
PvdL Orchestrates the Assembly of the Nonribosomal Peptide Synthetases Involved in Pyoverdine Biosynthesis in Pseudomonas aeruginosa

 , ,
, , 
Abstract
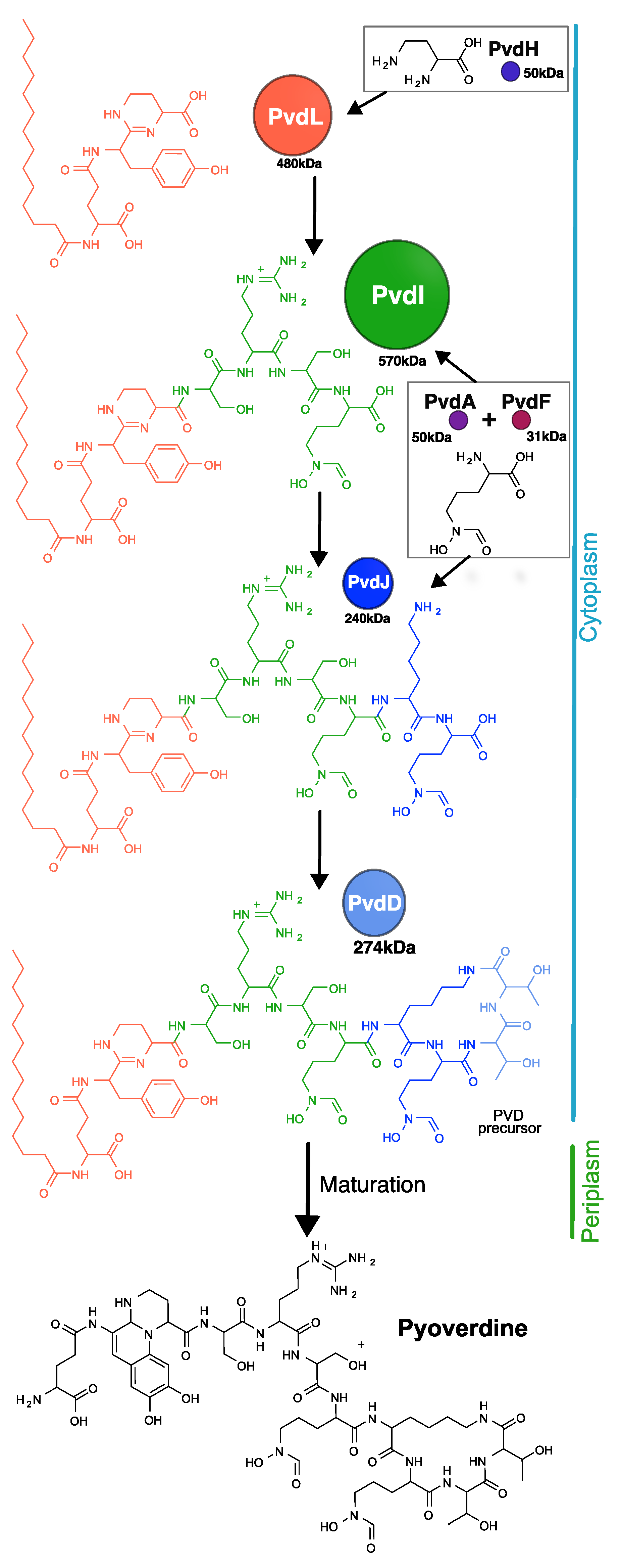
1. Introduction
2. Results
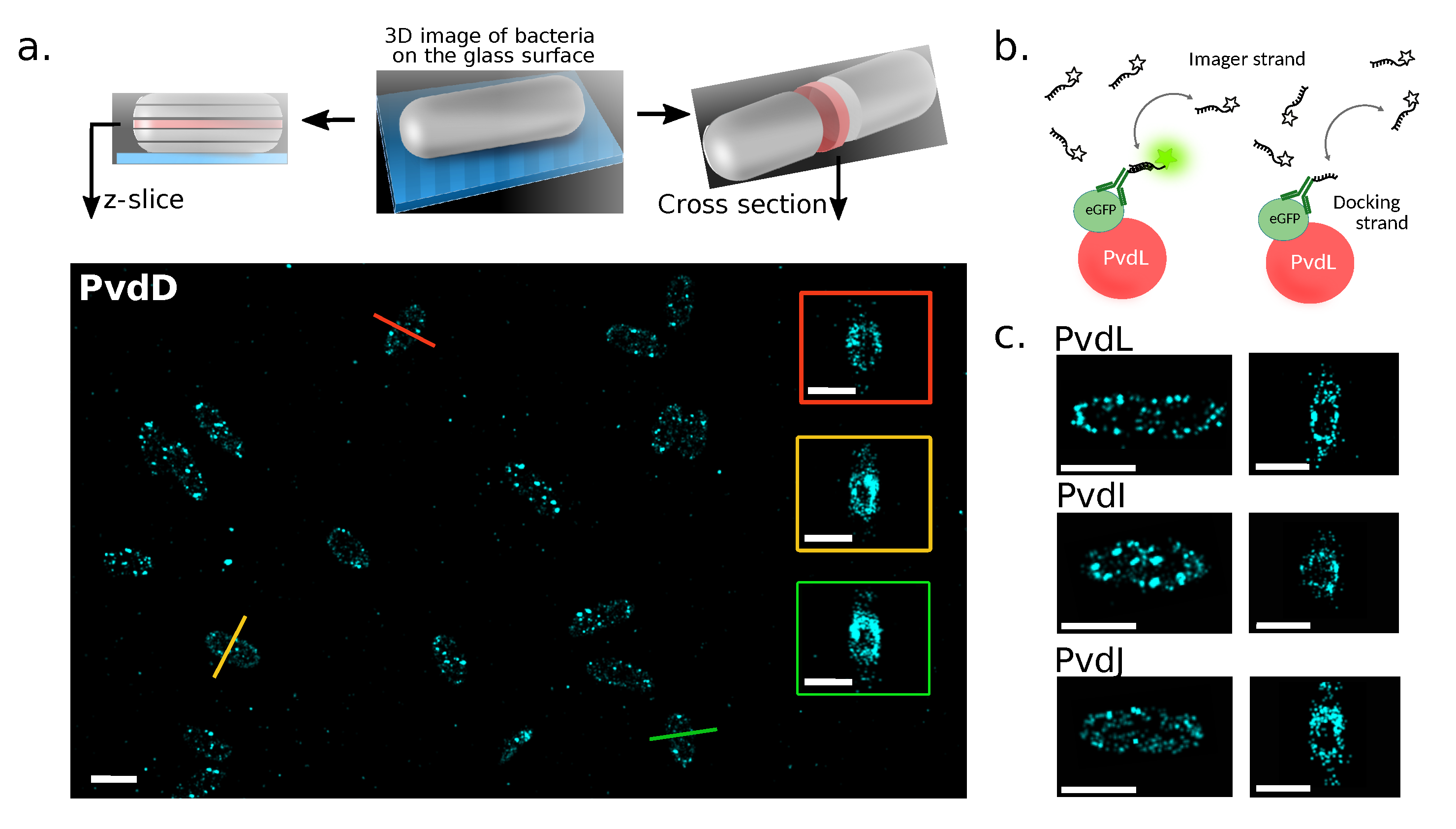
2.1. Localizations of NRPSs in Cells
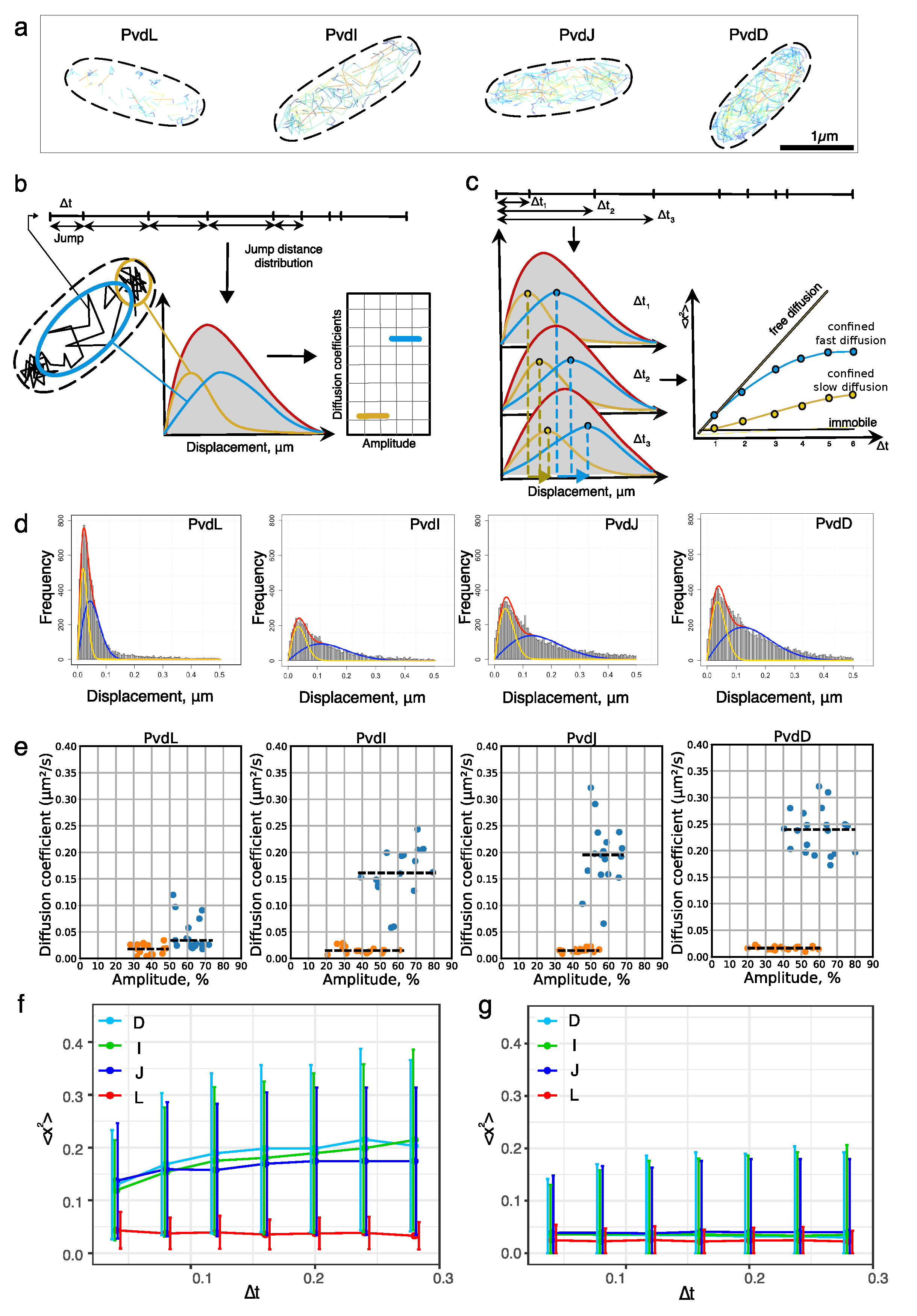
2.2. Diffusion of NRPSs in Living Cells
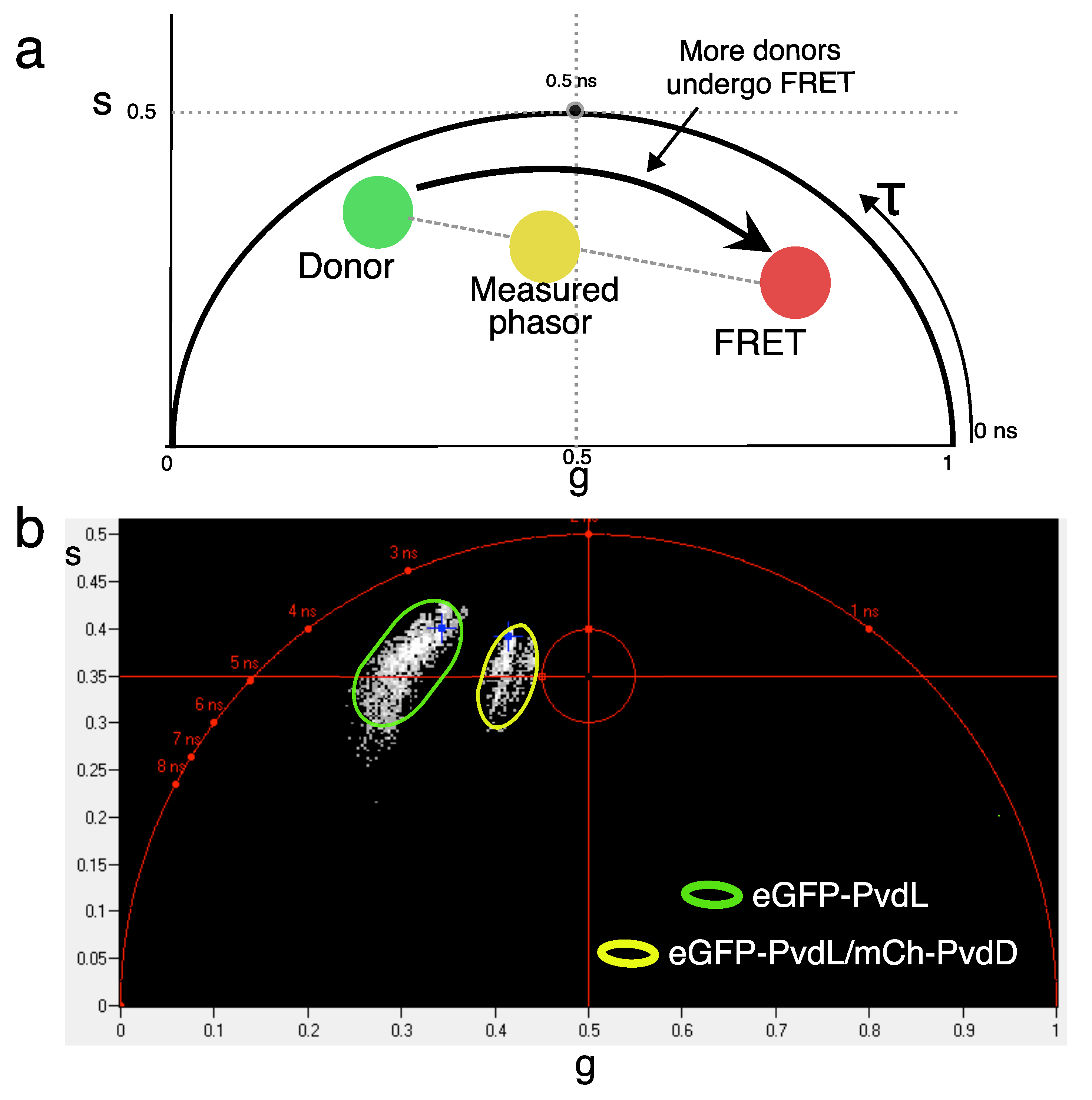
2.3. NRPS-NRPS Interactions
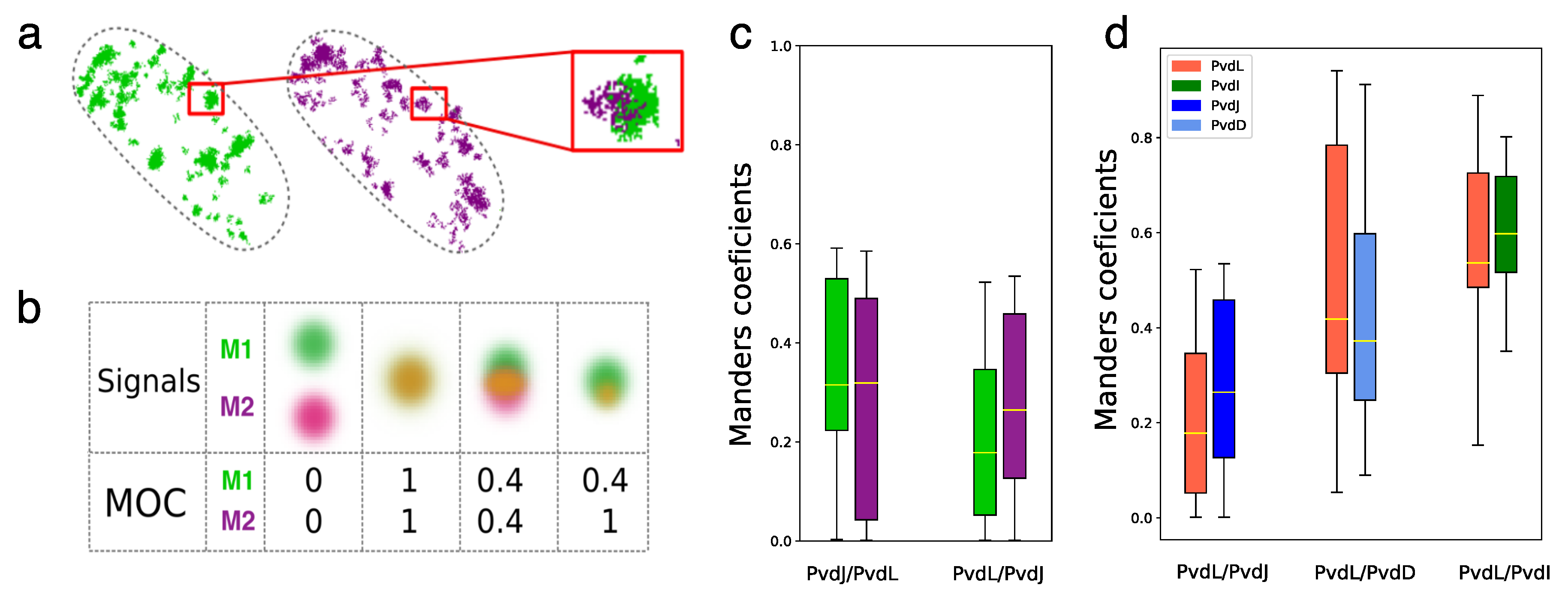
2.4. Co-Localizations of NRPSs
3. Discussion
4. Materials and Methods
4.1. Bacteria Mutants, Cell Culture, and Labeling
4.2. Microscopy and Analysis
4.3. FLIM-FRET
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NRPS | Nonribosomal peptide synthetase |
| FLIM-FRET | Förster resonance energy transfer measured by fluorescence lifetime imaging |
| IQR | Interquartile range |
| MSD | Mean square displacement |
| JD | Jump distance |
| OD | Optical density |
References
- Crone, S.; Vives-Flórez, M.; Kvich, L.; Saunders, A.M.; Malone, M.; Nicolaisen, M.H.; Martínez-García, E.; Rojas-Acosta, C.; Catalina Gomez-Puerto, M.; Calum, H.; et al. The environmental occurrence of Pseudomonas aeruginosa. Apmis 2020, 128, 220–231. [Google Scholar] [CrossRef]
- Vining, L.C. Secondary metabolism, inventive evolution and biochemical diversity—A review. Gene 1992, 115, 135–140. [Google Scholar] [CrossRef]
- Gross, H.; Loper, J.E. Genomics of secondary metabolite production by Pseudomonas spp. Nat. Prod. Rep. 2009, 26, 1408–1446. [Google Scholar] [CrossRef]
- Dell, M.; Dunbar, K.L.; Hertweck, C. Ribosome-independent peptide biosynthesis: The challenge of a unifying nomenclature. Nat. Prod. Rep. 2021, 39, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Gulick, A.M. Nonribosomal peptide synthetase biosynthetic clusters of ESKAPE pathogens. Nat. Prod. Rep. 2017, 34, 981–1009. [Google Scholar] [CrossRef] [PubMed]
- Süssmuth, R.D.; Mainz, A. Nonribosomal Peptyde Synthesis—Principles and Pprospects. Angew. Chem. 2017, 56, 3770–3821. [Google Scholar] [CrossRef] [PubMed]
- Finking, R.; Marahiel, M.A. Biosynthesis of nonribosomal peptides. Annu. Rev. Microbiol. 2004, 58, 453–488. [Google Scholar] [CrossRef]
- Schalk, I.J.; Rigouin, C.; Godet, J. An overview of siderophore biosynthesis among fluorescent Pseudomonads and new insights into their complex cellular organization. Environ. Microbiol. 2020, 22, 1447–1466. [Google Scholar] [CrossRef]
- Loaces, I.; Ferrando, L.; Scavino, A.F. Dynamics, Diversity and Function of Endophytic Siderophore-Producing Bacteria in Rice. Microb. Ecol. 2011, 61, 606–618. [Google Scholar] [CrossRef]
- Schalk, I.J.; Perraud, Q. Pseudomonas aeruginosa and its multiple strategies to access iron. Environ. Microbiol. 2023, 25, 811–831. [Google Scholar] [CrossRef]
- Imperi, F.; Massai, F.; Facchini, M.; Frangipani, E.; Visaggio, D.; Leoni, L.; Bragonzi, A.; Visca, P. Repurposing the antimycotic drug flucytosine for suppression of Pseudomonas aeruginosa pathogenicity. Proc. Natl. Acad. Sci. USA 2013, 110, 7458–7463. [Google Scholar] [CrossRef] [PubMed]
- Kirienko, D.R.; Kang, D.; Kirienko, N.V. Novel pyoverdine inhibitors mitigate Pseudomonas aeruginosa pathogenesis. Front. Microbiol. 2019, 10, 3317. [Google Scholar] [CrossRef] [PubMed]
- Sexton, D.J.; Schuster, M. Nutrient limitation determines the fitness of cheaters in bacterial siderophore cooperation. Nat. Commun. 2017, 8, 230. [Google Scholar] [CrossRef] [PubMed]
- Weigert, M.; Kümmerli, R. The physical boundaries of public goods cooperation between surface-attached bacterial cells. Proc. R. Soc. B Biol. Sci. 2017, 284, 20170631. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.; Özkaya, Ö.; Kümmerli, R. Bacterial siderophores in community and host interactions. Nat. Rev. Microbiol. 2020, 18, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Ringel, M.T.; Brüser, T. The biosynthesis of pyoverdines. Microb. Cell 2018, 5, 424–437. [Google Scholar] [CrossRef] [PubMed]
- Imperi, F.; Visca, P. Subcellular localization of the pyoverdine biogenesis machinery of Pseudomonas aeruginosa: A membrane-associated “siderosome”. FEBS Lett. 2013, 587, 3387–3391. [Google Scholar] [CrossRef] [PubMed]
- Gasser, V.; Guillon, L.; Cunrath, O.; Schalk, I.J. Cellular organization of siderophore biosynthesis in Pseudomonas aeruginosa: Evidence for siderosomes. J. Inorg. Biochem. 2015, 148, 27–34. [Google Scholar] [CrossRef]
- Guillon, L.; Altenburger, S.; Graumann, P.L.; Schalk, I.J. Deciphering protein dynamics of the siderophore pyoverdine pathway in Pseudomonas aeruginosa. PLoS ONE 2013, 8, e79111. [Google Scholar] [CrossRef]
- Gasser, V.; Malrieu, M.; Forster, A.; Mély, Y.; Schalk, I.J.; Godet, J. In cellulo FRET-FLIM and single molecule tracking reveal the supra-molecular organization of the pyoverdine bio-synthetic enzymes in Pseudomonas aeruginosa. Q. Rev. Biophys. 2020, 53, e1. [Google Scholar] [CrossRef]
- Qiu, X.Y.; Xie, S.S.; Min, L.; Wu, X.M.; Zhu, L.Y.; Zhu, L. Spatial organization of enzymes to enhance synthetic pathways in microbial chassis: A systematic review. Microb. Cell Factories 2018, 17, 120. [Google Scholar] [CrossRef] [PubMed]
- Llopis, P.M.; Jackson, A.F.; Sliusarenko, O.; Surovtsev, I.; Heinritz, J.; Emonet, T.; Jacobs-Wagner, C. Spatial organization of the flow of genetic information in bacteria. Nature 2010, 466, 77–81. [Google Scholar] [CrossRef]
- Surovtsev, I.V.; Jacobs-Wagner, C. Subcellular Organization: A Critical Feature of Bacterial Cell Replication. Cell 2018, 172, 1271–1293. [Google Scholar] [CrossRef] [PubMed]
- Manko, H.; Normant, V.; Perraud, Q.; Steffan, T.; Gasser, V.; Boutant, E.; Réal, É.; Schalk, I.J.; Mély, Y.; Godet, J. Flim-fret measurements of protein-protein interactions in live bacteria. J. Vis. Exp. 2020, 2020, 1–21. [Google Scholar] [CrossRef]
- Jungmann, R.; Avendaño, M.S.; Woehrstein, J.B.; Dai, M.; Shih, W.M.; Yin, P. Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nat. Methods 2014, 11, 313–318. [Google Scholar] [CrossRef]
- Schnitzbauer, J.; Strauss, M.T.; Schlichthaerle, T.; Schueder, F.; Jungmann, R. Super-resolution microscopy with DNA-PAINT. Nat. Protoc. 2017, 12, 1198–1228. [Google Scholar] [CrossRef] [PubMed]
- Neeli-Venkata, R.; Martikainen, A.; Gupta, A.; Gonçalves, N.; Fonseca, J.; Ribeiro, A.S. Robustness of the process of nucleoid exclusion of protein aggregates in Escherichia coli. J. Bacteriol. 2016, 198, 898–906. [Google Scholar] [CrossRef]
- Bakshi, S.; Siryaporn, A.; Goulian, M.; Weisshaar, J.C. Superresolution imaging of ribosomes and RNA polymerase in live Escherichia coli cells. Mol. Microbiol. 2012, 85, 21–38. [Google Scholar] [CrossRef]
- Manley, S.; Gillette, J.M.; Lippincott-Schwartz, J. Single-Particle Tracking Photoactivated Localization Microscopy for Mapping Single-Molecule Dynamics. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2010; Volume 475, pp. 109–120. [Google Scholar] [CrossRef]
- Tinevez, J.Y.; Perry, N.; Schindelin, J.; Hoopes, G.M.; Reynolds, G.D.; Laplantine, E.; Bednarek, S.Y.; Shorte, S.L.; Eliceiri, K.W. TrackMate: An open and extensible platform for single-particle tracking. Methods 2017, 115, 80–90. [Google Scholar] [CrossRef]
- Ershov, D.; Phan, M.S.; Pylvänäinen, J.W.; Rigaud, S.U.; Le Blanc, L.; Charles-Orszag, A.; Conway, J.R.; Laine, R.F.; Roy, N.H.; Bonazzi, D.; et al. TrackMate 7: Integrating state-of-the-art segmentation algorithms into tracking pipelines. Nat. Methods 2022, 19, 829–832. [Google Scholar] [CrossRef]
- Weimann, L.; Ganzinger, K.A.; McColl, J.; Irvine, K.L.; Davis, S.J.; Gay, N.J.; Bryant, C.E.; Klenerman, D. A Quantitative Comparison of Single-Dye Tracking Analysis Tools Using Monte Carlo Simulations. PLoS ONE 2013, 8, e64287. [Google Scholar] [CrossRef] [PubMed]
- Rowland, D.J.; Tuson, H.H.; Biteen, J.S. Resolving Fast, Confined Diffusion in Bacteria with Image Correlation Spectroscopy. Biophys. J. 2016, 110, 2241–2251. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, A.; Day, R.N. Visualizing protein interactions in living cells using digitized GFP imaging and FRET microscopy. In Methods in Cell Biology; Elsevier: Amsterdam, The Netherlands, 1999; Volume 58, pp. 293–314. [Google Scholar] [CrossRef]
- Manders, E.M.; Verbeek, F.J.; Aten, J.A. Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 1993, 169, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Levet, F.; Julien, G.; Galland, R.; Butler, C.; Beghin, A.; Chazeau, A.; Hoess, P.; Ries, J.; Giannone, G.; Sibarita, J.B. A tessellation-based colocalization analysis approach for single-molecule localization microscopy. Nat. Commun. 2019, 10, 2379. [Google Scholar] [CrossRef] [PubMed]
- Conrado, R.J.; Varner, J.D.; DeLisa, M.P. Engineering the spatial organization of metabolic enzymes: Mimicking nature’s synergy. Curr. Opin. Biotechnol. 2008, 19, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Kang, W.; Ma, T.; Liu, M.; Qu, J.; Liu, Z.; Zhang, H.; Shi, B.; Fu, S.; Ma, J.; Lai, L.T.F.; et al. Modular enzyme assembly for enhanced cascade biocatalysis and metabolic flux. Nat. Commun. 2019, 10, 4248. [Google Scholar] [CrossRef] [PubMed]
- Hannauer, M.; Schäfer, M.; Hoegy, F.; Gizzi, P.; Wehrung, P.; Mislin, G.L.; Budzikiewicz, H.; Schalk, I.J. Biosynthesis of the pyoverdine siderophore of Pseudomonas aeruginosa involves precursors with a myristic or a myristoleic acid chain. FEBS Lett. 2012, 586, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Pawson, T.; Scott, J.D. Signaling through scaffold, anchoring, and adaptor proteins. Science 1997, 278, 2075–2080. [Google Scholar] [CrossRef]
- Meneely, K.M.; Barr, E.W.; Bollinger, J.M.; Lamb, A.L. Kinetic mechanism of ornithine hydroxylase (PvdA) from Pseudomonas aeruginosa: Substrate triggering of O2 addition but not flavin reduction. Biochemistry 2009, 48, 4371–4376. [Google Scholar] [CrossRef]
- Wheeldon, I.; Minteer, S.D.; Banta, S.; Barton, S.C.; Atanassov, P.; Sigman, M. Substrate channelling as an approach to cascade reactions. Nat. Chem. 2016, 8, 299–309. [Google Scholar] [CrossRef]
- Winsor, G.L.; Griffiths, E.J.; Lo, R.; Dhillon, B.K.; Shay, J.A.; Brinkman, F.S. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res. 2016, 44, D646–D653. [Google Scholar] [CrossRef] [PubMed]
- Schalk, I.J. Siderophore–antibiotic conjugates: Exploiting iron uptake to deliver drugs into bacteria. Clin. Microbiol. Infect. 2018, 24, 801–802. [Google Scholar] [CrossRef] [PubMed]
- Ovesný, M.; Křížek, P.; Borkovec, J.; Švindrych, Z.; Hagen, G.M. ThunderSTORM: A comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics 2014, 30, 2389–2390. [Google Scholar] [CrossRef] [PubMed]
- Ries, J. SMAP: A modular super-resolution microscopy analysis platform for SMLM data. Nat. Methods 2020, 17, 870–872. [Google Scholar] [CrossRef]
- Godet, J.; Mély, Y. Exploring protein-protein interactions with large differences in protein expression levels using FLIM-FRET. Methods Appl. Fluoresc. 2020, 8, 14007. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain Name | (ns) | (ns) | FRET Efficiency |
|---|---|---|---|
| eGFP-PvdL/PvdJ-mCherry | 2.3 | 2.3 | 0.00 |
| PvdJ-eGFP/mCherry-PvdL | 2.3 | 2.3 | 0.00 |
| eGFP-PvdL/mCherry-PvdD | 2.3 | 2.0 | 0.13 |
| eGFP-PvdL/PvdI-mCherry | 2.3 | 2.2 | 0.04 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manko, H.; Steffan, T.; Gasser, V.; Mély, Y.; Schalk, I.; Godet, J. PvdL Orchestrates the Assembly of the Nonribosomal Peptide Synthetases Involved in Pyoverdine Biosynthesis in Pseudomonas aeruginosa. Int. J. Mol. Sci. 2024, 25, 6013. https://doi.org/10.3390/ijms25116013
Manko H, Steffan T, Gasser V, Mély Y, Schalk I, Godet J. PvdL Orchestrates the Assembly of the Nonribosomal Peptide Synthetases Involved in Pyoverdine Biosynthesis in Pseudomonas aeruginosa. International Journal of Molecular Sciences. 2024; 25(11):6013. https://doi.org/10.3390/ijms25116013
Chicago/Turabian StyleManko, Hanna, Tania Steffan, Véronique Gasser, Yves Mély, Isabelle Schalk, and Julien Godet. 2024. "PvdL Orchestrates the Assembly of the Nonribosomal Peptide Synthetases Involved in Pyoverdine Biosynthesis in Pseudomonas aeruginosa" International Journal of Molecular Sciences 25, no. 11: 6013. https://doi.org/10.3390/ijms25116013
APA StyleManko, H., Steffan, T., Gasser, V., Mély, Y., Schalk, I., & Godet, J. (2024). PvdL Orchestrates the Assembly of the Nonribosomal Peptide Synthetases Involved in Pyoverdine Biosynthesis in Pseudomonas aeruginosa. International Journal of Molecular Sciences, 25(11), 6013. https://doi.org/10.3390/ijms25116013