
Multi-Omics Analysis of a Chromosome Segment Substitution Line Reveals a New Regulation Network for Soybean Seed Storage Profile

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Analysis
2.1. Selection of a CSSL with High Protein Content and Low FA Content
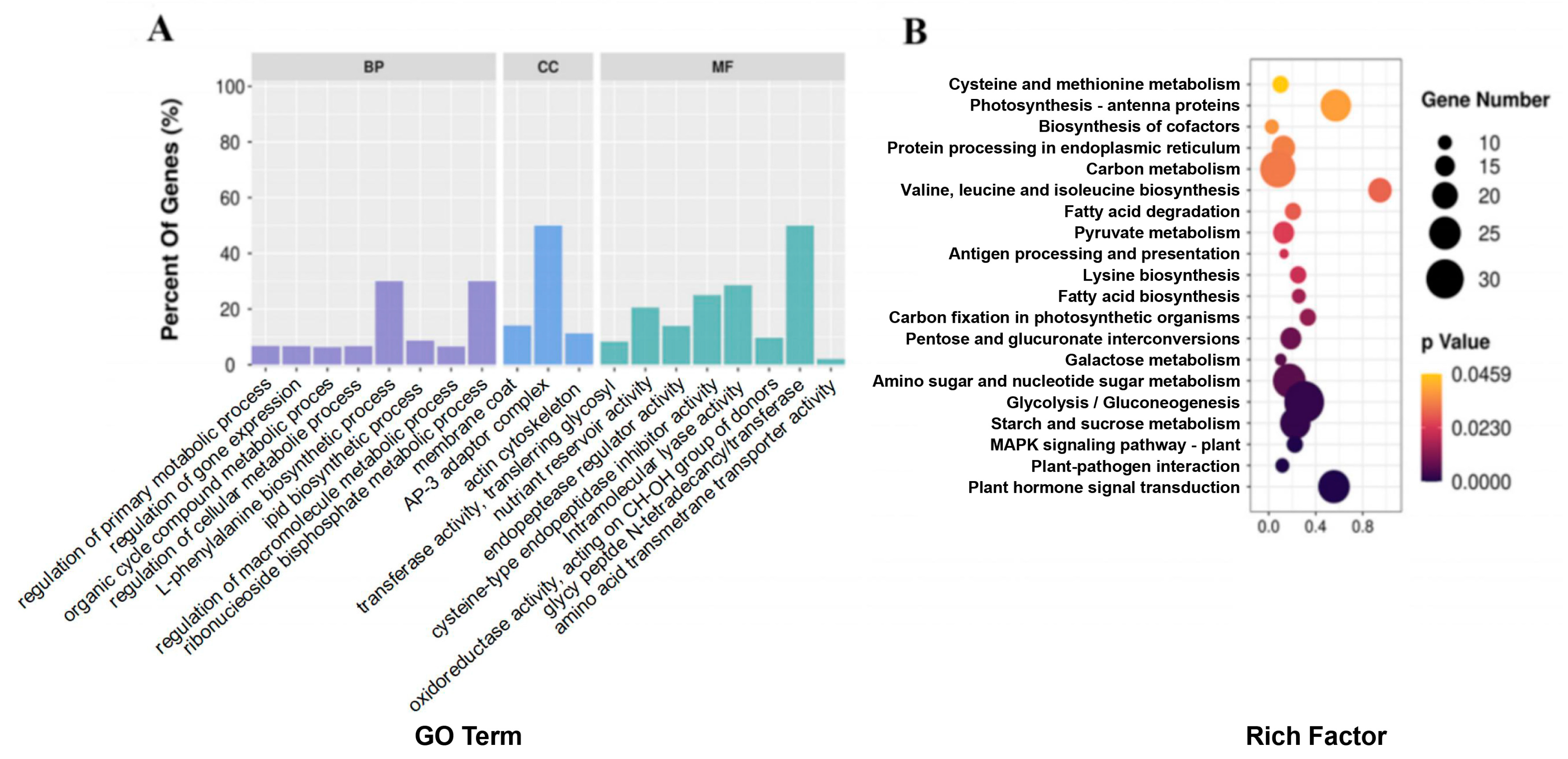
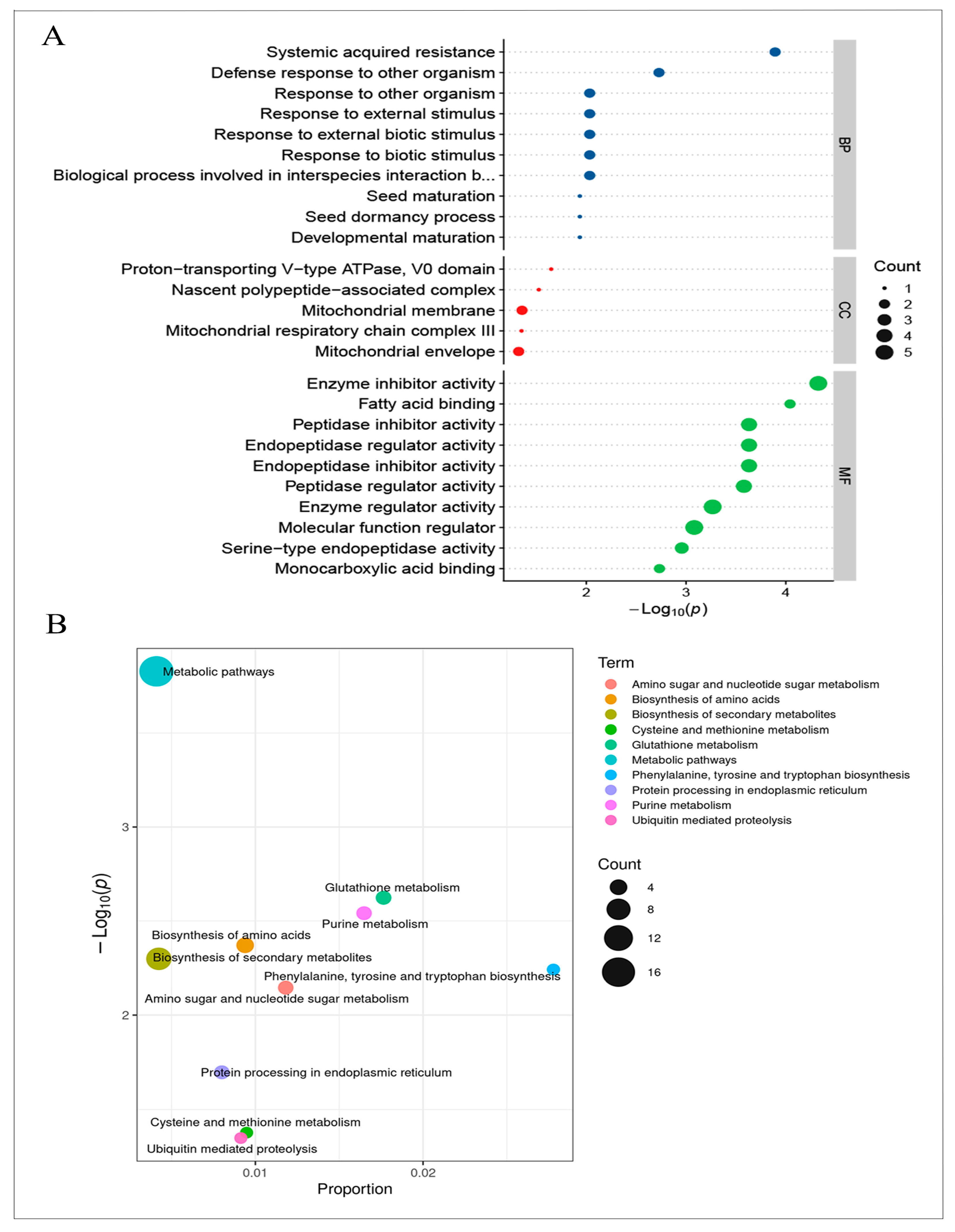
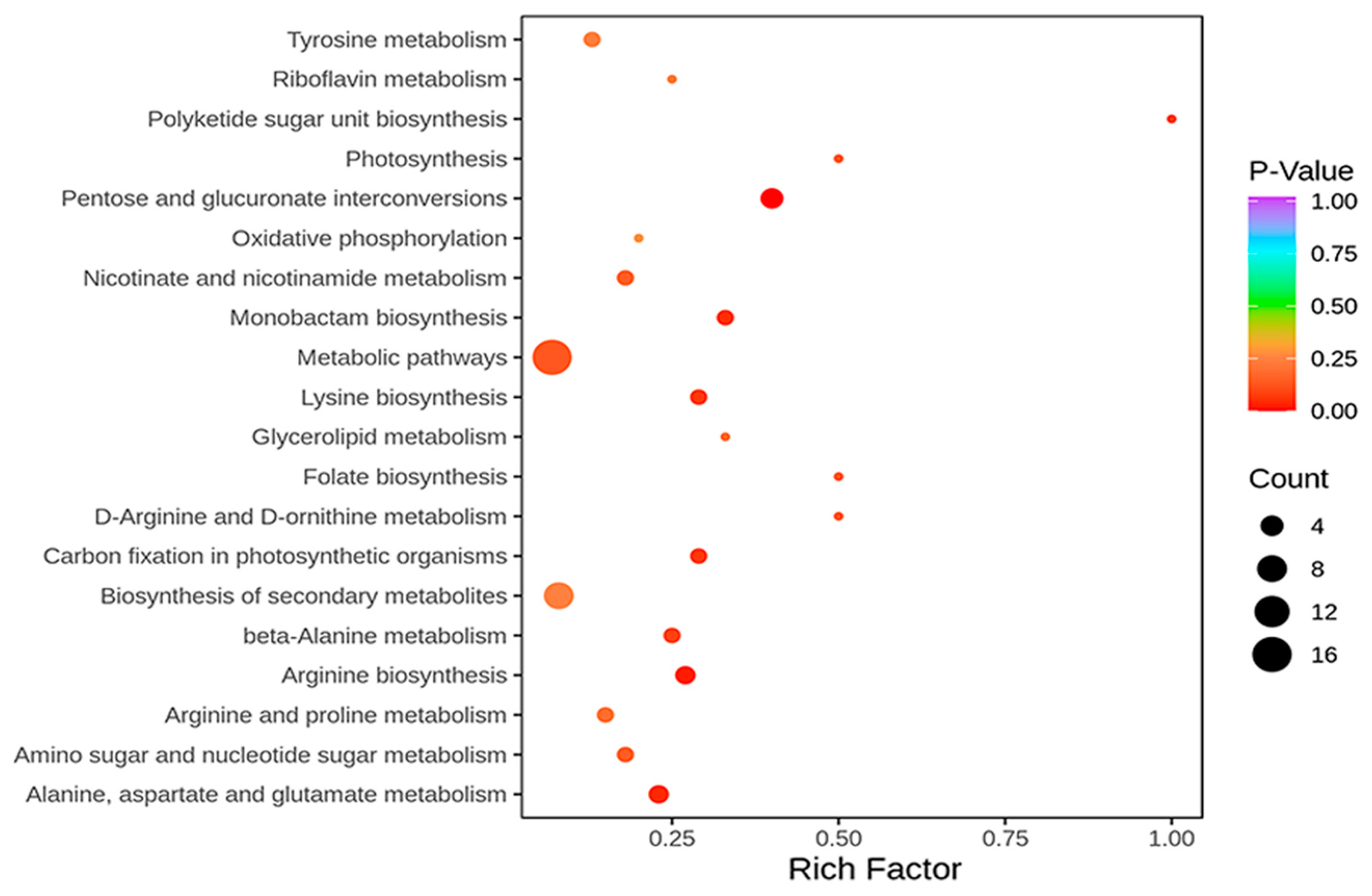
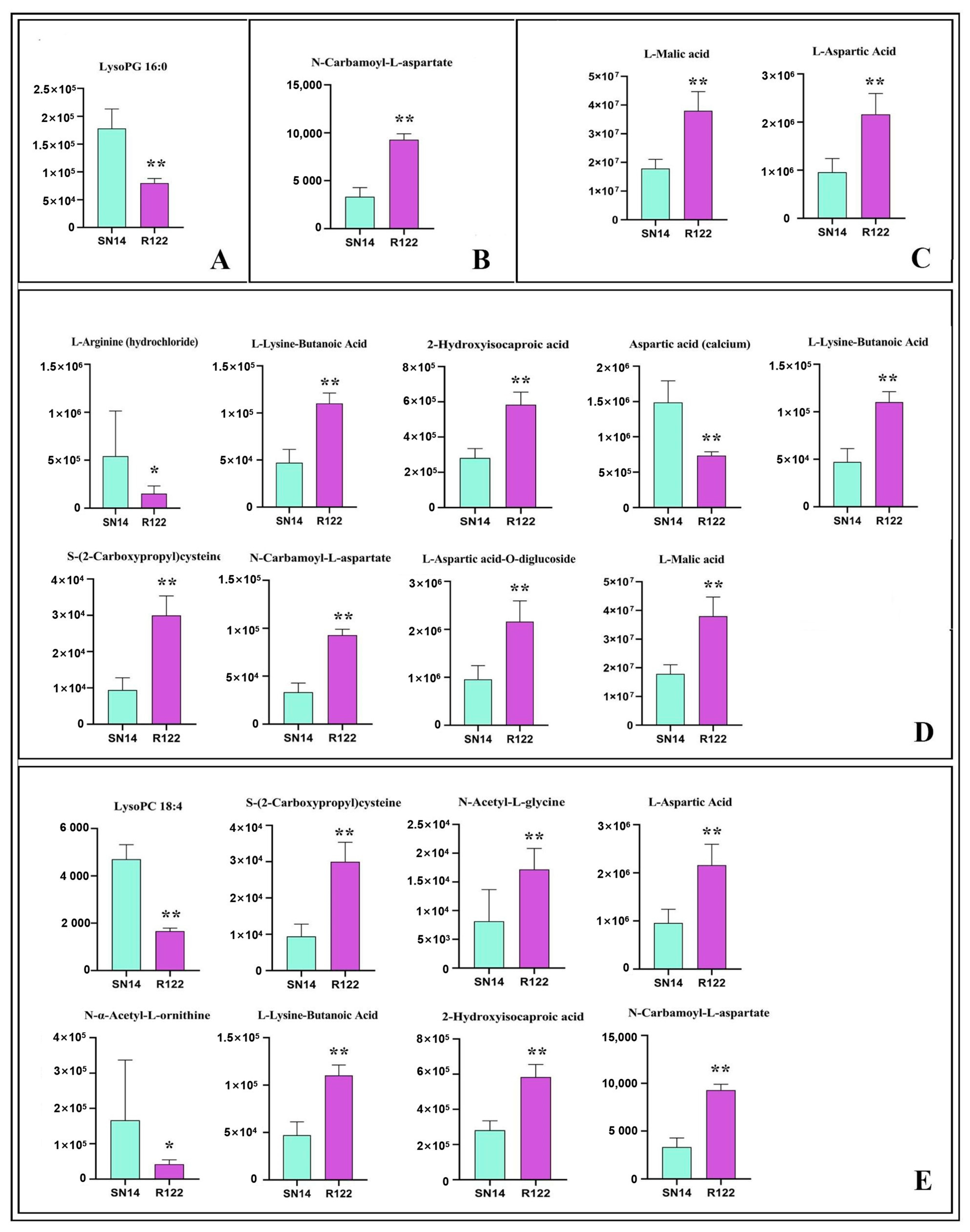
2.2. RNA-Seq, Proteomics, and Metabolomics Analyses of R122 and SN14
2.3. Integrated Analysis of Transcriptomics, Proteomics, and Metabolomics
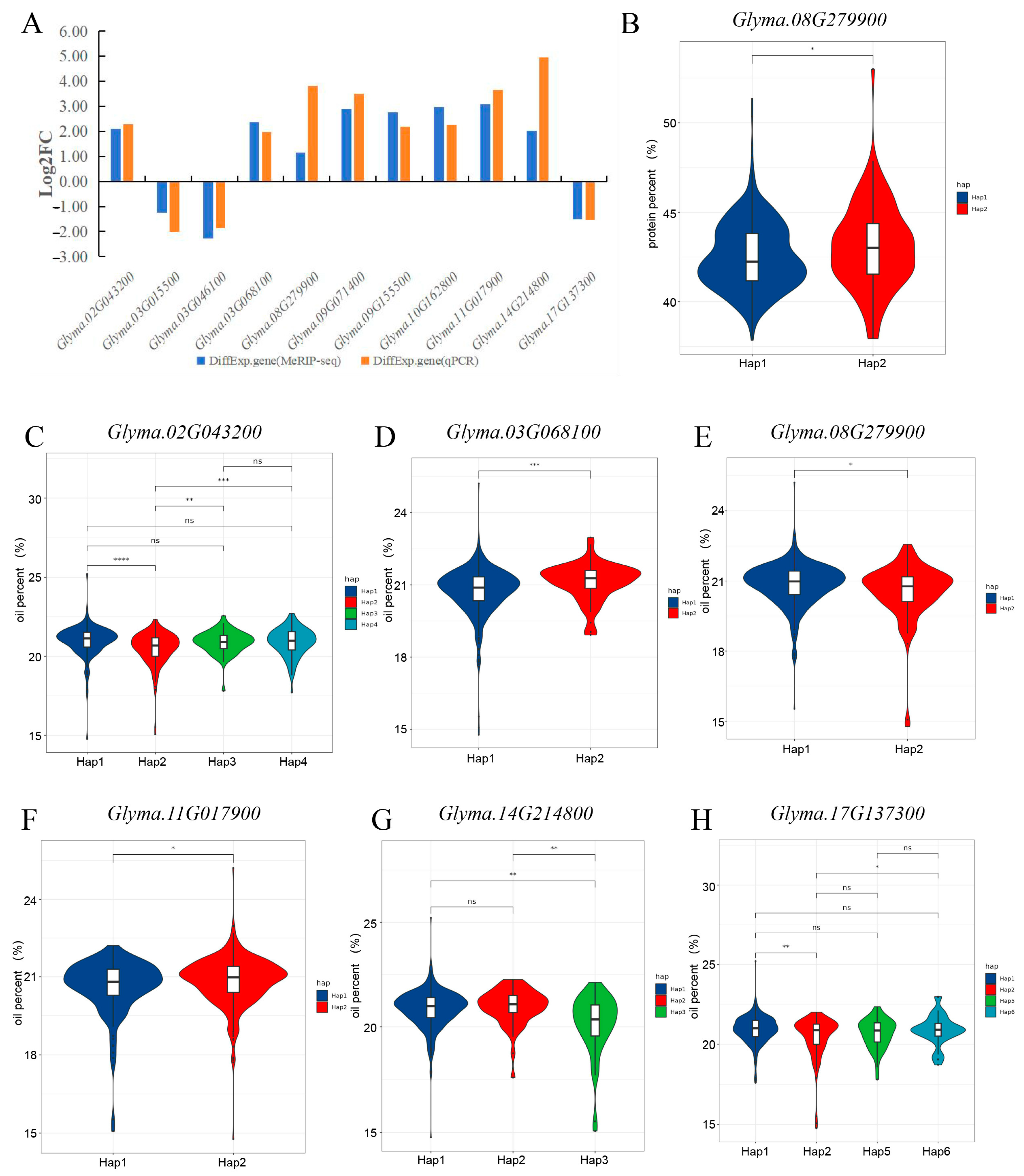
2.4. Haplotype Analyses of Candidate Gene
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Fatty Acid Profile and Seed Storage Protein Content Analyses
4.3. RNA-Seq Analysis
4.4. Proteomics Analysis
4.5. Metabolomics Analysis
4.6. qRT−PCR Validation of Candidate Genes
4.7. Statistical Analysis
4.8. Haplotype Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gupta, S.K.; Manjaya, J.G. Advances in improvement of soybean seed composition traits using genetic, genomic and biotechnological approaches. Euphytica 2022, 218, 99. [Google Scholar] [CrossRef]
- Zhang, H.; Goettel, W.; Song, Q.; Jiang, H.; Hu, Z.; Wang, M.L.; An, Y.C. Selection of GmSWEET39 for oil and protein improvement in soybean. PLoS Genet. 2020, 16, e1009114. [Google Scholar] [CrossRef] [PubMed]
- Abedi, E.; Sahari, M.A. Long-chain polyunsaturated fatty acid sources and evaluation of their nutritional and functional properties. Food Sci. Nutr. 2014, 2, 443–463. [Google Scholar] [CrossRef] [PubMed]
- Jokić, S.; SudaR, R.; Svilović, S.; Vidovic, S.S.; Bilić, M.; Velić, D.; Jurković, V. Fatty acid composition of oil obtained from soybeans by extraction with supercritical carbon dioxide. Czech J. Food Sci. 2013, 31, 116–125. [Google Scholar] [CrossRef]
- Yao, Y.; You, Q.; Duan, G.; Ren, J.; Chu, S.; Zhao, J.; Li, X.; Zhou, X.; Jiao, Y. Quantitative trait loci analysis of seed oil content and composition of wild and cultivated soybean. BMC Plant Biol. 2020, 20, 51. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.F. Soybeans: Improvement, production, and uses. Seed Compos. 2004, 16, 621–677. [Google Scholar]
- Lu, L.; Wei, W.; Li, Q.T.; Bian, X.H.; Lu, X.; Hu, Y.; Cheng, T.; Wang, Z.Y.; Jin, M.; Tao, J.J.; et al. A transcriptional regulatory module controls lipid accumulation in soybean. New Phytol. 2021, 231, 661–678. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Cao, P.; Cui, Y.; Liu, D.; Li, J.; Zhao, Y.; Yang, S.; Zhang, B.; Zhou, R.; Sun, M.; et al. Enhanced production of seed oil with improved fatty acid composition by overexpressing NAD+-dependent glycerol-3-phosphate dehydrogenase in soybean. J. Integr. Plant Biol. 2021, 63, 1036–1053. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Shanklin, J. Triacylglycerol metabolism, function, and accumulation in plant vegetative tissues. Annu. Rev. Plant Biol. 2016, 67, 179–206. [Google Scholar] [CrossRef]
- Taurino, M.; Costantini, S.; De Domenico, S.; Stefanelli, F.; Ruano, G.; Delgadillo, M.O.; Sánchez-Serrano, J.J.; Sanmartín, M.; Santino, A.; Rojo, E.; et al. SEIPIN proteins mediate lipid droplet biogenesis to promote pollen transmission and reduce seed dormancy. Plant Physiol. 2018, 176, 1531–1546. [Google Scholar] [CrossRef]
- Jun, W.; Lin, L.; Yong, G.; Wang, Y.H.; Zhang, L.; Jin, L.G.; Guan, R.X.; Liu, Z.X.; Wang, L.L.; Chang, R.Z.; et al. A dominant locus, qBSC-1, controls β subunit content of seed storage protein in soybean (Glycine max (L.) Merri.). J. Integr. Agric. 2014, 13, 1854–1864. [Google Scholar]
- Magni, C.; Sessa, F.; Capraro, J.; Duranti, M.; Maffioli, E.; Scarafoni, A. Structural and functional insights into the basic globulin 7S of soybean seeds by using trypsin as a molecular probe. Biochem. Biophys. Res. Commun. 2018, 496, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Niu, Y.; Zhang, G.; Feng, Z.; Bo, Y.; Lian, J.; Wang, B.; Gong, Y. Genome sequencing and population resequencing provide insights into the genetic basis of domestication and diversity of vegetable soybean. Hortic. Res. 2022, 9, uhab052. [Google Scholar] [CrossRef] [PubMed]
- Wan, T.F.; Shao, G.H.; Shan, X.C.; Zeng, N.Y.; Lam, H.M. Correlation between AS1 gene expression and seed protein contents in different soybean (Glycine max [L.] Merr.) cultivars. Plant Biol. 2006, 8, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Nguyen, V.; Liu, J.; Fu, W.; Chen, C.; Yu, K.; Cui, Y. Mutagenesis of seed storage protein genes in Soybean using CRISPR/Cas9. BMC Res. Notes 2019, 12, 176. [Google Scholar] [CrossRef]
- Li, L.; Wurtele, E.S. The QQS orphan gene of Arabidopsis modulates carbon and nitrogen allocation in soybean. Plant Biotechnol. J. 2015, 13, 177–187. [Google Scholar] [CrossRef]
- O’Conner, S.; Zheng, W.; Qi, M.; Kandel, Y.; Fuller, R.; Whitham, S.A.; Li, L. GmNF-YC4-2 increases protein, exhibits broad disease resistance and expedites maturity in soybean. Int. J. Mol. Sci. 2021, 22, 3586. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Xu, M.; Zhang, H.; He, D.; Kong, Y.; Chen, L.; Song, H. Transcriptome and proteome analyses of the molecular mechanisms associated with coix seed nutritional quality in the process of breeding. Food Chem. 2019, 272, 549–558. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, W.; Zhou, Y.; Ji, P.; Wan, Y.; Shi, S.; Yang, Z.; Gong, Y.; Mu, F.; Chen, S. Integrative analysis of metabolome and transcriptome reveals the improvements of seed quality in vegetable soybean (Glycine max (L.) Merr.). Phytochemistry 2022, 200, 113216. [Google Scholar] [CrossRef]
- Sharmin, R.A.; Bhuiyan, M.R.; Lv, W.; Yu, Z.; Chang, F.; Kong, J.; Bhat, J.A.; Zhao, T. RNA-Seq based transcriptomic analysis revealed genes associated with seed-flooding tolerance in wild soybean (Glycine soja Sieb. & Zucc.). Environ. Exp. Bot. 2020, 171, 103906. [Google Scholar]
- Wang, X.; Song, S.; Wang, X.; Liu, J.; Dong, S. Transcriptomic and metabolomic analysis of seedling-stage soybean responses to PEG-simulated drought stress. Int. J. Mol. Sci. 2022, 23, 6869. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hou, H.; Zhang, D.; Zhu, B. Transcriptomic and Metabolomic Analysis of Soybean Nodule Number Improvements with the Use of Water-Soluble Humic Materials. J. Agric. Food Chem. 2022, 71, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, D.; Surapaneni, M.; Mesapogu, S.; Neelamraju, S. Development and use of chromosome segment substitution lines as a genetic resource for crop improvement. Theor. Appl. Genet. 2019, 132, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; He, Q.; Yang, H.; Xiang, S.; Zhao, T.; Gai, J. Development of a chromosome segment substitution line population with wild soybean (Glycine soja Sieb. et Zucc.) as donor parent. Euphytica 2013, 189, 293–307. [Google Scholar] [CrossRef]
- Yang, H.; Wang, W.; He, Q.; Xiang, S.; Tian, D.; Zhao, T.; Gai, J. Identifying a wild allele conferring small seed size, high protein content and low oil content using chromosome segment substitution lines in soybean. Theor. Appl. Genet. 2019, 132, 2793–2807. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.N.; Zhang, Z.G.; Kang, Q.L.; Yu, S.; Wang, J.Q.; Chen, L.; Liu, Y.R.; Ma, C.; Zhu, R.S.; Zhu, Y.X.; et al. Characterization of chromosome segment substitution lines reveals candidate genes associated with the nodule number in soybean. J. Integr. Agric. 2022, 21, 2197–2210. [Google Scholar] [CrossRef]
- PAN, W.J.; Xue, H.; HUANG, S.Y.; Yu, J.Y.; Zhao, Y.; Qu, K.; Zhang, Z.X.; Yin, Z.G.; Qi, H.D.; Yu, G.L.; et al. Identification of candidate genes related to soluble sugar contents in soybean seeds using multiple genetic analyses. J. Integr. Agric. 2022, 21, 1886–1902. [Google Scholar] [CrossRef]
- Xin, D.; Qi, Z.; Jiang, H.; Hu, Z.; Zhu, R.; Hu, J.; Han, H.; Hu, G.; Liu, C.; Chen, Q. QTL location and epistatic effect analysis of 100-seed weight using wild soybean (Glycine soja Sieb. & Zucc.) chromosome segment substitution lines. PLoS ONE 2016, 11, e0149380. [Google Scholar]
- Han, X.; Shangguan, J.; Wang, Z.; Li, Y.; Fan, J.; Ren, A.; Zhao, M. Spermidine regulates mitochondrial function by enhancing eIF5A hypusination and contributes to reactive oxygen species production and ganoderic acid biosynthesis in Ganoderma lucidum. Appl. Environ. Microbiol. 2022, 88, e02037-21. [Google Scholar] [CrossRef]
- Gao, C.; Yu, C.K.; Qu, S.; San, M.W.; Li, K.Y.; Lo, S.W.; Jiang, L.W. The Golgi-localized Arabidopsis endomembrane protein12 contains both endoplasmic reticulum export and Golgi retention signals at its C terminus. Plant Cell 2012, 24, 2086–2104. [Google Scholar] [CrossRef]
- Stryiński, R.; Mateos, J.; Pascual, S.; Gonzalez, A.F.; Gallardo, J.M.; Łopieńska-Biernat, E.; Medina, I.; Carrera, M. Proteome profiling of L3 and L4 Anisakis simplex development stages by TMT-based quantitative proteomics. J. Proteom. 2019, 201, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, H.; Li, Y.; Zhang, Q.; Liu, Q.; Zhai, H.; Zhao, N.; Yang, Y.; He, S. Plastidial Phosphoglucomutase (pPGM) Overexpression Increases the Starch Content of Transgenic Sweet Potato Storage Roots. Genes 2022, 13, 2234. [Google Scholar] [CrossRef] [PubMed]
- Bonaventure, G.; Salas, J.J.; Pollard, M.R.; Ohlrogge, J.B. Disruption of the FATB gene in Arabidopsis demonstrates an essential role of saturated fatty acids in plant growth. Plant Cell 2003, 15, 1020–1033. [Google Scholar] [CrossRef] [PubMed]
- Rueckert, D.G.; Schmidt, K. Lipid transfer proteins. Chem. Phys. Lipids 1990, 56, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Jawahir, V.; Zolman, B.K. Long chain acyl CoA synthetase 4 catalyzes the first step in peroxisomal indole-3-butyric acid to IAA conversion. Plant Physiol. 2021, 185, 120–136. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Tan, Y.; Zhao, P.; Ren, Z. Seipin: A key factor for nuclear lipid droplet generation and lipid homeostasis. Int. J. Mol. Sci. 2020, 21, 8208. [Google Scholar] [CrossRef] [PubMed]
- Islam, N.; Krishnan, H.B.; Natarajan, S. Proteomic profiling of fast neutron-induced soybean mutant unveiled pathways associated with increased seed protein content. J. Proteome Res. 2020, 19, 3936–3944. [Google Scholar] [CrossRef] [PubMed]
- Kawakatsu, T.; Takaiwa, F. Cereal seed storage protein synthesis: Fundamental processes for recombinant protein production in cereal grains. Plant Biotechnol. J. 2010, 8, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.; Powell, A.F.; Mirzaei, M.; Wang, L.J.; Movahed, N.; Miller, J.K.; Piñeros, M.A.; Jander, G. Indole-3-glycerolphosphate synthase, a branchpoint for the biosynthesis of tryptophan, indole, and benzoxazinoids in maize. Plant J. 2021, 106, 245–257. [Google Scholar] [CrossRef]
- Qi, Z.; Guo, C.; Li, H.; Qiu, H.; Li, H.; Jong, C.; Yu, G.; Zhang, Y.; Hu, L.; Wu, X.; et al. Natural variation in Fatty Acid 9 is a determinant of fatty acid and protein content. Plant Biotechnol. J. 2024, 22, 759–773. [Google Scholar] [CrossRef]
- Qin, P.; Wang, T.; Luo, Y. A review on plant-based proteins from soybean: Health benefits and soy product development. J. Agric. Food Res. 2022, 7, 100265. [Google Scholar] [CrossRef]
- Liu, P.; Luo, J.; Zheng, Q.; Chen, Q.; Zhai, N.; Xu, S.; Xu, Y.; Jin, L.; Xu, G.; Lu, X.; et al. Integrating transcriptome and metabolome reveals molecular networks involved in genetic and environmental variation in tobacco. DNA Res. 2020, 27, dsaa006. [Google Scholar] [CrossRef]
- Kandoi, D.; Ruhil, K.; Govindjee, G.; Tripathy, B.C. Overexpression of cytoplasmic C4 Flaveria bidentis carbonic anhydrase in C3 Arabidopsis thaliana increases amino acids, photosynthetic potential, and biomass. Plant Biotechnol. J. 2022, 20, 1518–1532. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Sakoda, K.; Fukayama, H.; Kondo, E.; Suzuki, Y.; Makino, A.; Terashima, I.; Yamori, W. Overexpression of both Rubisco and Rubisco activase rescues rice photosynthesis and biomass under heat stress. Plant Cell Environ. 2021, 44, 2308–2320. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Zhang, Z.; Wang, Z.; Yu, J.; Qin, H.; Mao, X.; Jiang, H.; Xin, D.; Yin, Z.; Zhu, R.; et al. Meta-analysis and transcriptome profiling reveal hub genes for soybean seed storage composition during seed development. Plant Cell Environ. 2018, 41, 2109–2127. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.X.; Li, Q.T.; Liu, Y.F.; Zhang, F.X.; Ma, B.; Zhang, W.K.; Man, W.Q.; Du, W.G.; Wang, G.D.; Chen, S.Y.; et al. Soybean GmbZIP123 gene enhances lipid content in the seeds of transgenic Arabidopsis plants. J. Exp. Bot. 2013, 64, 4329–4341. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Rickert, D.A.; Deak, N.A.; Aldin, E.D.; Recknor, J.J.; Johnson, L.A.; Murphy, P.A. Comparison of Kjeldahl and Dumas methods for determining protein contents of soybean products. J. Am. Oil Chem. Soc. 2003, 80, 1169–1173. [Google Scholar] [CrossRef]
- Srivastava, N.; Rathour, R.; Jha, S.; Pandey, K.; Srivastava, M.; Thakur, V.K.; Sengar, R.S.; Gupta, V.K.; Mazumder, P.B.; Khan, A.F.; et al. Microbial beta glucosidase enzymes: Recent advances in biomass conversation for biofuels application. Biomolecules 2019, 9, 220. [Google Scholar] [CrossRef]
- Li, W.; Wen, L.; Chen, Z.; Zhang, Z.; Pang, X.; Deng, Z.; Liu, T.; Guo, Y. Study on metabolic variation in whole grains of four proso millet varieties reveals metabolites important for antioxidant properties and quality traits. Food Chem. 2021, 357, 129791. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jong, C.; Yu, Z.; Zhang, Y.; Choe, K.; Uh, S.; Kim, K.; Jong, C.; Cha, J.; Kim, M.; Kim, Y.; et al. Multi-Omics Analysis of a Chromosome Segment Substitution Line Reveals a New Regulation Network for Soybean Seed Storage Profile. Int. J. Mol. Sci. 2024, 25, 5614. https://doi.org/10.3390/ijms25115614
Jong C, Yu Z, Zhang Y, Choe K, Uh S, Kim K, Jong C, Cha J, Kim M, Kim Y, et al. Multi-Omics Analysis of a Chromosome Segment Substitution Line Reveals a New Regulation Network for Soybean Seed Storage Profile. International Journal of Molecular Sciences. 2024; 25(11):5614. https://doi.org/10.3390/ijms25115614
Chicago/Turabian StyleJong, Cholnam, Zhenhai Yu, Yu Zhang, Kyongho Choe, Songrok Uh, Kibong Kim, Chol Jong, Jinmyong Cha, Myongguk Kim, Yunchol Kim, and et al. 2024. "Multi-Omics Analysis of a Chromosome Segment Substitution Line Reveals a New Regulation Network for Soybean Seed Storage Profile" International Journal of Molecular Sciences 25, no. 11: 5614. https://doi.org/10.3390/ijms25115614
APA StyleJong, C., Yu, Z., Zhang, Y., Choe, K., Uh, S., Kim, K., Jong, C., Cha, J., Kim, M., Kim, Y., Han, X., Yang, M., Xu, C., Hu, L., Chen, Q., Liu, C., & Qi, Z. (2024). Multi-Omics Analysis of a Chromosome Segment Substitution Line Reveals a New Regulation Network for Soybean Seed Storage Profile. International Journal of Molecular Sciences, 25(11), 5614. https://doi.org/10.3390/ijms25115614