
The Importance of Large-Scale Genomic Studies to Unravel Genetic Risk Factors for Autism
 , and
, and
Abstract
:1. Introduction
2. Delineation of Locus and Gene Discovery in ASD
2.1. Chromosomal Rearrangements and Monogenic Syndromes Associated with ASD
2.2. Linkage and Candidate Gene Analysis
2.3. Microarray-Based Copy Number Variation Analysis
2.4. Whole Exome and Genome Sequencing
2.5. Genome-Wide Association Studies (GWAS)
3. Challenges and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zeidan, J.; Fombonne, E.; Scorah, J.; Ibrahim, A.; Durkin, M.S.; Saxena, S.; Yusuf, A.; Shih, A.; Elsabbagh, M. Global prevalence of autism: A systematic review update. Autism Res. 2022, 15, 778–790. [Google Scholar] [CrossRef]
- Maenner, M.J.; Warren, Z.; Williams, A.R.; Amoakohene, E.; Bakian, A.V.; Bilder, D.A.; Durkin, M.S.; Fitzgerald, R.T.; Furnier, S.M.; Hughes, M.M.; et al. Prevalence and Characteristics of Autism Spectrum Disorder among Children Aged 8 Years-Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2020. MMWR Surveill Summ. 2023, 72, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gaugler, T.; Klei, L.; Sanders, S.J.; Bodea, C.A.; Goldberg, A.P.; Lee, A.B.; Mahajan, M.; Manaa, D.; Pawitan, Y.; Reichert, J.; et al. Most genetic risk for autism resides with common variation. Nat. Genet. 2014, 46, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Larsson, H.; Hultman, C.M.; Reichenberg, A. The familial risk of autism. JAMA 2014, 311, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Tick, B.; Bolton, P.; Happé, F.; Rutter, M.; Rijsdijk, F. Heritability of autism spectrum disorders: A meta-analysis of twin studies. J. Child. Psychol. Psychiatry 2016, 57, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Yip, B.H.K.; Windham, G.C.; Sourander, A.; Francis, R.; Yoffe, R.; Glasson, E.; Mahjani, B.; Suominen, A.; Leonard, H.; et al. Association of Genetic and Environmental Factors with Autism in a 5-Country Cohort. JAMA Psychiatry 2019, 76, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, D.H.; Sowinski, J.; Lord, C.; Iversen, P.; Shestack, J.; Jones, P.; Ducat, L.; Spence, S.J. The autism genetic resource exchange: A resource for the study of autism and related neuropsychiatric conditions. Am. J. Hum. Genet. 2001, 69, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Lajonchere, C.M.; AGRE Consortium. Changing the landscape of autism research: The autism genetic resource exchange. Neuron 2010, 68, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Hu-Lince, D.; Craig, D.W.; Huentelman, M.J.; Stephan, D.A. The Autism Genome Project: Goals and strategies. Am. J. Pharmacogenomics 2005, 5, 233–246. [Google Scholar] [CrossRef]
- Fischbach, G.D.; Lord, C. The Simons Simplex Collection: A resource for identification of autism genetic risk factors. Neuron 2010, 68, 192–195. [Google Scholar] [CrossRef]
- Sullivan, P.F. The psychiatric GWAS consortium: Big science comes to psychiatry. Neuron 2010, 68, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.F.; Agrawal, A.; Bulik, C.M.; Andreassen, O.A.; Borglum, A.D.; Breen, G.; Cichon, S.; Edenberg, H.J.; Faraone, S.V.; Gelernter, J.; et al. Psychiatric Genomics: An Update and an Agenda. Am. J. Psychiatry 2018, 175, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.D.; Daly, M.J.; Devlin, B.; Lehner, T.; Roeder, K.; State, M.W. The autism sequencing consortium: Large-scale, high-throughput sequencing in autism spectrum disorders. Neuron 2012, 76, 1052–1056. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, C.B.; Bybjerg-Grauholm, J.; Pedersen, M.G.; Grove, J.; Agerbo, E.; Bækvad-Hansen, M.; Poulsen, J.B.; Hansen, C.S.; McGrath, J.J.; Als, T.D.; et al. The iPSYCH2012 case-cohort sample: New directions for unravelling genetic and environmental architectures of severe mental disorders. Mol. Psychiatry 2018, 23, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Yuen, R.K.C.; Merico, D.; Bookman, M.; Howe, J.L.; Thiruvahindrapuram, B.; Patel, R.V.; Whitney, J.; Deflaux, N.; Bingham, J.; Wang, Z.; et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 2017, 20, 602–611. [Google Scholar] [CrossRef] [PubMed]
- SPARK Consortium. SPARK: A US Cohort of 50,000 Families to Accelerate Autism Research. Neuron 2018, 97, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.-Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef]
- Fu, J.M.; Satterstrom, F.K.; Peng, M.; Brand, H.; Collins, R.L.; Dong, S.; Wamsley, B.; Klei, L.; Wang, L.; Hao, S.P.; et al. Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat. Genet. 2022, 54, 1320–1331. [Google Scholar] [CrossRef]
- Trost, B.; Thiruvahindrapuram, B.; Chan, A.J.; Engchuan, W.; Higginbotham, E.J.; Howe, J.L.; Loureiro, L.O.; Reuter, M.S.; Roshandel, D.; Whitney, J.; et al. Genomic architecture of autism from comprehensive whole-genome sequence annotation. Cell 2022, 185, 4409–4427.e18. [Google Scholar] [CrossRef]
- Zhou, X.; Feliciano, P.; Shu, C.; Wang, T.; Astrovskaya, I.; Hall, J.B.; Obiajulu, J.U.; Wright, J.R.; Murali, S.C.; Xu, S.X.; et al. Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate-risk genes. Nat. Genet. 2022, 54, 1305–1319. [Google Scholar] [CrossRef] [PubMed]
- Autism Spectrum Disorders Working Group of the Psychiatric Genomics Consortium. Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol. Autism. 2017, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Matoba, N.; Liang, D.; Sun, H.; Aygün, N.; McAfee, J.C.; Davis, J.E.; Raffield, L.M.; Qian, H.; Piven, J.; Li, Y.; et al. Common genetic risk variants identified in the SPARK cohort support DDHD2 as a candidate risk gene for autism. Transl. Psychiatry 2020, 10, 265. [Google Scholar] [CrossRef] [PubMed]
- Weiner, D.J.; iPSYCH-Broad Autism Group; Wigdor, E.M.; Ripke, S.; Walters, R.K.; Kosmicki, J.A.; Grove, J.; Samocha, K.E.; Goldstein, J.I.; Okbay, A.; et al. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat. Genet. 2017, 49, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Klei, L.; McClain, L.L.; Mahjani, B.; Panayidou, K.; De Rubeis, S.; Grahnat, A.-C.S.; Karlsson, G.; Lu, Y.; Melhem, N.; Xu, X.; et al. How rare and common risk variation jointly affect liability for autism spectrum disorder. Mol. Autism. 2021, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Antaki, D.; Guevara, J.; Maihofer, A.X.; Klein, M.; Gujral, M.; Grove, J.; Carey, C.E.; Hong, O.; Arranz, M.J.; Hervas, A.; et al. A phenotypic spectrum of autism is attributable to the combined effects of rare variants, polygenic risk and sex. Nat. Genet. 2022, 54, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Gillberg, C.; Wahlström, J. Chromosome abnormalities in infantile autism and other childhood psychoses: A population study of 66 cases. Dev. Med. Child. Neurol. 1985, 27, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.-H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Pieretti, M.; Zhang, F.P.; Fu, Y.H.; Warren, S.T.; Oostra, B.A.; Caskey, C.; Nelson, D.L. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 1991, 66, 817–822. [Google Scholar] [CrossRef]
- Ashley, C.T., Jr.; Wilkinson, K.D.; Reines, D.; Warren, S.T. FMR1 protein: Conserved RNP family domains and selective RNA binding. Science 1993, 262, 563–566. [Google Scholar] [CrossRef]
- European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 1993, 75, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- van Slegtenhorst, M.; de Hoogt, R.; Hermans, C.; Nellist, M.; Janssen, B.; Verhoef, S.; Lindhout, D.; Ouweland, A.V.; Halley, D.; Young, J.; et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997, 277, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Nyholt, D.R.; Magnussen, P.; Parano, E.; Pavone, P.; Geschwind, D.; Lord, C.; Iversen, P.; Hoh, J.; The Autism Genetic Resource Exchange Consortium; et al. A genomewide screen for autism susceptibility loci. Am. J. Hum. Genet. 2001, 69, 327–340. [Google Scholar] [CrossRef]
- Yonan, A.L.; Alarcón, M.; Cheng, R.; Magnusson, P.K.; Spence, S.J.; Palmer, A.A.; Grunn, A.; Juo, S.-H.H.; Terwilliger, J.D.; Liu, J.; et al. A genomewide screen of 345 families for autism-susceptibility loci. Am. J. Hum. Genet. 2003, 73, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, C.W.; Goedken, R.; Vieland, V.J. Effects of updating linkage evidence across subsets of data: Reanalysis of the autism genetic resource exchange data set. Am. J. Hum. Genet. 2005, 76, 688–695. [Google Scholar] [CrossRef] [PubMed]
- McCauley, J.L.; Olson, L.M.; Dowd, M.; Amin, T.; Steele, A.; Blakely, R.; Folstein, S.; Haines, J.; Sutcliffe, J. Linkage and association analysis at the serotonin transporter (SLC6A4) locus in a rigid-compulsive subset of autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2004, 127B, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, J.S.; Delahanty, R.J.; Prasad, H.C.; McCauley, J.L.; Han, Q.; Jiang, L.; Li, C.; Folstein, S.E.; Blakely, R.D. Allelic heterogeneity at the serotonin transporter locus (SLC6A4) confers susceptibility to autism and rigid-compulsive behaviors. Am. J. Hum. Genet. 2005, 77, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Cheslack-Postava, K.; Fallin, M.D.; Avramopoulos, D.; Connors, S.L.; Zimmerman, A.W.; Eberhart, C.G.; Newschaffer, C.J. beta2-Adrenergic receptor gene variants and risk for autism in the AGRE cohort. Mol. Psychiatry 2007, 12, 283–291. [Google Scholar] [CrossRef]
- Jamain, S.; Quach, H.; Betancur, C.; Råstam, M.; Colineaux, C.; Gillberg, I.C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gillberg, C.; et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef]
- Sebat, J.; Lakshmi, B.; Malhotra, D.; Troge, J.; Lese-Martin, C.; Walsh, T.; Yamrom, B.; Yoon, S.; Krasnitz, A.; Kendall, J.; et al. Strong association of de novo copy number mutations with autism. Science 2007, 316, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.R.; Noor, A.; Vincent, J.B.; Lionel, A.C.; Feuk, L.; Skaug, J.; Shago, M.; Moessner, R.; Pinto, D.; Ren, Y.; et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008, 82, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; Ercan-Sencicek, A.G.; Hus, V.; Luo, R.; Murtha, M.T.; Moreno-De-Luca, D.; Chu, S.H.; Moreau, M.P.; Gupta, A.R.; Thomson, S.A.; et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 2011, 70, 863–885. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Ronemus, M.; Yamrom, B.; Lee, Y.-H.; Leotta, A.; Kendall, J.; Marks, S.; Lakshmi, B.; Pai, D.; Ye, K.; et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 2011, 70, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Girirajan, S.; Dennis, M.Y.; Baker, C.; Malig, M.; Coe, B.P.; Campbell, C.D.; Mark, K.; Vu, T.H.; Alkan, C.; Cheng, Z.; et al. Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. Am. J. Hum. Genet. 2013, 92, 221–237. [Google Scholar] [CrossRef]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Leppa, V.M.; Kravitz, S.N.; Martin, C.L.; Andrieux, J.; Le Caignec, C.; Martin-Coignard, D.; DyBuncio, C.; Sanders, S.J.; Lowe, J.K.; Cantor, R.M.; et al. Rare Inherited and De Novo CNVs Reveal Complex Contributions to ASD Risk in Multiplex Families. Am. J. Hum. Genet. 2016, 99, 540–554. [Google Scholar] [CrossRef] [PubMed]
- Robinson, E.B.; Lichtenstein, P.; Anckarsäter, H.; Happé, F.; Ronald, A. Examining and interpreting the female protective effect against autistic behavior. Proc. Natl. Acad. Sci. USA 2013, 110, 5258–5262. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Deriziotis, P.; Lee, C.; Vives, L.; Schwartz, J.J.; Girirajan, S.; Karakoc, E.; MacKenzie, A.P.; Ng, S.B.; Baker, C.; et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 2011, 43, 585–589. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Neale, B.M.; Kou, Y.; Liu, L.; Ma’Ayan, A.; Samocha, K.E.; Sabo, A.; Lin, C.-F.; Stevens, C.; Wang, L.-S.; Makarov, V.; et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012, 485, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.-H.; Narzisi, G.; Leotta, A.; et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Sanders, S.J.; Liu, L.; De Rubeis, S.; Lim, E.T.; Sutcliffe, J.S.; Schellenberg, G.D.; Gibbs, R.A.; Daly, M.J.; Buxbaum, J.D.; et al. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet. 2013, 9, e1003671. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Stessman, H.A.; Boyle, E.A.; Witherspoon, K.T.; Martin, B.; Lee, C.; Vives, L.; Baker, C.; Hiatt, J.B.; Nickerson, D.A.; et al. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat. Commun. 2014, 5, 5595. [Google Scholar] [CrossRef]
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S.; et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef]
- Feliciano, P.; Zhou, X.; Astrovskaya, I.; Turner, T.N.; Wang, T.; Brueggeman, L.; Barnard, R.; Hsieh, A.; Snyder, L.G.; Eichler, E.E.; et al. Exome sequencing of 457 autism families recruited online provides evidence for autism risk genes. NPJ Genom. Med. 2019, 4, 19. [Google Scholar] [CrossRef]
- Willsey, A.J.; Sanders, S.J.; Li, M.; Dong, S.; Tebbenkamp, A.T.; Muhle, R.A.; Reilly, S.K.; Lin, L.; Fertuzinhos, S.; Miller, J.A.; et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 2013, 155, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Parikshak, N.N.; Luo, R.; Zhang, A.; Won, H.; Lowe, J.K.; Chandran, V.; Horvath, S.; Geschwind, D.H. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 2013, 155, 1008–1021. [Google Scholar] [CrossRef] [PubMed]
- Velmeshev, D.; Schirmer, L.; Jung, D.; Haeussler, M.; Perez, Y.; Mayer, S.; Bhaduri, A.; Goyal, N.; Rowitch, D.H.; Kriegstein, A.R. Single-cell genomics identifies cell type-specific molecular changes in autism. Science 2019, 364, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Moyses-Oliveira, M.; De Esch, C.E.F.; Bhavsar, R.; Nuttle, X.; Li, A.; Yu, A.; Burt, N.D.; Erdin, S.; Fu, J.M.; et al. Convergent coexpression of autism-associated genes suggests some novel risk genes may not be detectable in large-scale genetic studies. Cell Genom. 2023, 3, 100277. [Google Scholar] [CrossRef]
- Doan, R.N.; Lim, E.T.; De Rubeis, S.; Betancur, C.; Cutler, D.J.; Chiocchetti, A.G.; Overman, L.M.; Soucy, A.; Goetze, S.; Autism Sequencing Consortium; et al. Recessive gene disruptions in autism spectrum disorder. Nat. Genet. 2019, 51, 1092–1098. [Google Scholar] [CrossRef]
- Freed, D.; Pevsner, J. The Contribution of Mosaic Variants to Autism Spectrum Disorder. PLoS Genet. 2016, 12, e1006245. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Yang, X.; Li, Z.; Wang, S.; Zhang, Z.; Ye, A.Y.; Yan, L.; Yang, C.; Wu, Q.; Li, J.; et al. Postzygotic single-nucleotide mosaicisms contribute to the etiology of autism spectrum disorder and autistic traits and the origin of mutations. Hum. Mutat. 2017, 38, 1002–1013. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.T.; Uddin, M.; De Rubeis, S.; Chan, Y.; Kamumbu, A.S.; Zhang, X.; D’Gama, A.M.; Kim, S.N.; Hill, R.S.; Goldberg, A.P.; et al. Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat. Neurosci. 2017, 20, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Krupp, D.R.; Barnard, R.A.; Duffourd, Y.; Evans, S.A.; Mulqueen, R.M.; Bernier, R.; Rivière, J.-B.; Fombonne, E.; O’roak, B.J. Exonic Mosaic Mutations Contribute Risk for Autism Spectrum Disorder. Am. J. Hum. Genet. 2017, 101, 369–390. [Google Scholar] [CrossRef]
- Jiang, Y.H.; Yuen, R.K.; Jin, X.; Wang, M.; Chen, N.; Wu, X.; Ju, J.; Mei, J.; Shi, Y.; He, M.; et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am. J. Hum. Genet. 2013, 93, 249–263. [Google Scholar] [CrossRef]
- Yuen, R.K.; Thiruvahindrapuram, B.; Merico, D.; Walker, S.; Tammimies, K.; Hoang, N.; Chrysler, C.; Nalpathamkalam, T.; Pellecchia, G.; Liu, Y.; et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med. 2015, 21, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Turner, T.N.; Hormozdiari, F.; Duyzend, M.H.; McClymont, S.A.; Hook, P.W.; Iossifov, I.; Raja, A.; Baker, C.; Hoekzema, K.; Stessman, H.A.; et al. Genome Sequencing of Autism-Affected Families Reveals Disruption of Putative Noncoding Regulatory DNA. Am. J. Hum. Genet. 2016, 98, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Yuen, R.K.; Merico, D.; Cao, H.; Pellecchia, G.; Alipanahi, B.; Thiruvahindrapuram, B.; Tong, X.; Sun, Y.; Cao, D.; Zhang, T.; et al. Genome-wide characteristics of de novo mutations in autism. NPJ Genom. Med. 2016, 1, 160271–1602710. [Google Scholar] [CrossRef] [PubMed]
- Brandler, W.M.; Antaki, D.; Gujral, M.; Kleiber, M.L.; Whitney, J.; Maile, M.S.; Hong, O.; Chapman, T.R.; Tan, S.; Tandon, P.; et al. Paternally inherited cis-regulatory structural variants are associated with autism. Science 2018, 360, 327–331. [Google Scholar] [CrossRef] [PubMed]
- An, J.Y.; Lin, K.; Zhu, L.; Werling, D.M.; Dong, S.; Brand, H.; Wang, H.Z.; Zhao, X.; Schwartz, G.B.; Collins, R.L.; et al. Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science 2018, 362, eaat6576. [Google Scholar] [CrossRef] [PubMed]
- Werling, D.M.; Brand, H.; An, J.Y.; Stone, M.R.; Zhu, L.; Glessner, J.T.; Collins, R.L.; Dong, S.; Layer, R.M. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat. Genet. 2018, 50, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Ruzzo, E.K.; Pérez-Cano, L.; Jung, J.Y.; Wang, L.-K.; Kashef-Haghighi, D.; Hartl, C.; Singh, C.; Xu, J.; Hoekstra, J.N.; Leventhal, O.; et al. Inherited and De Novo Genetic Risk for Autism Impacts Shared Networks. Cell 2019, 178, 850–866.e26. [Google Scholar] [CrossRef]
- Zhou, J.; Park, C.Y.; Theesfeld, C.L.; Wong, A.K.; Yuan, Y.; Scheckel, C.; Fak, J.J.; Funk, J.; Yao, K.; Tajima, Y.; et al. Whole-genome deep-learning analysis identifies contribution of noncoding mutations to autism risk. Nat. Genet. 2019, 51, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.; Engchuan, W.; Nguyen, C.M.; Thiruvahindrapuram, B.; Dolzhenko, E.; Backstrom, I.; Mirceta, M.; Mojarad, B.A.; Yin, Y.; Dov, A.; et al. Genome-wide detection of tandem DNA repeats that are expanded in autism. Nature 2020, 586, 80–86. [Google Scholar] [CrossRef]
- Mitra, I.; Huang, B.; Mousavi, N.; Ma, N.; Lamkin, M.; Yanicky, R.; Shleizer-Burko, S.; Lohmueller, K.E.; Gymrek, M. Patterns of de novo tandem repeat mutations and their role in autism. Nature 2021, 589, 246–250. [Google Scholar] [CrossRef]
- Wilfert, A.B.; Turner, T.N.; Murali, S.C.; Hsieh, P.; Sulovari, A.; Wang, T.; Coe, B.P.; Guo, H.; Hoekzema, K.; Bakken, T.E.; et al. Recent ultra-rare inherited variants implicate new autism candidate risk genes. Nat. Genet. 2021, 53, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Klei, L.; Sanders, S.J.; Murtha, M.T.; Hus, V.; Lowe, J.K.; Willsey, A.J.; Moreno-De-Luca, D.; Yu, T.W.; Fombonne, E.; Geschwind, D.; et al. Common genetic variants, acting additively, are a major source of risk for autism. Mol. Autism. 2012, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.A.; Arking, D.E.; Gene Discovery Project of Johns Hopkins & the Autism Consortium; Daly, M.J.; Chakravarti, A. A genome-wide linkage and association scan reveals novel loci for autism. Nature 2009, 461, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, H.; Ma, D.; Bucan, M.; Glessner, J.T.; Abrahams, B.S.; Salyakina, D.; Imielinski, M.; Bradfield, J.P.; Sleiman, P.M.; et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 2009, 459, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Anney, R.; Klei, L.; Pinto, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; Sykes, N.; Pagnamenta, A.T.; et al. A genome-wide scan for common alleles affecting risk for autism. Hum. Mol. Genet. 2010, 19, 4072–4082. [Google Scholar] [CrossRef] [PubMed]
- Anney, R.; Klei, L.; Pinto, D.; Almeida, J.; Bacchelli, E.; Baird, G.; Bolshakova, N.; Bölte, S.; Bolton, P.F.; Bourgeron, T.; et al. Individual common variants exert weak effects on the risk for autism spectrum disorders. Hum. Mol. Genet. 2012, 21, 4781–4792. [Google Scholar] [CrossRef] [PubMed]
- Torrico, B.; Chiocchetti, A.G.; Bacchelli, E.; Trabetti, E.; Hervás, A.; Franke, B.; Buitelaar, J.K.; Rommelse, N.; Yousaf, A.; Duketis, E.; et al. Lack of replication of previous autism spectrum disorder GWAS hits in European populations. Autism Res. 2017, 10, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, E.; Grove, J.; Shapland, C.Y.; Demontis, D.; Burgess, S.; Rai, D.; Børglum, A.D.; Pourcain, B.S. Discordant associations of educational attainment with ASD and ADHD implicate a polygenic form of pleiotropy. Nat. Commun. 2021, 12, 6534. [Google Scholar] [CrossRef]
- Mattheisen, M.; Grove, J.; Als, T.D.; Martin, J.; Voloudakis, G.; Meier, S.; Demontis, D.; Bendl, J.; Walters, R.; Carey, C.E.; et al. Identification of shared and differentiating genetic architecture for autism spectrum disorder, attention-deficit hyperactivity disorder and case subgroups. Nat. Genet. 2022, 54, 1470–1478. [Google Scholar] [CrossRef]
- Warrier, V.; Zhang, X.; Reed, P.; Havdahl, A.; Moore, T.M.; Cliquet, F.; Leblond, C.S.; Rolland, T.; Rosengren, A.; Rowitch, D.H.; et al. Genetic correlates of phenotypic heterogeneity in autism. Nat. Genet. 2022, 54, 1293–1304. [Google Scholar] [CrossRef]
- Clarke, T.K.; Lupton, M.K.; Fernandez-Pujals, A.M.; Starr, J.; Davies, G.; Cox, S.; Pattie, A.; Liewald, D.C.; Hall, L.S.; MacIntyre, D.J.; et al. Common polygenic risk for autism spectrum disorder (ASD) is associated with cognitive ability in the general population. Mol. Psychiatry 2016, 21, 419–425. [Google Scholar] [CrossRef]
- Cirnigliaro, M.; Chang, T.S.; Arteaga, S.A.; Perez-Cano, L.; Ruzzo, E.K.; Gordon, A.; Bicks, L.K.; Jung, J.-Y.; Lowe, J.K.; Wall, D.P.; et al. The contributions of rare inherited and polygenic risk to ASD in multiplex families. Proc. Natl. Acad. Sci. USA 2023, 120, e2215632120. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Bryois, J.; Kim, A.; Dobbyn, A.; Huckins, L.M.; Munoz-Manchado, A.B.; Ruderfer, D.M.; Genovese, G.; Fromer, M.; Xu, X.; et al. Integrated Bayesian analysis of rare exonic variants to identify risk genes for schizophrenia and neurodevelopmental disorders. Genome Med. 2017, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Simmons, S.K.; Guo, A.; Shetty, A.S.; Ko, M.; Nguyen, L.; Jokhi, V.; Robinson, E.; Oyler, P.; Curry, N.; et al. In vivo Perturb-Seq reveals neuronal and glial abnormalities associated with autism risk genes. Science 2020, 370, eaaz6063. [Google Scholar] [CrossRef] [PubMed]
- Willsey, H.R.; Exner, C.R.T.; Xu, Y.; Everitt, A.; Sun, N.; Wang, B.; Dea, J.; Schmunk, G.; Zaltsman, Y.; Teerikorpi, N.; et al. Parallel in vivo analysis of large-effect autism genes implicates cortical neurogenesis and estrogen in risk and resilience. Neuron 2021, 109, 1409. [Google Scholar] [CrossRef]
- Cederquist, G.Y.; Tchieu, J.; Callahan, S.J.; Ramnarine, K.; Ryan, S.; Zhang, C.; Rittenhouse, C.; Zeltner, N.; Chung, S.Y.; Zhou, T.; et al. A Multiplex Human Pluripotent Stem Cell Platform Defines Molecular and Functional Subclasses of Autism-Related Genes. Cell Stem Cell. 2020, 27, 35–49.e6. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Fleck, J.S.; Martins-Costa, C.; Burkard, T.R.; Themann, J.; Stuempflen, M.; Peer, A.M.; Vertesy, A.; Littleboy, J.B.; Esk, C.; et al. Single-cell brain organoid screening identifies developmental defects in autism. Nature 2023, 621, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Yao, D.; Imaizumi, K.; Chen, X.; Kelley, K.W.; Reis, N.; Thete, M.V.; McKinney, A.A.; Kulkarni, S.; Panagiotakos, G.; et al. Assembloid CRISPR screens reveal impact of disease genes in human neurodevelopment. Nature. 2023, 622, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Jaudon, F.; Thalhammer, A.; Zentilin, L.; Cingolani, L.A. CRISPR-mediated activation of autism gene Itgb3 restores cortical network excitability via mGluR5 signaling. Mol. Ther. Nucleic Acids 2022, 29, 462–480. [Google Scholar] [CrossRef]
- Tamura, S.; Nelson, A.D.; Spratt, P.W.E.; Kyoung, H.; Zhou, X.; Li, Z.; Zhao, J.; Holden, S.S.; Sahagun, A.; Keeshen, C.M.; et al. CRISPR activation rescues abnormalities in SCN2A haploinsufficiency-associated autism spectrum disorder. bioRxiv 2022. [Google Scholar] [CrossRef]
- Krishnan, A.; Zhang, R.; Yao, V.; Theesfeld, C.L.; Wong, A.K.; Tadych, A.; Volfovsky, N.; Packer, A.; Lash, A.; Troyanskaya, O.G. Genome-wide prediction and functional characterization of the genetic basis of autism spectrum disorder. Nat. Neurosci. 2016, 19, 1454–1462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shen, Y. A Cell Type-Specific Expression Signature Predicts Haploinsufficient Autism-Susceptibility Genes. Hum. Mutat. 2017, 38, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Afshar, S.; Rajadhyaksha, A.M.; Potash, J.B.; Han, S. A Machine Learning Approach to Predicting Autism Risk Genes: Validation of Known Genes and Discovery of New Candidates. Front. Genet. 2020, 11, 500064. [Google Scholar] [CrossRef] [PubMed]
- Brueggeman, L.; Koomar, T.; Michaelson, J.J. Forecasting risk gene discovery in autism with machine learning and genome-scale data. Sci. Rep. 2020, 10, 4569. [Google Scholar] [CrossRef]

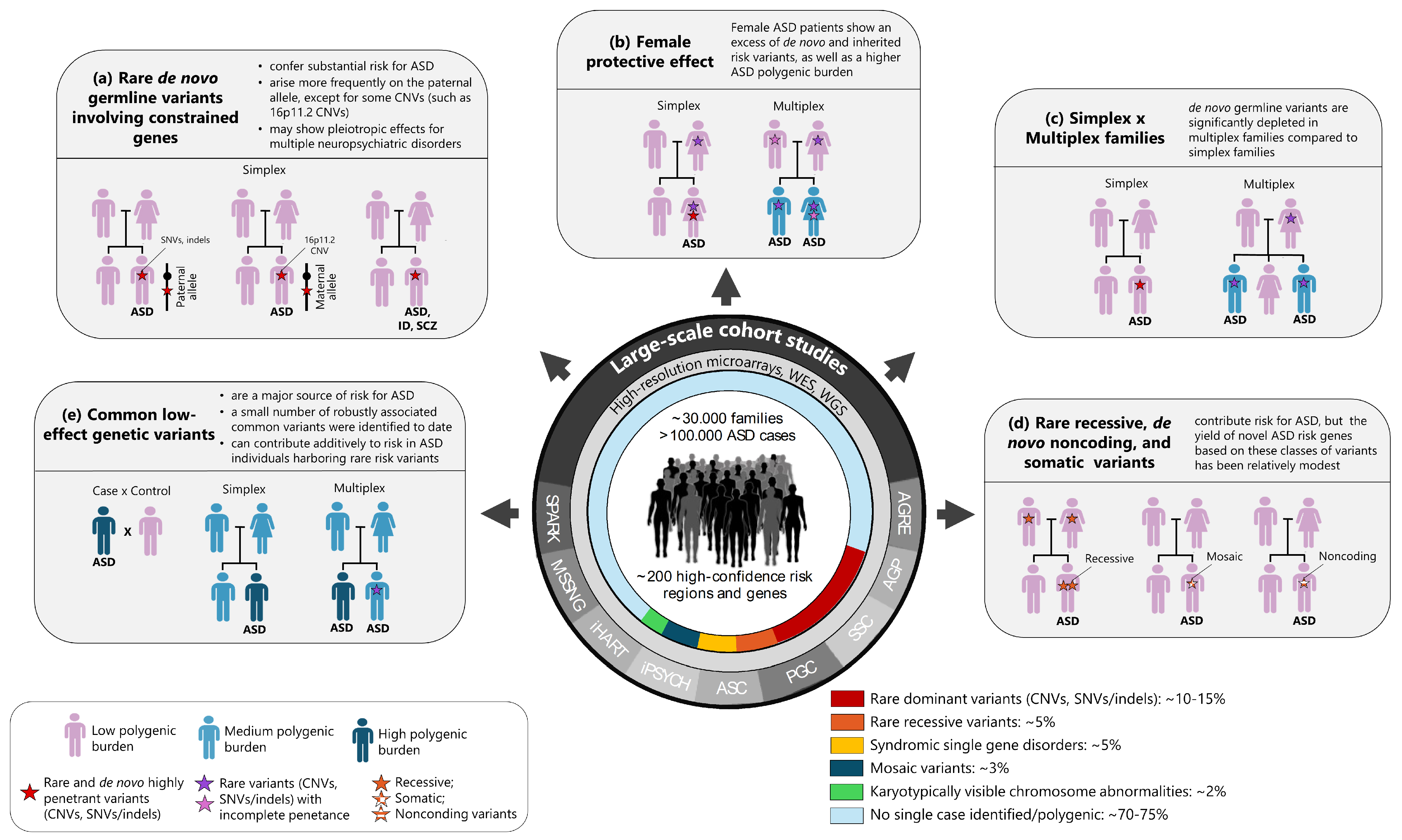
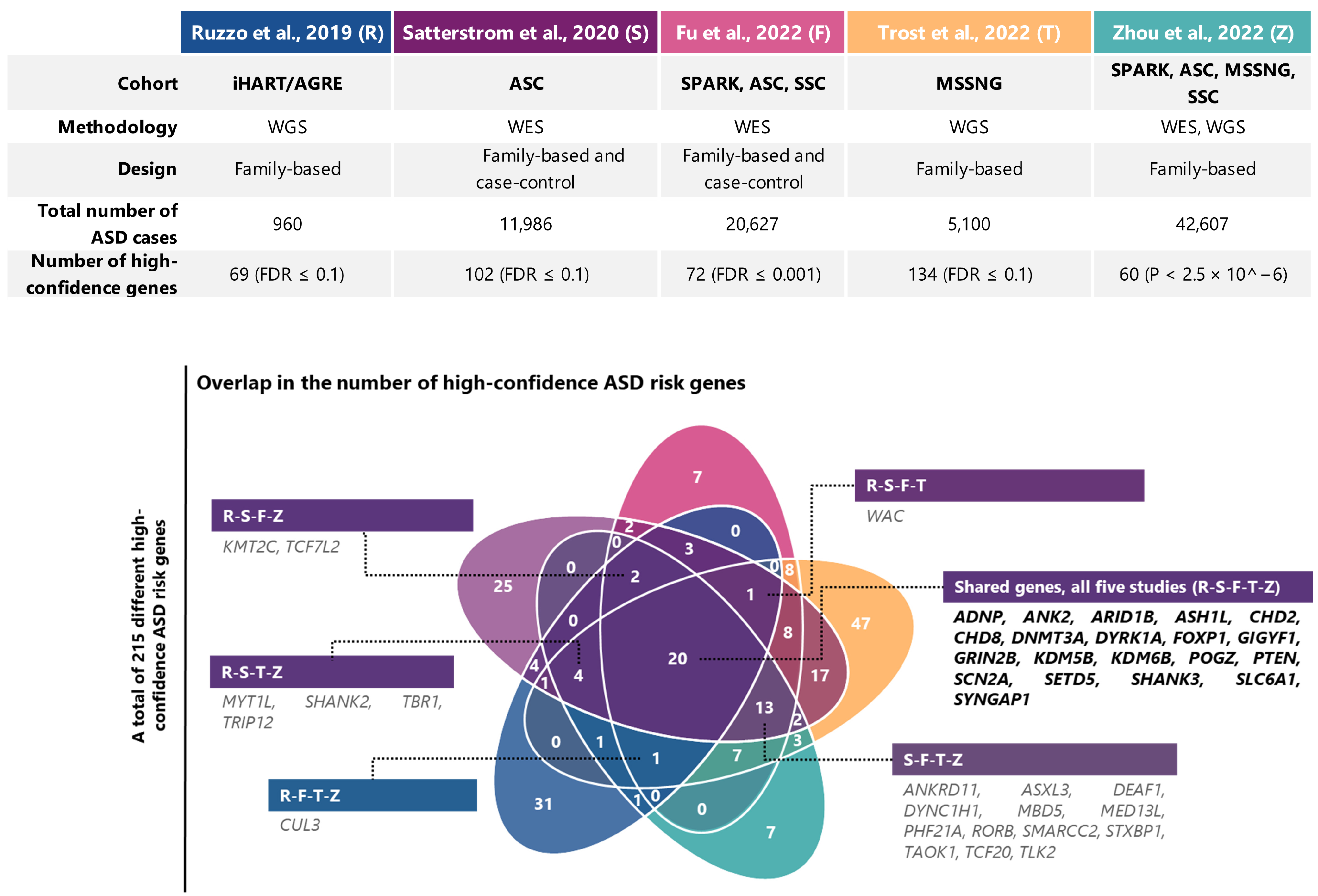
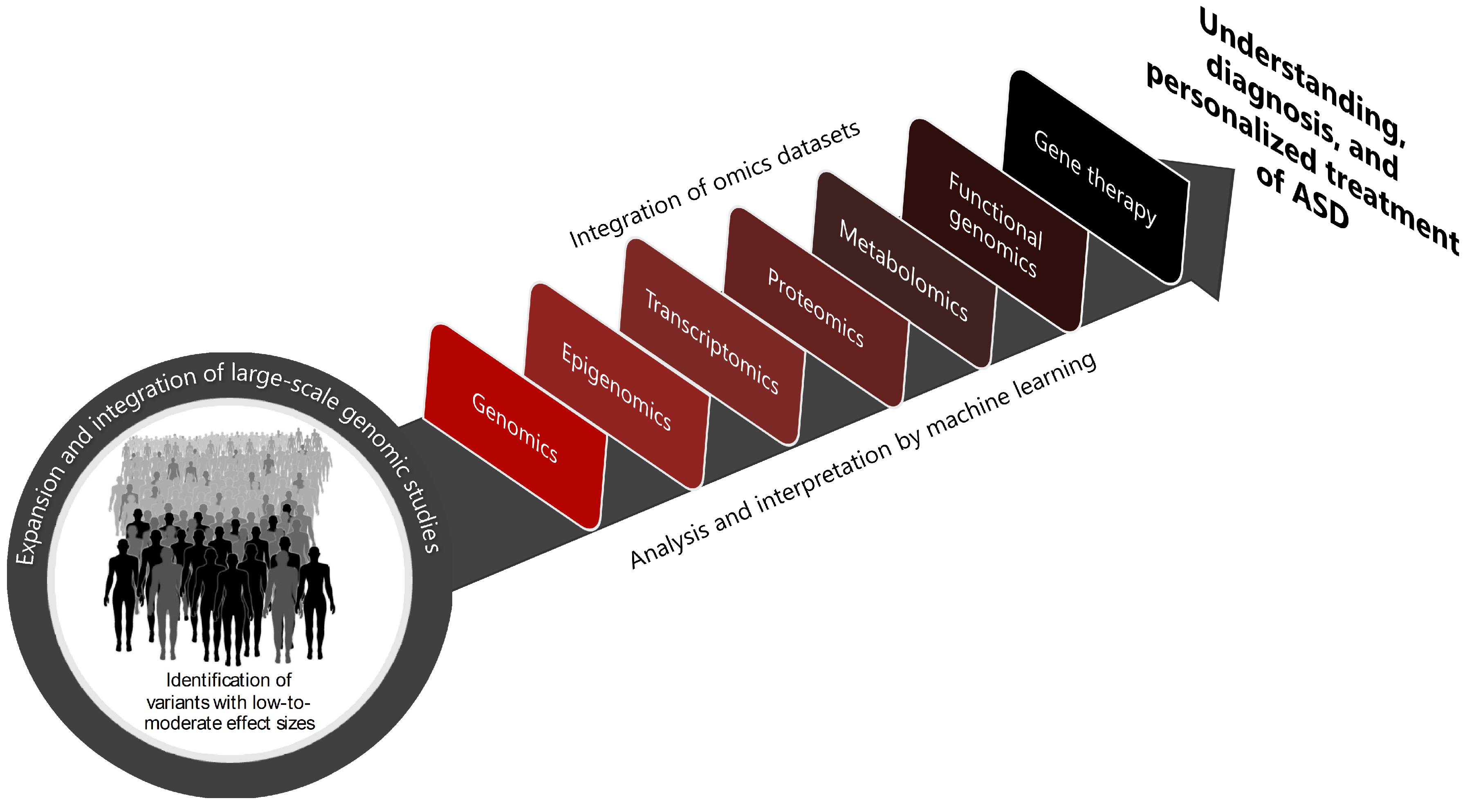

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort/Consortium Studies | References and Website | History | Study Design | Current Size | Genomic Datasets |
|---|---|---|---|---|---|
| Autism Genetic Resource Exchange (AGRE) | [7,8] https://www.autismspeaks.org/about-agre (accessed on 20 May 2024) | Established in 1997 by Cure Autism Now (CAN), AGRE is currently funded by the National Institute of Mental Health and Autism Speaks, which merged with CAN in 2006. AGRE houses a collection of biomaterials and genetic and phenotypic data from families with ASD, made available to the greater scientific community | Simplex and multiplex families | ~2000 families | Genome-wide genotyping, WES, WGS |
| Autism Genome Project (AGP) a | [9] https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000267.v5.p2 (accessed on 20 May 2024) | Established in 2004 by the National Alliance for Autism Research and the National Institutes of Health, AGP is now coordinated by Autism Speaks. The project involves more than 50 centers in North America and Europe that have pooled their DNA samples in a collaborative effort to examine families for SNPs and CNVs affecting risk for ASD | Simplex and multiplex families | ~2600 families | Genome-wide genotyping |
| Simons Simplex Collection (SSC) | [10] https://www.sfari.org/resource/simons-simplex-collection/ (accessed on 20 May 2024) | Established in 2008 by the Simons Foundation Autism Research Initiative, SSC is a repository of clinical and genetic data from simplex ASD families designed to facilitate the discovery of rare and de novo variation | Simplex families | ~2600 families | Genome-wide genotyping, WES, WGS |
| Psychiatric Genomics Consortium (PGC)–ASD Working Group | [11,12] http://www.med.unc.edu/pgc (accessed on 20 May 2024) | Founded in 2007 by Dr. Patrick Sullivan with the aim of conducting statistically rigorous and comprehensive GWAS meta-analysis for common psychiatric disorders. The ASD Working Group was founded at the start of PGC, in 2008, and currently includes 48 investigators from 10 countries | Simplex families, case-control | ~18,000 ASD cases b | Genome-wide genotyping |
| Autism Sequencing Consortium (ASC) c | [13] https://genome.emory.edu/ASC/ (accessed on 20 May 2024) | Founded in 2010 by Dr. Joseph D. Buxbaum, the ASC is a large-scale international genomic consortium integrating several ASD cohorts and WES data from over 100 investigators. The project is funded by the National Institute of Mental Health with additional support from the National Human Genome Research Institute | Simplex families, case-control | ~12,000 ASD cases | WES |
| Integrative Psychiatric Research (iPSYCH) | [14] https://ipsych.dk/en/about-ipsych (accessed on 20 May 2024) | Founded in 2012 by six leading Danish researchers in the fields of psychiatry and genetics, the project consists of a large Danish population-based case-cohort sample aimed at unraveling the genetic and environmental architecture of five of the most serious mental disorders (schizophrenia, ASD, ADHD, bipolar disorder, and depression) | Case-control | ~15,000 ASD cases d | Genome-wide genotyping, WES |
| Hartwell Autism Research and Technology Initiative (iHART)/AGRE e | https://thehartwellfoundation.com/iHART.shtml (accessed on 20 May 2024) | Launched in 2014 by the Hartwell Foundation, the iHART database contains WGS data from multiplex families of the AGRE collection | Multiplex families | ~1000 families (~4500 ASD cases | WGS |
| MSSNG a | [15] https://research.mss.ng/ (accessed on 20 May 2024) | Launched in 2014, the project is a collaboration between Autism Speaks, Verily, DNAstack, Hospital for Sick Children, and the research community. Its primary goal was to create the world’s largest WGS database of 10,000 individuals with ASD and their families with deep phenotyping | Simplex and multiplex families | 4258 families (5100 ASD cases) | WGS |
| Simons Foundation Powering Autism Research for Knowledge (SPARK) | [16] https://www.sfari.org/resource/spark/ (accessed on 20 May 2024) | Established in 2015 by the Simons Foundation Autism Research Initiative, the project aims to establish a permanent repository of genetic samples from 50,000 families with ASD living in the U.S. | Simplex and multiplex families | ~50,000 ASD cases | Genome-wide genotyping, WES, WGS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nóbrega, I.d.S.; Teles e Silva, A.L.; Yokota-Moreno, B.Y.; Sertié, A.L. The Importance of Large-Scale Genomic Studies to Unravel Genetic Risk Factors for Autism. Int. J. Mol. Sci. 2024, 25, 5816. https://doi.org/10.3390/ijms25115816
Nóbrega IdS, Teles e Silva AL, Yokota-Moreno BY, Sertié AL. The Importance of Large-Scale Genomic Studies to Unravel Genetic Risk Factors for Autism. International Journal of Molecular Sciences. 2024; 25(11):5816. https://doi.org/10.3390/ijms25115816
Chicago/Turabian StyleNóbrega, Isabella de Sousa, André Luíz Teles e Silva, Bruno Yukio Yokota-Moreno, and Andréa Laurato Sertié. 2024. "The Importance of Large-Scale Genomic Studies to Unravel Genetic Risk Factors for Autism" International Journal of Molecular Sciences 25, no. 11: 5816. https://doi.org/10.3390/ijms25115816