehler, S.; Ising, C.; Schermer, B.; Kann, M.; Benzing, T.; Brinkkoetter, P.T. Quantitative Deep Mapping of the Cultured Podocyte Proteome Uncovers Shifts in Proteostatic Mechanisms during Differentiation. Am. J. Physiol. Physiol. 2016, 311, C404–C417. [Google Scholar] [CrossRef] [PubMed]
- Rinschen, M.M.; Gödel, M.; Grahammer, F.; Zschiedrich, S.; Helmstädter, M.; Kretz, O.; Zarei, M.; Braun, D.A.; Dittrich, S.; Pahmeyer, C.; et al. A Multi-Layered Quantitative In Vivo Expression Atlas of the Podocyte Unravels Kidney Disease Candidate Genes. Cell Rep. 2018, 23, 2495–2508. [Google Scholar] [CrossRef]
- Rinschen, M.M.; Saez-Rodriguez, J. The Tissue Proteome in the Multi-Omic Landscape of Kidney Disease. Nat. Rev. Nephrol. 2021, 17, 205–219. [Google Scholar] [CrossRef]
- Koehler, S.; Brinkkoetter, P.; Rinschen, M.M. Podocyte Proteome Analysis Reveals Stress Responses in Glomerular Sclerosis. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Koehler, S.; Kuczkowski, A.; Kuehne, L.; Jüngst, C.; Hoehne, M.; Grahammer, F.; Eddy, S.; Kretzler, M.; Beck, B.B.; Höhfeld, J.; et al. Proteome Analysis of Isolated Podocytes Reveals Stress Responses in Glomerular Sclerosis. J. Am. Soc. Nephrol. 2020, 31, 544–559. [Google Scholar] [CrossRef] [PubMed]
- Chhuon, C.; Herrera-Marcos, L.V.; Zhang, S.-Y.; Charrière-Bertrand, C.; Jung, V.; Lipecka, J.; Savas, B.; Nasser, N.; Pawlak, A.; Boulmerka, H.; et al. Proteomics of Plasma and Plasma-Treated Podocytes: Application to Focal and Segmental Glomerulosclerosis. Int. J. Mol. Sci. 2023, 24, 12124. [Google Scholar] [CrossRef] [PubMed]
- Haas, M. Glomerular Disease Pathology in the Era of Proteomics: From Pattern to Pathogenesis. J. Am. Soc. Nephrol. 2018, 29, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING Database in 2023: Protein–Protein Association Networks and Functional Enrichment Analyses for Any Sequenced Genome of Interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Itoh, M.; Nakadate, K.; Horibata, Y.; Matsusaka, T.; Xu, J.; Hunziker, W.; Sugimoto, H. The Structural and Functional Organization of the Podocyte Filtration Slits Is Regulated by Tjp1/ZO-1. PLoS ONE 2014, 9, e106621. [Google Scholar] [CrossRef] [PubMed]
- Blaine, J.; Dylewski, J. Regulation of the Actin Cytoskeleton in Podocytes. Cells 2020, 9, 1700. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.E.; Jones, N. Nephrin Signaling in the Podocyte: An Updated View of Signal Regulation at the Slit Diaphragm and Beyond. Front. Endocrinol. 2018, 9, 302. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Zhang, Y.; Zhuang, J.; Bi, Y.; Xu, H.; Shen, Q.; Liu, J.; Fu, H.; Wang, J.; Feng, C.; et al. The Important Roles and Molecular Mechanisms of Annexin A2 Autoantibody in Children with Nephrotic Syndrome. Ann. Transl. Med. 2021, 9, 1452. [Google Scholar] [CrossRef]
- Su, S.-C.; Maxwell, S.A.; Bayless, K.J. Annexin 2 Regulates Endothelial Morphogenesis by Controlling AKT Activation and Junctional Integrity. J. Biol. Chem. 2010, 285, 40624–40634. [Google Scholar] [CrossRef] [PubMed]
- Yamada, A.; Irie, K.; Hirota, T.; Ooshio, T.; Fukuhara, A.; Takai, Y. Involvement of the Annexin II-S100A10 Complex in the Formation of E-Cadherin-Based Adherens Junctions in Madin-Darby Canine Kidney Cells. J. Biol. Chem. 2005, 280, 6016–6027. [Google Scholar] [CrossRef] [PubMed]
- Heyraud, S.; Jaquinod, M.; Durmort, C.; Dambroise, E.; Concord, E.; Schaal, J.P.; Huber, P.; Gulino-Debrac, D. Contribution of Annexin 2 to the Architecture of Mature Endothelial Adherens Junctions. Mol. Cell. Biol. 2008, 28, 1657–1668. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.J.; Moss, S.E. Annexin 2 Has a Dual Role as Regulator and Effector of V-Src in Cell Transformation. J. Biol. Chem. 2009, 284, 10202–10210. [Google Scholar] [CrossRef] [PubMed]
- Babbin, B.A.; Parkos, C.A.; Mandell, K.J.; Winfree, L.M.; Laur, O.; Ivanov, A.I.; Nusrat, A. Annexin 2 Regulates Intestinal Epithelial Cell Spreading and Wound Closure through Rho-Related Signaling. Am. J. Pathol. 2007, 170, 951–966. [Google Scholar] [CrossRef] [PubMed]
- Barwe, S.P.; Anilkumar, G.; Moon, S.Y.; Zheng, Y.; Whitelegge, J.P.; Rajasekaran, S.A.; Rajasekaran, A.K. Novel Role for Na,K-ATPase in Phosphatidylinositol 3-Kinase Signaling and Suppression of Cell Motility. Mol. Biol. Cell 2005, 16, 1082–1094. [Google Scholar] [CrossRef] [PubMed]
- De Graauw, M.; Tijdens, I.; Smeets, M.B.; Hensbergen, P.J.; Deelder, A.M.; van de Water, B. Annexin A2 Phosphorylation Mediates Cell Scattering and Branching Morphogenesis via Cofilin Activation. Mol. Cell. Biol. 2008, 28, 1029–1040. [Google Scholar] [CrossRef]
- Yung, S.; Cheung, K.F.; Zhang, Q.; Chan, T.M. Anti-DsDNA Antibodies Bind to Mesangial Annexin II in Lupus Nephritis. J. Am. Soc. Nephrol. 2010, 21, 1912–1927. [Google Scholar] [CrossRef]
- Caster, D.J.; Korte, E.A.; Merchant, M.L.; Klein, J.B.; Wilkey, D.W.; Rovin, B.H.; Birmingham, D.J.; Harley, J.B.; Cobb, B.L.; Namjou, B.; et al. Autoantibodies Targeting Glomerular Annexin A2 Identify Patients with Proliferative Lupus Nephritis. PROTEOMICS Clin. Appl. 2015, 9, 1012–1020. [Google Scholar] [CrossRef]
- Tesch, S.; Abdirama, D.; Grießbach, A.-S.; Brand, H.A.; Goerlich, N.; Humrich, J.Y.; Bacher, P.; Hiepe, F.; Riemekasten, G.; Enghard, P. Identification and Characterization of Antigen-Specific CD4+ T Cells Targeting Renally Expressed Antigens in Human Lupus Nephritis with Two Independent Methods. Sci. Rep. 2020, 10, 21312. [Google Scholar] [CrossRef]
- Ka, S.-M.; Rifai, A.; Chen, J.-H.; Cheng, C.-W.; Shui, H.-A.; Lee, H.-S.; Lin, Y.-F.; Hsu, L.-F.; Chen, A. Glomerular Crescent-Related Biomarkers in a Murine Model of Chronic Graft versus Host Disease. Nephrol. Dial. Transplant. 2006, 21, 288–298. [Google Scholar] [CrossRef]
- Salle, V.; Cordonnier, C.; Schmidt, J.; Mazière, C.; Smail, A.; Attencourt, C.; Mabille, M.P.; Mazière, J.C.; Makdassi, R.; Choukroun, G.; et al. Vascular Expression of Annexin A2 in Lupus Nephritis. J. Clin. Pathol. 2016, 69, 533–536. [Google Scholar] [CrossRef]
- Zhou, Y.; Xiao, L.; Tang, S. Annexin A2 and FTH1 Are Potential Biomarkers for Lupus Nephritis. Exp. Ther. Med. 2018, 16, 3766–3776. [Google Scholar] [CrossRef]
- Lin, L.; Wu, C.; Hu, K. Tissue Plasminogen Activator Activates NF-ΚB through a Pathway Involving Annexin A2/CD11b and Integrin-Linked Kinase. J. Am. Soc. Nephrol. 2012, 23, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Hu, K. Tissue-Type Plasminogen Activator Modulates Macrophage M2 to M1 Phenotypic Change through Annexin A2-Mediated NF-ΚB Pathway. Oncotarget 2017, 8, 88094–88103. [Google Scholar] [CrossRef]
- Renner, B.; Tong, H.H.; Laskowski, J.; Jonscher, K.; Goetz, L.; Woolaver, R.; Hannan, J.; Li, Y.X.; Hourcade, D.; Pickering, M.C.; et al. Annexin A2 Enhances Complement Activation by Inhibiting Factor H. J. Immunol. 2016, 196, 1355–1365. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, H.; Anderson, J.M.; Kerjaschki, D.; Farquhar, M.G. The Altered Glomerular Filtration Slits Seen in Puromycin Aminonucleoside Nephrosis and Protamine Sulfate-Treated Rats Contain the Tight Junction Protein ZO-1. Am. J. Pathol. 1992, 141, 805–816. [Google Scholar] [PubMed]
- Rincon-Choles, H.; Vasylyeva, T.L.; Pergola, P.E.; Bhandari, B.; Bhandari, K.; Zhang, J.-H.; Wang, W.; Gorin, Y.; Barnes, J.L.; Abboud, H.E. ZO-1 Expression and Phosphorylation in Diabetic Nephropathy. Diabetes 2006, 55, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kang, Y.S.; Dai, C.; Kiss, L.P.; Wen, X.; Liu, Y. Epithelial-to-Mesenchymal Transition Is a Potential Pathway Leading to Podocyte Dysfunction and Proteinuria. Am. J. Pathol. 2008, 172, 299–308. [Google Scholar] [CrossRef]
- Arif, E.; Wagner, M.C.; Johnstone, D.B.; Wong, H.N.; George, B.; Pruthi, P.A.; Lazzara, M.J.; Nihalani, D. Motor Protein Myo1c Is a Podocyte Protein That Facilitates the Transport of Slit Diaphragm Protein Neph1 to the Podocyte Membrane. Mol. Cell. Biol. 2011, 31, 2134–2150. [Google Scholar] [CrossRef]
- Evangelista, M.; Klebl, B.M.; Tong, A.H.Y.; Webb, B.A.; Leeuw, T.; Leberer, E.; Whiteway, M.; Thomas, D.Y.; Boone, C. A Role for Myosin-I in Actin Assembly through Interactions with Vrp1p, Bee1p, and the Arp2/3 Complex. J. Cell Biol. 2000, 148, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.S.; Powell, B.C.; Cheney, R.E. A Millennial Myosin Census. Mol. Biol. Cell 2001, 12, 780–794. [Google Scholar] [CrossRef]
- Soldati, T. Unconventional Myosins, Actin Dynamics and Endocytosis: A Ménage à Trois? Traffic 2003, 4, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Murshed, M.; Smyth, N.; Miosge, N.; Karolat, J.; Krieg, T.; Paulsson, M.; Nischt, R. The Absence of Nidogen 1 Does Not Affect Murine Basement Membrane Formation. Mol. Cell. Biol. 2000, 20, 7007–7012. [Google Scholar] [CrossRef]
- Schymeinsky, J.; Nedbal, S.; Miosge, N.; Pöschl, E.; Rao, C.; Beier, D.R.; Skarnes, W.C.; Timpl, R.; Bader, B.L. Gene Structure and Functional Analysis of the Mouse Nidogen-2 Gene: Nidogen-2 Is Not Essential for Basement Membrane Formation in Mice. Mol. Cell. Biol. 2002, 22, 6820–6830. [Google Scholar] [CrossRef] [PubMed]
- Lennon, R.; Randles, M.J.; Humphries, M.J. The Importance of Podocyte Adhesion for a Healthy Glomerulus. Front. Endocrinol. 2014, 5, 160. [Google Scholar] [CrossRef]
- Katz, A.; Fish, A.J.; Kleppel, M.M.; Hagen, S.G.; Michael, A.F.; Butkowski, R.J. Renal Entactin (Nidogen): Isolation, Characterization and Tissue Distribution. Kidney Int. 1991, 40, 643–652. [Google Scholar] [CrossRef]
- Bülow, R.D.; Boor, P. Extracellular Matrix in Kidney Fibrosis: More Than Just a Scaffold. J. Histochem. Cytochem. 2019, 67, 643–661. [Google Scholar] [CrossRef]
- Sachs, N.; Kreft, M.; van den Bergh Weerman, M.A.; Beynon, A.J.; Peters, T.A.; Weening, J.J.; Sonnenberg, A. Kidney Failure in Mice Lacking the Tetraspanin CD151. J. Cell Biol. 2006, 175, 33–39. [Google Scholar] [CrossRef]
- Kreidberg, J.A.; Donovan, M.J.; Goldstein, S.L.; Rennke, H.; Shepherd, K.; Jones, R.C.; Jaenisch, R. Alpha 3 Beta 1 Integrin Has a Crucial Role in Kidney and Lung Organogenesis. Development 1996, 122, 3537–3547. [Google Scholar] [CrossRef]
- Kreidberg, J.A.; Symons, J.M. Integrins in Kidney Development, Function, and Disease. Am. J. Physiol. Physiol. 2000, 279, F233–F242. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, N.; Margadant, C.; Kevelam, S.H.; Lilien, M.R.; Oosterveld, M.J.S.; Kreft, M.; van Eerde, A.M.; Pfundt, R.; Terhal, P.A.; van der Zwaag, B.; et al. Gain of Glycosylation in Integrin A3 Causes Lung Disease and Nephrotic Syndrome. J. Clin. Investig. 2012, 122, 4375–4387. [Google Scholar] [CrossRef] [PubMed]
- Has, C.; Spartà, G.; Kiritsi, D.; Weibel, L.; Moeller, A.; Vega-Warner, V.; Waters, A.; He, Y.; Anikster, Y.; Esser, P.; et al. Integrin α3 Mutations with Kidney, Lung, and Skin Disease. N. Engl. J. Med. 2012, 366, 1508–1514. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, A.; Zent, R. Integrins in Kidney Disease. J. Am. Soc. Nephrol. 2013, 24, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-C.; Chen, C.-A.; Guh, J.-Y.; Chang, J.-M.; Shin, S.-J.; Lai, Y.-H. Altering Expression of A3β1 Integrin on Podocytes of Human and Rats with Diabetes. Life Sci. 2000, 67, 2345–2353. [Google Scholar] [CrossRef] [PubMed]
- Mullins, R.D.; Heuser, J.A.; Pollard, T.D. The Interaction of Arp2/3 Complex with Actin: Nucleation, High Affinity Pointed End Capping, and Formation of Branching Networks of Filaments. Proc. Natl. Acad. Sci. USA 1998, 95, 6181–6186. [Google Scholar] [CrossRef] [PubMed]
- Sever, S. Role of Actin Cytoskeleton in Podocytes. Pediatr. Nephrol. 2021, 36, 2607–2614. [Google Scholar] [CrossRef] [PubMed]
- Swaney, K.F.; Li, R. Function and Regulation of the Arp2/3 Complex during Cell Migration in Diverse Environments. Curr. Opin. Cell Biol. 2016, 42, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, K.; Morita, H.; Suetsugu, S.; Kuraba, S.; Numata, Y.; Yamamoto, Y.; Inui, K.; Ideura, T.; Wakisaka, N.; Nakano, K.; et al. Actin -Related Protein 3 (Arp3) Is Mutated in Proteinuric BUF/Mna Rats. Mamm. Genome 2008, 19, 41–50. [Google Scholar] [CrossRef]
- Michalczyk, K.; Ziman, M. Nestin Structure and Predicted Function in Cellular Cytoskeletal Organisation. Histol. Histopathol. 2005, 20, 665–671. [Google Scholar] [CrossRef]
- Chen, J.; Boyle, S.; Zhao, M.; Su, W.; Takahashi, K.; Davis, L.; DeCaestecker, M.; Takahashi, T.; Breyer, M.D.; Hao, C.-M. Differential Expression of the Intermediate Filament Protein Nestin during Renal Development and Its Localization in Adult Podocytes. J. Am. Soc. Nephrol. 2006, 17, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Chen, J.; Yang, H.; You, L.; Xu, L.; Wang, X.; Li, R.; Gao, L.; Gu, Y.; Lin, S.; et al. Expression of Nestin in the Podocytes of Normal and Diseased Human Kidneys. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1761–R1767. [Google Scholar] [CrossRef] [PubMed]
- Wagner, N.; Wagner, K.-D.; Scholz, H.; Kirschner, K.M.; Schedl, A. Intermediate Filament Protein Nestin Is Expressed in Developing Kidney and Heart and Might Be Regulated by the Wilms’ Tumor Suppressor Wt1. Am. J. Physiol. Integr. Comp. Physiol. 2006, 291, R779–R787. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, Y.; Hao, J.; Liu, S.; Liu, Q.; Zhao, S.; Shi, Y.; Duan, H. Nestin Protects Mouse Podocytes against High Glucose-induced Apoptosis by a Cdk5-dependent Mechanism. J. Cell. Biochem. 2012, 113, 3186–3196. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Guo, H.; Miao, X.; Xu, J.; Yang, R.; Zhao, L.; Liu, J.; Yang, L.; Gao, F.; Zhang, W.; et al. Nestin Protects Podocyte from Injury in Lupus Nephritis by Mitophagy and Oxidative Stress. Cell Death Dis. 2020, 11, 319. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Glassock, R.J.; Fervenza, F.C. Focal Segmental Glomerulosclerosis: Towards a Better Understanding for the Practicing Nephrologist. Nephrol. Dial. Transplant. 2015, 30, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Müller-Deile, J.; Schenk, H.; Schiffer, M. Minimal-Change-Glomerulonephritis Und Fokal-Segmentale Glomerulosklerose. Internist 2019, 60, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.Z.; Kopp, J.B. Focal Segmental Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2017, 12, 502–517. [Google Scholar] [CrossRef] [PubMed]
- Mavrogeorgis, E.; Mischak, H.; Beige, J.; Latosinska, A.; Siwy, J. Understanding Glomerular Diseases through Proteomics. Expert Rev. Proteom. 2021, 18, 137–157. [Google Scholar] [CrossRef]
- Sigdel, T.K.; Piehowski, P.D.; Roy, S.; Liberto, J.; Hansen, J.R.; Swensen, A.C.; Zhao, R.; Zhu, Y.; Rashmi, P.; Schroeder, A.; et al. Near-Single-Cell Proteomics Profiling of the Proximal Tubular and Glomerulus of the Normal Human Kidney. Front. Med. 2020, 7, 499. [Google Scholar] [CrossRef]
- Bărar, A.A.; Pralea, I.E.; Berindan-Neagoe, I.; Pirlog, R.; Nutu, A.; Maslyennikov, Y.; Potra, A.R.; Iuga, C.A.; Kacso, I.M. Proteomic Patterns in Glomerular Research, a Laser Capture Microdissection and Liquid Chromatography-Tandem Mass Spectrometry Approach. Rev. Rom. Med. Lab. 2023, 31, 263–274. [Google Scholar] [CrossRef]
- Hughes, C.S.; Moggridge, S.; Müller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-Pot, Solid-Phase-Enhanced Sample Preparation for Proteomics Experiments. Nat. Protoc. 2019, 14, 68–85. [Google Scholar] [CrossRef] [PubMed]
- Distler, U.; Kuharev, J.; Navarro, P.; Tenzer, S. Label-Free Quantification in Ion Mobility–Enhanced Data-Independent Acquisition Proteomics. Nat. Protoc. 2016, 11, 795–812. [Google Scholar] [CrossRef] [PubMed]
- Thul, P.J.; Åkesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A Subcellular Map of the Human Proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bărar, A.A.; Pralea, I.-E.; Maslyennikov, Y.; Munteanu, R.; Berindan-Neagoe, I.; Pîrlog, R.; Rusu, I.; Nuțu, A.; Rusu, C.C.; Moldovan, D.T.; et al. Minimal Change Disease: Pathogenetic Insights from Glomerular Proteomics. Int. J. Mol. Sci. 2024, 25, 5613. https://doi.org/10.3390/ijms25115613
Bărar AA, Pralea I-E, Maslyennikov Y, Munteanu R, Berindan-Neagoe I, Pîrlog R, Rusu I, Nuțu A, Rusu CC, Moldovan DT, et al. Minimal Change Disease: Pathogenetic Insights from Glomerular Proteomics. International Journal of Molecular Sciences. 2024; 25(11):5613. https://doi.org/10.3390/ijms25115613
Chicago/Turabian StyleBărar, Andrada Alina, Ioana-Ecaterina Pralea, Yuriy Maslyennikov, Raluca Munteanu, Ioana Berindan-Neagoe, Radu Pîrlog, Ioana Rusu, Andreea Nuțu, Crina Claudia Rusu, Diana Tania Moldovan, and et al. 2024. "Minimal Change Disease: Pathogenetic Insights from Glomerular Proteomics" International Journal of Molecular Sciences 25, no. 11: 5613. https://doi.org/10.3390/ijms25115613
APA StyleBărar, A. A., Pralea, I.-E., Maslyennikov, Y., Munteanu, R., Berindan-Neagoe, I., Pîrlog, R., Rusu, I., Nuțu, A., Rusu, C. C., Moldovan, D. T., Potra, A. R., Tirinescu, D., Ticala, M., Elec, F. I., Iuga, C. A., & Kacso, I. M. (2024). Minimal Change Disease: Pathogenetic Insights from Glomerular Proteomics. International Journal of Molecular Sciences, 25(11), 5613. https://doi.org/10.3390/ijms25115613