
Adenosine and Its Receptors in the Pathogenesis and Treatment of Inflammatory Skin Diseases


Abstract
1. Introduction
2. Background of Adenosine and Its Receptor
2.1. Biochemistry of Adenosine: Source, Regulation, and Uptake
2.2. Overview of Adenosine Receptors (A1, A2A, A2B, and A3) and Their General Functions
3. Molecular Mechanisms of Adenosine
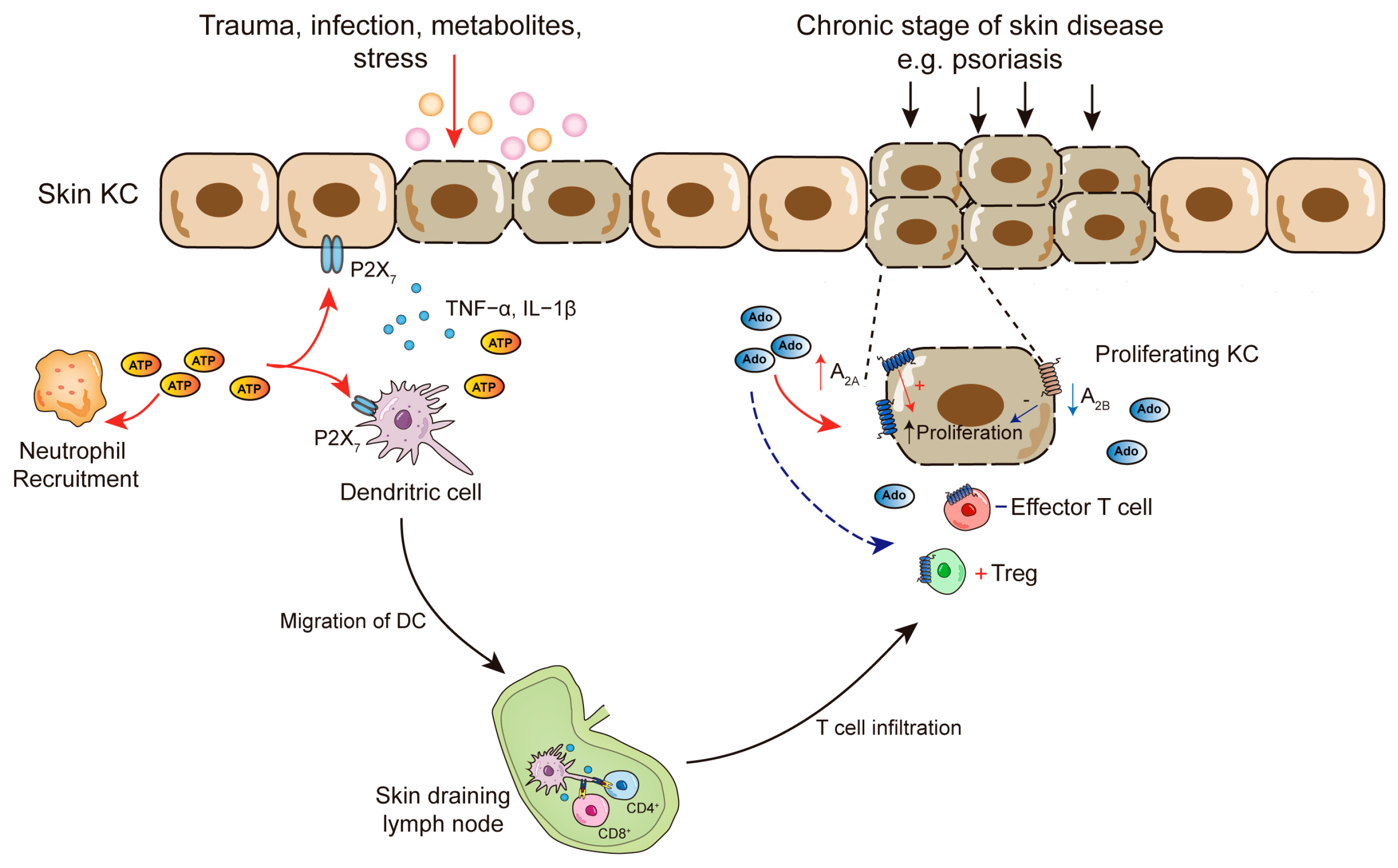
3.1. Adenosine-Induced Actions in Skin Cells
3.1.1. Adenosine in Keratinocytes and Fibroblasts
3.1.2. Adenosine in Melanocytes
3.1.3. Adenosine in Innate Immune Cells
3.1.4. Adenosine in T Cells
3.2. Interaction with Other Inflammatory Mediators and Pathways
4. Adenosine in Skin Pathology
4.1. Psoriasis
4.2. Skin Inflammation and Anergy
5. Clinical Evidence and Studies
5.1. P2 Receptors
5.2. A1 Adenosine Receptors
5.3. A2A Adenosine Receptors
5.4. A2B Adenosine Receptors
5.5. A3 Adenosine Receptors
6. Overview of Current Treatments Interacting with Adenosine Pathways
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Drury, A.N.; Szent-Györgyi, A. The Physiological Activity of Adenine Compounds with Especial Reference to Their Action upon the Mammalian Heart. J. Physiol. 1929, 68, 213–237. [Google Scholar] [CrossRef] [PubMed]
- Newby, A.C.; Holmquist, C.A.; Illingworth, J.; Pearson, J.D. The Control of Adenosine Concentration in Polymorphonuclear Leucocytes, Cultured Heart Cells and Isolated Perfused Heart from the Rat. Biochem. J. 1983, 214, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Bajgar, A.; Kucerova, K.; Jonatova, L.; Tomcala, A.; Schneedorferova, I.; Okrouhlik, J.; Dolezal, T. Extracellular Adenosine Mediates a Systemic Metabolic Switch during Immune Response. PLoS Biol. 2015, 13, e1002135. [Google Scholar] [CrossRef] [PubMed]
- Angello, D.A.; Berne, R.M.; Coddington, N.M. Adenosine and Insulin Mediate Glucose Uptake in Normoxic Rat Hearts by Different Mechanisms. Am. J. Physiol. 1993, 265, H880–H885. [Google Scholar] [CrossRef] [PubMed]
- Mainwaring, R.D.; Mentzer, R.M. Effects of Dipyridamole on Myocardial Glucose Uptake in the Newborn Lamb. J. Surg. Res. 1986, 40, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Cronstein, B.N.; Kramer, S.B.; Weissmann, G.; Hirschhorn, R. Adenosine: A Physiological Modulator of Superoxide Anion Generation by Human Neutrophils. J. Exp. Med. 1983, 158, 1160–1177. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.V.; Suman, S.; Goruganthu, M.U.L.; Tchekneva, E.E.; Guan, S.; Arasada, R.R.; Antonucci, A.; Piao, L.; Ilgisonis, I.; Bobko, A.A.; et al. Improving Combination Therapies: Targeting A2B-Adenosine Receptor to Modulate Metabolic Tumor Microenvironment and Immunosuppression. J. Natl. Cancer Inst. 2023, 115, 1404–1419. [Google Scholar] [CrossRef] [PubMed]
- Cronstein, B.; Sitkovsky, M. Adenosine and Adenosine Receptors in the Pathogenesis and Treatment of Rheumatic Diseases. Nat. Rev. Rheumatol. 2017, 13, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-H.; Chen, T.; Zhang, X.; Ma, X.-L.; Shi, H.-S. Small Molecule Inhibitors Targeting the Cancers. MedComm 2022, 3, e181. [Google Scholar] [CrossRef]
- IJzerman, A.P.; Jacobson, K.A.; Müller, C.E.; Cronstein, B.N.; Cunha, R.A. International Union of Basic and Clinical Pharmacology. CXII: Adenosine Receptors: A Further Update. Pharmacol. Rev. 2022, 74, 340–372. [Google Scholar] [CrossRef]
- Gammelgaard, O.L.; Terp, M.G.; Renn, C.; Labrijn, A.F.; Hamaker, O.; Nielsen, A.Y.; Vever, H.; Hansen, S.W.; Gjerstorff, M.F.; Müller, C.E.; et al. Targeting Two Distinct Epitopes on Human CD73 with a Bispecific Antibody Improves Anticancer Activity. J. Immunother. Cancer 2022, 10, e004554. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Knight, G.; Greig, A. Purinergic Signaling in Healthy and Diseased Skin. J. Investig. Dermatol. 2012, 132, 526–546. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; Burnstock, G.; Kennedy, C.; King, B.F.; North, R.A.; Séguéla, P.; Voigt, M.; Humphrey, P.P. International Union of Pharmacology. XXIV. Current Status of the Nomenclature and Properties of P2X Receptors and Their Subunits. Pharmacol. Rev. 2001, 53, 107–118. [Google Scholar] [PubMed]
- Burnstock, G. Purine and Pyrimidine Receptors. Cell. Mol. Life Sci. CMLS 2007, 64, 1471–1483. [Google Scholar] [CrossRef]
- Möser, G.H.; Schrader, J.; Deussen, A. Turnover of Adenosine in Plasma of Human and Dog Blood. Am. J. Physiol. 1989, 256, C799–C806. [Google Scholar] [CrossRef] [PubMed]
- Vekaria, R.M.; Unwin, R.J.; Shirley, D.G. Intraluminal ATP Concentrations in Rat Renal Tubules. J. Am. Soc. Nephrol. JASN 2006, 17, 1841–1847. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, S.A.; Beal, P.R.; Yao, S.Y.M.; King, A.E.; Cass, C.E.; Young, J.D. The Equilibrative Nucleoside Transporter Family, SLC29. Pflug. Arch. 2004, 447, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Haskó, G.; Linden, J.; Cronstein, B.; Pacher, P. Adenosine Receptors: Therapeutic Aspects for Inflammatory and Immune Diseases. Nat. Rev. Drug Discov. 2008, 7, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; Chern, Y.; Franco, R.; Sitkovsky, M. Aspects of the General Biology of Adenosine A2A Signaling. Prog. Neurobiol. 2007, 83, 263–276. [Google Scholar] [CrossRef]
- Borea, P.; Varani, K.; Vincenzi, F.; Baraldi, P.; Tabrizi, M.; Merighi, S.; Gessi, S. The A3 Adenosine Receptor: History and Perspectives. Pharmacol. Rev. 2015, 67, 74–102. [Google Scholar] [CrossRef]
- Lee, H.-S.; Chung, H.-J.; Lee, H.W.; Jeong, L.S.; Lee, S.K. Suppression of Inflammation Response by a Novel A₃ Adenosine Receptor Agonist Thio-Cl-IB-MECA through Inhibition of Akt and NF-κB Signaling. Immunobiology 2011, 216, 997–1003. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, Y.; Cui, L.; Shi, Y.; Guo, C. Advances in the Pathogenesis of Psoriasis: From Keratinocyte Perspective. Cell Death Dis. 2022, 13, 81. [Google Scholar] [CrossRef]
- Andrés, R.M.; Terencio, M.C.; Arasa, J.; Payá, M.; Valcuende-Cavero, F.; Navalón, P.; Montesinos, M.C. Adenosine A2A and A2B Receptors Differentially Modulate Keratinocyte Proliferation: Possible Deregulation in Psoriatic Epidermis. J. Investig. Dermatol. 2017, 137, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Borea, P.; Varani, K.; Gessi, S. Deregulation of Adenosine Receptors in Psoriatic Epidermis: An Option for Therapeutic Treatment. J. Investig. Dermatol. 2017, 137, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Braun, M.; Lelieur, K.; Kietzmann, M. Purinergic Substances Promote Murine Keratinocyte Proliferation and Enhance Impaired Wound Healing in Mice. Wound Repair Regen. 2006, 14, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Mirandola, P.; Milani, D.; Varani, K.; Gessi, S.; Klotz, K.-N.; Leung, E.; Baraldi, P.G.; Borea, P.A. Adenosine Receptors as Mediators of Both Cell Proliferation and Cell Death of Cultured Human Melanoma Cells. J. Investig. Dermatol. 2002, 119, 923–933. [Google Scholar] [CrossRef]
- Shaikh, G.; Cronstein, B. Signaling Pathways Involving Adenosine A2A and A2B Receptors in Wound Healing and Fibrosis. Purinergic Signal. 2016, 12, 191–197. [Google Scholar] [CrossRef]
- Karmouty-Quintana, H.; Molina, J.G.; Philip, K.; Bellocchi, C.; Gudenkauf, B.; Wu, M.; Chen, N.-Y.; Collum, S.D.; Ko, J.; Agarwal, S.K.; et al. The Antifibrotic Effect of A2B Adenosine Receptor Antagonism in a Mouse Model of Dermal Fibrosis. Arthritis Rheumatol. 2018, 70, 1673–1684. [Google Scholar] [CrossRef] [PubMed]
- Uong, A.; Zon, L.I. Melanocytes in Development and Cancer. J. Cell. Physiol. 2010, 222, 38–41. [Google Scholar] [CrossRef]
- Yang, F.; Brown, T.L.; Cornelius, J.; Babcock, G.F.; Das, P.K.; Boissy, R.E.; Le Poole, I.C. Altered Gene Expression in Melanocytes Exposed to 4-Tertiary Butyl Phenol (4-TBP): Upregulation of the A2b Adenosine Receptor1. J. Investig. Dermatol. 1999, 113, 725–731. [Google Scholar] [CrossRef]
- Rodríguez, C.I.; Setaluri, V. Cyclic AMP (cAMP) Signaling in Melanocytes and Melanoma. Arch. Biochem. Biophys. 2014, 563, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Bastiaens, M.T.; ter Huurne, J.A.C.; Kielich, C.; Gruis, N.A.; Westendorp, R.G.J.; Vermeer, B.J.; Bavinck, J.N.B. Melanocortin-1 Receptor Gene Variants Determine the Risk of Nonmelanoma Skin Cancer Independently of Fair Skin and Red Hair. Am. J. Hum. Genet. 2001, 68, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Nasti, T.H.; Timares, L. MC1R, Eumelanin and Pheomelanin: Their Role in Determining the Susceptibility to Skin Cancer. Photochem. Photobiol. 2015, 91, 188–200. [Google Scholar] [CrossRef]
- Mitra, D.; Luo, X.; Morgan, A.; Wang, J.; Hoang, M.P.; Lo, J.; Guerrero, C.R.; Lennerz, J.K.; Mihm, M.C.; Wargo, J.A.; et al. An Ultraviolet-Radiation-Independent Pathway to Melanoma Carcinogenesis in the Red Hair/Fair Skin Background. Nature 2012, 491, 449–453. [Google Scholar] [CrossRef]
- Bang, J.; Zippin, J.H. Cyclic Adenosine Monophosphate (cAMP) Signaling in Melanocyte Pigmentation and Melanomagenesis. Pigment Cell Melanoma Res. 2021, 34, 28–43. [Google Scholar] [CrossRef]
- Merighi, S.; Varani, K.; Gessi, S.; Cattabriga, E.; Iannotta, V.; Ulouglu, C.; Leung, E.; Borea, P.A. Pharmacological and Biochemical Characterization of Adenosine Receptors in the Human Malignant Melanoma A375 Cell Line. Br. J. Pharmacol. 2001, 134, 1215–1226. [Google Scholar] [CrossRef]
- Merighi, S.; Benini, A.; Mirandola, P.; Gessi, S.; Varani, K.; Leung, E.; MacLennan, S.; Baraldi, P.G.; Borea, P.A. A3 Adenosine Receptors Modulate Hypoxia-Inducible Factor-1a Expression in Human A375 Melanoma Cells. Neoplasia 2005, 7, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Lee, H.-E.; Im, M.; Lee, Y.; Kim, C.-D.; Lee, J.-H.; Seo, Y.-J. Effect of Adenosine on Melanogenesis in B16 Cells and Zebrafish. Ann. Dermatol. 2014, 26, 209–213. [Google Scholar] [CrossRef]
- Madi, L.; Rosenberg-Haggen, B.; Nyska, A.; Korenstein, R. Enhancing Pigmentation via Activation of A3 Adenosine Receptors in B16 Melanoma Cells and in Human Skin Explants. Exp. Dermatol. 2013, 22, 74–77. [Google Scholar] [CrossRef]
- Lorthois, I.; Asselineau, D.; Seyler, N.; Pouliot, R. Contribution of In Vivo and Organotypic 3D Models to Understanding the Role of Macrophages and Neutrophils in the Pathogenesis of Psoriasis. Mediat. Inflamm. 2017, 2017, 7215072. [Google Scholar] [CrossRef]
- Bullough, D.A.; Magill, M.J.; Mullane, K.M.; Firestein, G.S. Carbohydrate- and CD18-Dependent Neutrophil Adhesion to Cardiac Myocytes: Effects of Adenosine. Cardiovasc. Res. 1996, 32, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Cronstein, B.N.; Levin, R.I.; Philips, M.; Hirschhorn, R.; Abramson, S.B.; Weissmann, G. Neutrophil Adherence to Endothelium Is Enhanced via Adenosine A1 Receptors and Inhibited via Adenosine A2 Receptors. J. Immunol. 1992, 148, 2201–2206. [Google Scholar] [CrossRef] [PubMed]
- Rose, F.R.; Hirschhorn, R.; Weissmann, G.; Cronstein, B.N. Adenosine Promotes Neutrophil Chemotaxis. J. Exp. Med. 1988, 167, 1186–1194. [Google Scholar] [CrossRef] [PubMed]
- Cronstein, B.N.; Daguma, L.; Nichols, D.; Hutchison, A.J.; Williams, M. The Adenosine/Neutrophil Paradox Resolved: Human Neutrophils Possess Both A1 and A2 Receptors That Promote Chemotaxis and Inhibit O2 Generation, Respectively. J. Clin. Investig. 1990, 85, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Chen, Y.; Hirsh, M.I.; Yip, L.; Junger, W.G. A3 and P2Y2 Receptors Control the Recruitment of Neutrophils to the Lungs in a Mouse Model of Sepsis. Shock 2008, 30, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Corriden, R.; Inoue, Y.; Yip, L.; Hashiguchi, N.; Zinkernagel, A.; Nizet, V.; Insel, P.A.; Junger, W.G. ATP Release Guides Neutrophil Chemotaxis via P2Y2 and A3 Receptors. Science 2006, 314, 1792–1795. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.R.; Robb, C.T.; Perretti, M.; Rossi, A.G. The Role of Neutrophils in Inflammation Resolution. Semin. Immunol. 2016, 28, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Katayama, H. Development of Psoriasis by Continuous Neutrophil Infiltration into the Epidermis. Exp. Dermatol. 2018, 27, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-M.; Jin, H.-Z. Role of Neutrophils in Psoriasis. J. Immunol. Res. 2020, 2020, e3709749. [Google Scholar] [CrossRef]
- Rosa, G.; Mignogna, C. The Histopathology of Psoriasis. Reumatismo 2007, 59 (Suppl. 1), 46–48. [Google Scholar] [CrossRef]
- Antonioli, L.; Fornai, M.; Blandizzi, C.; Pacher, P.; Haskó, G. Adenosine Signaling and the Immune System: When a Lot Could Be Too Much. Immunol. Lett. 2019, 205, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Cooney, K.A.; Shin, E.Y.; Wang, L.; Deppen, J.N.; Ginn, S.C.; Levit, R.D. Adenosine from a Biologic Source Regulates Neutrophil Extracellular Traps (NETs). J. Leukoc. Biol. 2019, 105, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Magni, G.; Ceruti, S. Adenosine Signaling in Autoimmune Disorders. Pharmaceuticals 2020, 13, 260. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Erreni, M.; Allavena, P.; Porta, C. Macrophage Polarization in Pathology. Cell. Mol. Life Sci. CMLS 2015, 72, 4111–4126. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, C.J.; Pinhal-Enfield, G.; Elson, G.; Cronstein, B.N.; Hasko, G.; Outram, S.; Leibovich, S.J. The Adenosine-Dependent Angiogenic Switch of Macrophages to an M2-like Phenotype Is Independent of Interleukin-4 Receptor Alpha (IL-4Rα) Signaling. Inflammation 2013, 36, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, C.; Hossain, F.; Rodriguez, P.C.; Sierra, R.A.; Pannuti, A.; Osborne, B.A.; Minter, L.M.; Miele, L.; Morello, S. Adenosine A2A Receptor Stimulation Inhibits TCR-Induced Notch1 Activation in CD8+T-Cells. Front. Immunol. 2019, 10, 162. [Google Scholar] [CrossRef] [PubMed]
- Mastelic-Gavillet, B.; Navarro Rodrigo, B.; Décombaz, L.; Wang, H.; Ercolano, G.; Ahmed, R.; Lozano, L.E.; Ianaro, A.; Derré, L.; Valerio, M.; et al. Adenosine Mediates Functional and Metabolic Suppression of Peripheral and Tumor-Infiltrating CD8+ T Cells. J. Immunother. Cancer 2019, 7, 257. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Alabdullah, M.; Mahnke, K. Adenosine, Bridging Chronic Inflammation and Tumor Growth. Front. Immunol. 2023, 14, 1258637. [Google Scholar] [CrossRef]
- Leone, R.D.; Emens, L.A. Targeting Adenosine for Cancer Immunotherapy. J. Immunother. Cancer 2018, 6, 57. [Google Scholar] [CrossRef]
- Csóka, B.; Himer, L.; Selmeczy, Z.; Vizi, E.S.; Pacher, P.; Ledent, C.; Deitch, E.A.; Spolarics, Z.; Németh, Z.H.; Haskó, G. Adenosine A2A Receptor Activation Inhibits T Helper 1 and T Helper 2 Cell Development and Effector Function. FASEB J. 2008, 22, 3491. [Google Scholar] [CrossRef]
- Wilson, J.M.; Kurtz, C.C.; Black, S.G.; Ross, W.G.; Alam, M.S.; Linden, J.; Ernst, P.B. The A2B Adenosine Receptor Promotes Th17 Differentiation via Stimulation of Dendritic Cell IL-6. J. Immunol. 2011, 186, 6746–6752. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Du, C.; Lv, J.; Zhao, G.; Li, Z.; Wu, Z.; Haskó, G.; Xie, X. Blocking A2B Adenosine Receptor Alleviates Pathogenesis of Experimental Autoimmune Encephalomyelitis via Inhibition of IL-6 Production and Th17 Differentiation. J. Immunol. 2013, 190, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Zuo, A.; Shao, H.; Chen, M.; Kaplan, H.J.; Sun, D. Anti-Inflammatory or Proinflammatory Effect of an Adenosine Receptor Agonist on the Th17 Autoimmune Response Is Inflammatory Environment–Dependent. J. Immunol. 2014, 193, 5498–5505. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.S.; Cavanaugh, C.; Pereira, M.; Babu, U.; Williams, K. Susceptibility of Aging Mice to Listeriosis: Role of Anti-Inflammatory Responses with Enhanced Treg-Cell Expression of CD39/CD73 and Th-17 Cells. Int. J. Med. Microbiol. IJMM 2020, 310, 151397. [Google Scholar] [CrossRef] [PubMed]
- Zarek, P.E.; Huang, C.-T.; Lutz, E.R.; Kowalski, J.; Horton, M.R.; Linden, J.; Drake, C.G.; Powell, J.D. A2A Receptor Signaling Promotes Peripheral Tolerance by Inducing T-Cell Anergy and the Generation of Adaptive Regulatory T Cells. Blood 2008, 111, 251–259. [Google Scholar] [CrossRef]
- Ohta, A.; Kini, R.; Ohta, A.; Subramanian, M.; Madasu, M.; Sitkovsky, M. The Development and Immunosuppressive Functions of CD4(+) CD25(+) FoxP3(+) Regulatory T Cells Are under Influence of the Adenosine-A2A Adenosine Receptor Pathway. Front. Immunol. 2012, 3, 190. [Google Scholar] [CrossRef]
- Ujiie, H. Regulatory T Cells in Autoimmune Skin Diseases. Exp. Dermatol. 2019, 28, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.N.; Gouirand, V.; Macon, C.E.; Lowe, M.M.; Boothby, I.C.; Moreau, J.M.; Gratz, I.K.; Stoecklinger, A.; Weaver, C.T.; Sharpe, A.H.; et al. Regulatory T Cells in Skin Mediate Immune Privilege of the Hair Follicle Stem Cell Niche. Sci. Immunol. 2024, 9, eadh0152. [Google Scholar] [CrossRef]
- Da, M.; Chen, L.; Enk, A.; Mahnke, K. Tolerance to 2,4-Dinitrofluorobenzene–Induced Contact Hypersensitivity Is Mediated by CD73-Expressing Tissue-Homing Regulatory T Cells. J. Investig. Dermatol. 2023, 143, 1011–1022. [Google Scholar] [CrossRef]
- Rosenblum, M.D.; Gratz, I.K.; Paw, J.S.; Lee, K.; Marshak-Rothstein, A.; Abbas, A.K. Response to Self Antigen Imprints Regulatory Memory in Tissues. Nature 2011, 480, 538–542. [Google Scholar] [CrossRef]
- Flutter, B.; Nestle, F.O. TLRs to Cytokines: Mechanistic Insights from the Imiquimod Mouse Model of Psoriasis. Eur. J. Immunol. 2013, 43, 3138–3146. [Google Scholar] [CrossRef] [PubMed]
- Strass, S.; Geiger, J.; Martorelli, M.; Geiger, S.; Cloos, N.; Keppler, M.; Fischer, T.; Riexinger, L.; Schwamborn, A.; Guezguez, J.; et al. Isostearic Acid Is an Active Component of Imiquimod Formulations Used to Induce Psoriaform Disease Models. Inflammopharmacology 2023, 31, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Schön, M.P.; Schön, M.; Klotz, K.-N. The Small Antitumoral Immune Response Modifier Imiquimod Interacts with Adenosine Receptor Signaling in a TLR7- and TLR8-Independent Fashion. J. Investig. Dermatol. 2006, 126, 1338–1347. [Google Scholar] [CrossRef]
- Kasten, K.R.; Tschöp, J.; Goetzman, H.S.; England, L.G.; Dattilo, J.R.; Cave, C.M.; Seitz, A.P.; Hildeman, D.A.; Caldwell, C.C. T-cell Activation Differentially Mediates the Host Response to Sepsis. Shock 2010, 34, 377. [Google Scholar] [CrossRef] [PubMed]
- Gessi, S.; Merighi, S.; Borea, P.A. Targeting Adenosine Receptors to Prevent Inflammatory Skin Diseases. Exp. Dermatol. 2014, 23, 553–554. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Perez, J.; Killeen, M.; Yang, Y.; Carey, C.; Falo, L.; Mathers, A. Extracellular ATP and IL-23 Form a Local Inflammatory Circuit Leading to the Development of a Neutrophil-Dependent Psoriasiform Dermatitis. J. Investig. Dermatol. 2018, 138, 2595–2605. [Google Scholar] [CrossRef] [PubMed]
- Killeen, M.E.; Ferris, L.; Kupetsky, E.A.; Falo, L.; Mathers, A.R. Signaling through Purinergic Receptors for ATP Induces Human Cutaneous Innate and Adaptive Th17 Responses: Implications in the Pathogenesis of Psoriasis. J. Immunol. 2013, 190, 4324–4336. [Google Scholar] [CrossRef] [PubMed]
- Silva-Vilches, C.; Bolduan, V.; Alabdullah, M.; Steinbrink, K.; Probst, H.C.; Enk, A.; Mahnke, K. Topical Application of Adenosine A2-Type Receptor Agonists Prevents Contact Hypersensitivity Reactions in Mice by Affecting Skin Dendritic Cells. J. Investig. Dermatol. 2023, 143, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Stohl, L.; Zang, J.; Ding, W.; Manni, M.; Zhou, X.; Granstein, R. Norepinephrine and Adenosine-5′-Triphosphate Synergize in Inducing IL-6 Production by Human Dermal Microvascular Endothelial Cells. Cytokine 2013, 64, 605–612. [Google Scholar] [CrossRef]
- Weber, F.; Esser, P.; Müller, T.; Ganesan, J.; Pellegatti, P.; Simon, M.; Zeiser, R.; Idzko, M.; Jakob, T.; Martin, S. Lack of the Purinergic Receptor P2X7 Results in Resistance to Contact Hypersensitivity. J. Exp. Med. 2010, 207, 2609–2619. [Google Scholar] [CrossRef]
- Silva-Vilches, C.; Ring, S.; Schrader, J.; Clausen, B.E.; Probst, H.-C.; Melchior, F.; Schild, H.; Enk, A.; Mahnke, K. Production of Extracellular Adenosine by CD73+ Dendritic Cells Is Crucial for Induction of Tolerance in Contact Hypersensitivity Reactions. J. Investig. Dermatol. 2019, 139, 541–551. [Google Scholar] [CrossRef] [PubMed]
- da Silva, G.L.; Sperotto, N.D.M.; Borges, T.J.; Bonorino, C.; Takyia, C.M.; Coutinho-Silva, R.; Campos, M.M.; Zanin, R.F.; Morrone, F.B. P2X7 Receptor Is Required for Neutrophil Accumulation in a Mouse Model of Irritant Contact Dermatitis. Exp. Dermatol. 2013, 22, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Irrera, N.; Bitto, A.; Vaccaro, M.; Mannino, F.; Squadrito, V.; Pallio, G.; Arcoraci, V.; Minutoli, L.; Ieni, A.; Lentini, M.; et al. PDRN, a Bioactive Natural Compound, Ameliorates Imiquimod-Induced Psoriasis through NF-κB Pathway Inhibition and Wnt/β-Catenin Signaling Modulation. Int. J. Mol. Sci. 2020, 21, 1215. [Google Scholar] [CrossRef]
- Welihinda, A.; Ravikumar, P.; Kaur, M.; Mechanic, J.; Yadav, S.; Kang, G.; Amento, E. Positive Allosteric Modulation of A2AR Alters Immune Cell Responses and Ameliorates Psoriasis-Like Dermatitis in Mice. J. Investig. Dermatol. 2022, 142, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Marín-Castejón, A.; Marco-Bonilla, M.; Terencio, M.C.; Arasa, J.; Carceller, M.C.; Ferrandiz, M.L.; Noguera, M.A.; Andrés-Ejarque, R.; Montesinos, M.C. Adenosine A2B Receptor Agonist Improves Epidermal Barrier Integrity in a Murine Model of Epidermal Hyperplasia. Biomed. Pharmacother. 2024, 173, 116401. [Google Scholar] [CrossRef] [PubMed]
- López-Cano, M.; Filgaira, I.; Nolen, E.; Cabré, G.; Hernando, J.; Tosh, D.; Jacobson, K.; Soler, C.; Ciruela, F. Optical Control of Adenosine A3 Receptor Function in Psoriasis. Pharmacol. Res. 2021, 170, 105731. [Google Scholar] [CrossRef] [PubMed]
- Soni, S.; Lukhey, M.S.; Thawkar, B.S.; Chintamaneni, M.; Kaur, G.; Joshi, H.; Ramniwas, S.; Tuli, H.S. A Current Review on P2X7 Receptor Antagonist Patents in the Treatment of Neuroinflammatory Disorders: A Patent Review on Antagonists. Naunyn Schmiedebergs Arch. Pharmacol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, S.; Schnermann, J.; Noorbakhsh, F.; Henry, S.; Yong, V.W.; Winston, B.W.; Warren, K.; Power, C. A1 Adenosine Receptor Upregulation and Activation Attenuates Neuroinflammation and Demyelination in a Model of Multiple Sclerosis. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 1521–1529. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhang, W.; Guo, J.; Kong, F.; Zhou, S.; Chen, S.; Wang, Z.; Zang, D. Adenosine Binds Predominantly to Adenosine Receptor A1 Subtype in Astrocytes and Mediates an Immunosuppressive Effect. Brain Res. 2018, 1700, 47–55. [Google Scholar] [CrossRef]
- Tsutsui, S.; Vergote, D.; Shariat, N.; Warren, K.; Ferguson, S.S.G.; Power, C. Glucocorticoids Regulate Innate Immunity in a Model of Multiple Sclerosis: Reciprocal Interactions between the A1 Adenosine Receptor and Beta-Arrestin-1 in Monocytoid Cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2008, 22, 786–796. [Google Scholar] [CrossRef]
- Oliver, S.; Mathew, S.; Wilder, T.; Cronstein, B. Restraint Stress Fails to Modulate Cutaneous Hypersensitivity Responses in Mice Lacking the Adenosine A1 Receptor. Purinergic Signal. 2011, 7, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Boeynaems, J.-M. Purinergic Signalling and Immune Cells. Purinergic Signal. 2014, 10, 529–564. [Google Scholar] [CrossRef] [PubMed]
- Borea, P.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef] [PubMed]
- Uribe-Herranz, M.; Lian, L.; Hooper, K.; Milora, K.; Jensen, L. IL-1R1 Signaling Facilitates Munro’s Microabscess Formation in Psoriasiform Imiquimod-Induced Skin Inflammation. J. Investig. Dermatol. 2013, 133, 1541–1549. [Google Scholar] [CrossRef]
- Arasa, J.; Martos, P.; Terencio, M.C.; Valcuende-Cavero, F.; Montesinos, M.C. Topical Application of the Adenosine A2A Receptor Agonist CGS-21680 Prevents Phorbol-Induced Epidermal Hyperplasia and Inflammation in Mice. Exp. Dermatol. 2014, 23, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Perez-Aso, M.; Mediero, A.; Low, Y.; Levine, J.; Cronstein, B. Adenosine A2A Receptor Plays an Important Role in Radiation-Induced Dermal Injury. FASEB J. 2016, 30, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, G.; Zhang, J.; Perez-Aso, M.; Mediero, A.; Cronstein, B. Adenosine A2A Receptor Promotes Collagen Type III Synthesis via β-Catenin Activation in Human Dermal Fibroblasts. Br. J. Pharmacol. 2016, 173, 3279–3291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Corciulo, C.; Liu, H.; Wilder, T.; Ito, M.; Cronstein, B. Adenosine A2a Receptor Blockade Diminishes Wnt/β-Catenin Signaling in a Murine Model of Bleomycin-Induced Dermal Fibrosis. Am. J. Pathol. 2017, 187, 1935–1944. [Google Scholar] [CrossRef]
- Chen, M.; Liang, D.; Zuo, A.; Shao, H.; Kaplan, H.J.; Sun, D. An A2B Adenosine Receptor Agonist Promotes Th17 Autoimmune Responses in Experimental Autoimmune Uveitis (EAU) via Dendritic Cell Activation. PLoS ONE 2015, 10, e0132348. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Zuo, A.; Shao, H.; Chen, M.; Kaplan, H.J.; Sun, D. A2B Adenosine Receptor Activation Switches Differentiation of Bone Marrow Cells to a CD11c(+)Gr-1(+) Dendritic Cell Subset That Promotes the Th17 Response. Immun. Inflamm. Dis. 2015, 3, 360–373. [Google Scholar] [CrossRef]
- Shakya, A.; Naik, R.; Almasri, I.; Kaur, A. Role and Function of Adenosine and Its Receptors in Inflammation, Neuroinflammation, IBS, Autoimmune Inflammatory Disorders, Rheumatoid Arthritis and Psoriasis. Curr. Pharm. Des. 2019, 25, 2875–2891. [Google Scholar] [CrossRef] [PubMed]
- Kolachala, V.L.; Vijay-Kumar, M.; Dalmasso, G.; Yang, D.; Linden, J.; Wang, L.; Gewirtz, A.; Ravid, K.; Merlin, D.; Sitaraman, S.V. A2B Adenosine Receptor Gene Deletion Attenuates Murine Colitis. Gastroenterology 2008, 135, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Dascalu, A.; Matithyou, A.; Oron, Y.; Korenstein, R. A Hyperosmotic Stimulus Elevates Intracellular Calcium and Inhibits Proliferation of a Human Keratinocyte Cell Line. J. Investig. Dermatol. 2000, 115, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Ochaion, A.; Bar-Yehuda, S.; Cohen, S.; Barer, F.; Patoka, R.; Amital, H.; Reitblat, T.; Reitblat, A.; Ophir, J.; Konfino, I.; et al. The Anti-Inflammatory Target A(3) Adenosine Receptor Is over-Expressed in Rheumatoid Arthritis, Psoriasis and Crohn’s Disease. Cell. Immunol. 2009, 258, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Bar-Yehuda, S.; Silverman, M.H.; Kerns, W.D.; Ochaion, A.; Cohen, S.; Fishman, P. The Anti-Inflammatory Effect of A3 Adenosine Receptor Agonists: A Novel Targeted Therapy for Rheumatoid Arthritis. Expert Opin. Investig. Drugs 2007, 16, 1601–1613. [Google Scholar] [CrossRef] [PubMed]
- Varani, K.; Padovan, M.; Vincenzi, F.; Targa, M.; Trotta, F.; Govoni, M.; Borea, P.A. A2A and A3 Adenosine Receptor Expression in Rheumatoid Arthritis: Upregulation, Inverse Correlation with Disease Activity Score and Suppression of Inflammatory Cytokine and Metalloproteinase Release. Arthritis Res. Ther. 2011, 13, R197. [Google Scholar] [CrossRef] [PubMed]
- David, M.; Akerman, L.; Ziv, M.; Kadurina, M.; Gospodinov, D.; Pavlotsky, F.; Yankova, R.; Kouzeva, V.; Ramon, M.; Silverman, M.H.; et al. Treatment of Plaque-type Psoriasis with Oral CF101: Data from an Exploratory Randomized Phase 2 Clinical Trial. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.; Beyska-Rizova, S.; Gantcheva, M.; Simeonova, E.; Brezoev, P.; Celic, M.; Groppa, L.; Blicharski, T.; Selmanagic, A.; Kalicka-Dudzik, M.; et al. Efficacy and Safety of Piclidenoson in Plaque Psoriasis: Results from a Randomized Phase 3 Clinical Trial (COMFORT-1). J. Eur. Acad. Dermatol. Venereol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Fishman, P.; Cohen, S. The A3 Adenosine Receptor (A3AR): Therapeutic Target and Predictive Biological Marker in Rheumatoid Arthritis. Clin. Rheumatol. 2016, 35, 2359–2362. [Google Scholar] [CrossRef]
- Cohen, S.; Barer, F.; Itzhak, I.; Silverman, M.; Fishman, P. Inhibition of IL-17 and IL-23 in Human Keratinocytes by the A3 Adenosine Receptor Agonist Piclidenoson. J. Immunol. Res. 2018, 2018, 2310970. [Google Scholar] [CrossRef]
- Stemmer, S.M.; Manojlovic, N.S.; Marinca, M.V.; Petrov, P.; Cherciu, N.; Ganea, D.; Ciuleanu, T.E.; Pusca, I.A.; Beg, M.S.; Purcell, W.T.; et al. Namodenoson in Advanced Hepatocellular Carcinoma and Child-Pugh B Cirrhosis: Randomized Placebo-Controlled Clinical Trial. Cancers 2021, 13, 187. [Google Scholar] [CrossRef] [PubMed]
- David, M.; Gospodinov, D.; Gheorghe, N.; Mateev, G.; Rusinova, M.; Hristakieva, E.; Solovastru, L.; Patel, R.; Giurcaneanu, C.; Hitova, M.; et al. Treatment of Plaque-Type Psoriasis with Oral CF101: Data from a Phase II/III Multicenter, Randomized, Controlled Trial. J. Drugs Dermatol. 2016, 15, 931–938. [Google Scholar] [PubMed]
- Lerch, M.M.; Hansen, M.J.; van Dam, G.M.; Szymanski, W.; Feringa, B.L. Emerging Targets in Photopharmacology. Angew. Chem. Int. Ed. 2016, 55, 10978–10999. [Google Scholar] [CrossRef] [PubMed]
- Fries, J.F.; Ramey, D.R.; Singh, G.; Morfeld, D.; Bloch, D.A.; Raynauld, J.P. A Reevaluation of Aspirin Therapy in Rheumatoid Arthritis. Arch. Intern. Med. 1993, 153, 2465–2471. [Google Scholar] [CrossRef] [PubMed]
- Li, R.W.S.; Yang, C.; Sit, A.S.M.; Lin, S.Y.T.; Ho, E.Y.W.; Leung, G.P.H. Physiological and Pharmacological Roles of Vascular Nucleoside Transporters. J. Cardiovasc. Pharmacol. 2012, 59, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Cronstein, B.N.; Montesinos, M.C.; Weissmann, G. Salicylates and Sulfasalazine, but Not Glucocorticoids, Inhibit Leukocyte Accumulation by an Adenosine-Dependent Mechanism That Is Independent of Inhibition of Prostaglandin Synthesis and P105 of NFkappaB. Proc. Natl. Acad. Sci. USA 1999, 96, 6377–6381. [Google Scholar] [CrossRef] [PubMed]
- Barczyk, K.; Ehrchen, J.; Tenbrock, K.; Ahlmann, M.; Kneidl, J.; Viemann, D.; Roth, J. Glucocorticoids Promote Survival of Anti-Inflammatory Macrophages via Stimulation of Adenosine Receptor A3. Blood 2010, 116, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Bürger, C.; Shirsath, N.; Lang, V.; Diehl, S.; Kaufmann, R.; Weigert, A.; Han, Y.-Y.; Ringel, C.; Wolf, P. Blocking mTOR Signalling with Rapamycin Ameliorates Imiquimod-Induced Psoriasis in Mice. Acta Derm. Venereol. 2017, 97, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Hong, S.Y.; Wang, J.; Rehan, S.; Liu, W.; Peng, H.; Das, M.; Li, W.; Bhat, S.; Peiffer, B.; et al. Rapamycin-Inspired Macrocycles with New Target Specificity. Nat. Chem. 2019, 11, 254–263. [Google Scholar] [CrossRef]
- Balak, D.M.W.; Gerdes, S.; Parodi, A.; Salgado-Boquete, L. Long-Term Safety of Oral Systemic Therapies for Psoriasis: A Comprehensive Review of the Literature. Dermatol. Ther. 2020, 10, 589–613. [Google Scholar] [CrossRef]
- Koźmiński, P.; Halik, P.K.; Chesori, R.; Gniazdowska, E. Overview of Dual-Acting Drug Methotrexate in Different Neurological Diseases, Autoimmune Pathologies and Cancers. Int. J. Mol. Sci. 2020, 21, 3483. [Google Scholar] [CrossRef] [PubMed]
- Friedman, B.; Cronstein, B. Methotrexate Mechanism in Treatment of Rheumatoid Arthritis. Jt. Bone Spine Rev. Rhum. 2019, 86, 301–307. [Google Scholar] [CrossRef]
- Fernandez, C.A. Pharmacological Strategies for Mitigating Anti-TNF Biologic Immunogenicity in Rheumatoid Arthritis Patients. Curr. Opin. Pharmacol. 2023, 68, 102320. [Google Scholar] [CrossRef]
- Maksimovic, V.; Pavlovic-Popovic, Z.; Vukmirović, S.; Cvejic, J.; Mooranian, A.; Al-Salami, H.; Mikov, M.; Golocorbin-Kon, S. Molecular Mechanism of Action and Pharmacokinetic Properties of Methotrexate. Mol. Biol. Rep. 2020, 47, 4699–4708. [Google Scholar] [CrossRef]
- Chan, E.S.L.; Cronstein, B.N. Methotrexate—How Does It Really Work? Nat. Rev. Rheumatol. 2010, 6, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Min, G.; Kim, T.; Kim, J.; Cho, W.; Yang, J.; Ma, J. Inhibitory Effect of Isatis tinctoria L. Water Extract on DNCB-Induced Atopic Dermatitis in BALB/c Mice and HaCaT Cells. Chin. Med. 2022, 17, 66. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Reitman, M.L. Adenosine-Related Mechanisms in Non-Adenosine Receptor Drugs. Cells 2020, 9, 956. [Google Scholar] [CrossRef]
- Translational Therapeutics of Dipyridamole|Arteriosclerosis, Thrombosis, and Vascular Biology. Available online: https://www.ahajournals.org/doi/10.1161/atvbaha.107.160226 (accessed on 5 April 2024).
- Torres, T.; Puig, L. Apremilast: A Novel Oral Treatment for Psoriasis and Psoriatic Arthritis. Am. J. Clin. Dermatol. 2018, 19, 23–32. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Ligands | Receptor Selectivity | Mouse Model | Effect | Reference |
|---|---|---|---|---|
| KN-62 | P2X7 antagonist | CHS | Reduced reaction and IL-1 secretion by DCs | [80] |
| A438079 | P2X7 antagonist | croton oil-induced oedema | Impair of croton oil-induced oedema; reduced IL-1β production and neutrophil infiltration | [82] |
| A438079 | P2X7 antagonist | ATP analog and E-NTPDase inhibitors-induced psoriasiform dermatitis | Block of psoriasiform dermatitis and inflammatory response | [76] |
| PDRN | A2A agonist | imiquimod-induced mouse model | Inhibition of inflammatory response and restoration of normal skin architecture, decreased T cell recruitment, and a shift towards an anti-inflammatory cytokine profile | [83] |
| CGS 21680 | A2A agonist | CHS | Less-activated T cells and more anergic cells; reduced proinflammatory cytokines and chemokines in inflamed ear; reduced functional skin migratory DCs, which are also less functional | [78] |
| CGS 21680 | A2A agonist | phorbol-induced epidermal hyperplasia | Reduction in epidermal hyperplasia and promotion of collagen synthesis normalization of epidermal structure and enhancement of fibroblast proliferation in the dermis reduction of chemotactic mediator expression and NF-κB inhibition | [75] |
| AEA061 | positive allosteric modulator of A2A | imiquimod-induced psoriasis-like dermatitis mice model | Reduced ear swelling, skin thickness, erythema, scale formation, and inflammatory cytokine expression | [84] |
| BAY60–6583 | A2B agonist | CHS | Reduced ear swelling; suppressed activation and migration of skin migratory DCs | [78] |
| BAY60–6583 | A2B agonist | TPA-induced epidermal hyperplasia | Reduced skin inflammation; reduced leucocytes infiltration; preserved epidermal barrier integrity | [85] |
| MRS5698 | Photosensitive A3 agonist | IL-23 mouse model of psoriasis | Reduced skin swelling; cAMP reduction | [86] |
| Ligands | Receptor Selectivity | Indication | Phase | Reference |
|---|---|---|---|---|
| Poclidenoson | A3 agonist | Psoriasis | 3 | NCT00428974 |
| Poclidenoson | A3 agonist | Rheumatoid arthritis | 3 | NCT00428974 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Lei, X.; Mahnke, K. Adenosine and Its Receptors in the Pathogenesis and Treatment of Inflammatory Skin Diseases. Int. J. Mol. Sci. 2024, 25, 5810. https://doi.org/10.3390/ijms25115810
Chen L, Lei X, Mahnke K. Adenosine and Its Receptors in the Pathogenesis and Treatment of Inflammatory Skin Diseases. International Journal of Molecular Sciences. 2024; 25(11):5810. https://doi.org/10.3390/ijms25115810
Chicago/Turabian StyleChen, Luxia, Xuan Lei, and Karsten Mahnke. 2024. "Adenosine and Its Receptors in the Pathogenesis and Treatment of Inflammatory Skin Diseases" International Journal of Molecular Sciences 25, no. 11: 5810. https://doi.org/10.3390/ijms25115810
APA StyleChen, L., Lei, X., & Mahnke, K. (2024). Adenosine and Its Receptors in the Pathogenesis and Treatment of Inflammatory Skin Diseases. International Journal of Molecular Sciences, 25(11), 5810. https://doi.org/10.3390/ijms25115810