
Marine-Derived Phosphoeleganin and Its Semisynthetic Derivative Decrease IL6 Levels and Improve Insulin Signaling in Human Hepatocellular Carcinoma Cells

 , ,
, ,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Results
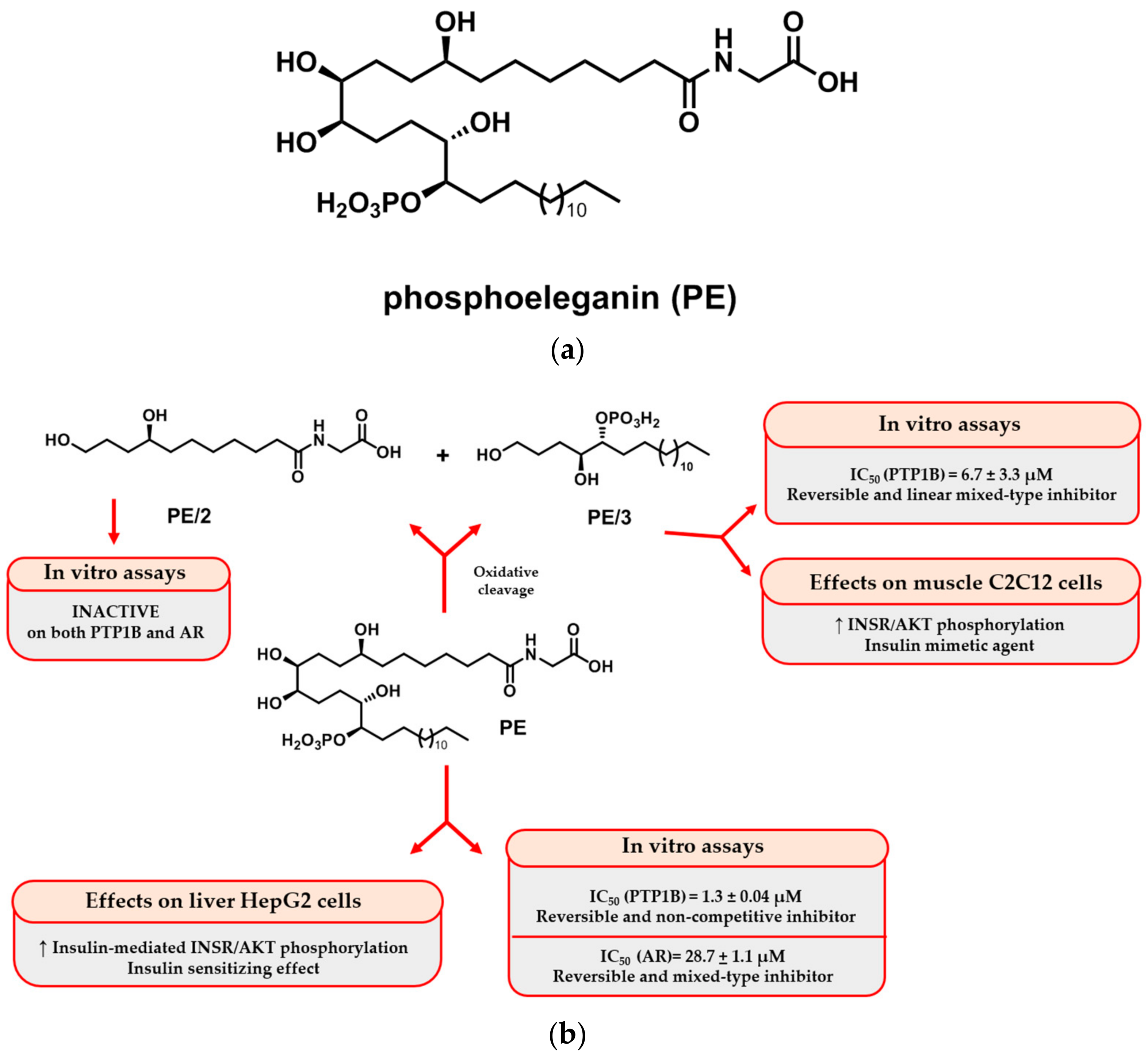
2.1. Isolation and Purification of PE and Semisynthetic Approach for Obtaining PE/2 and PE/3
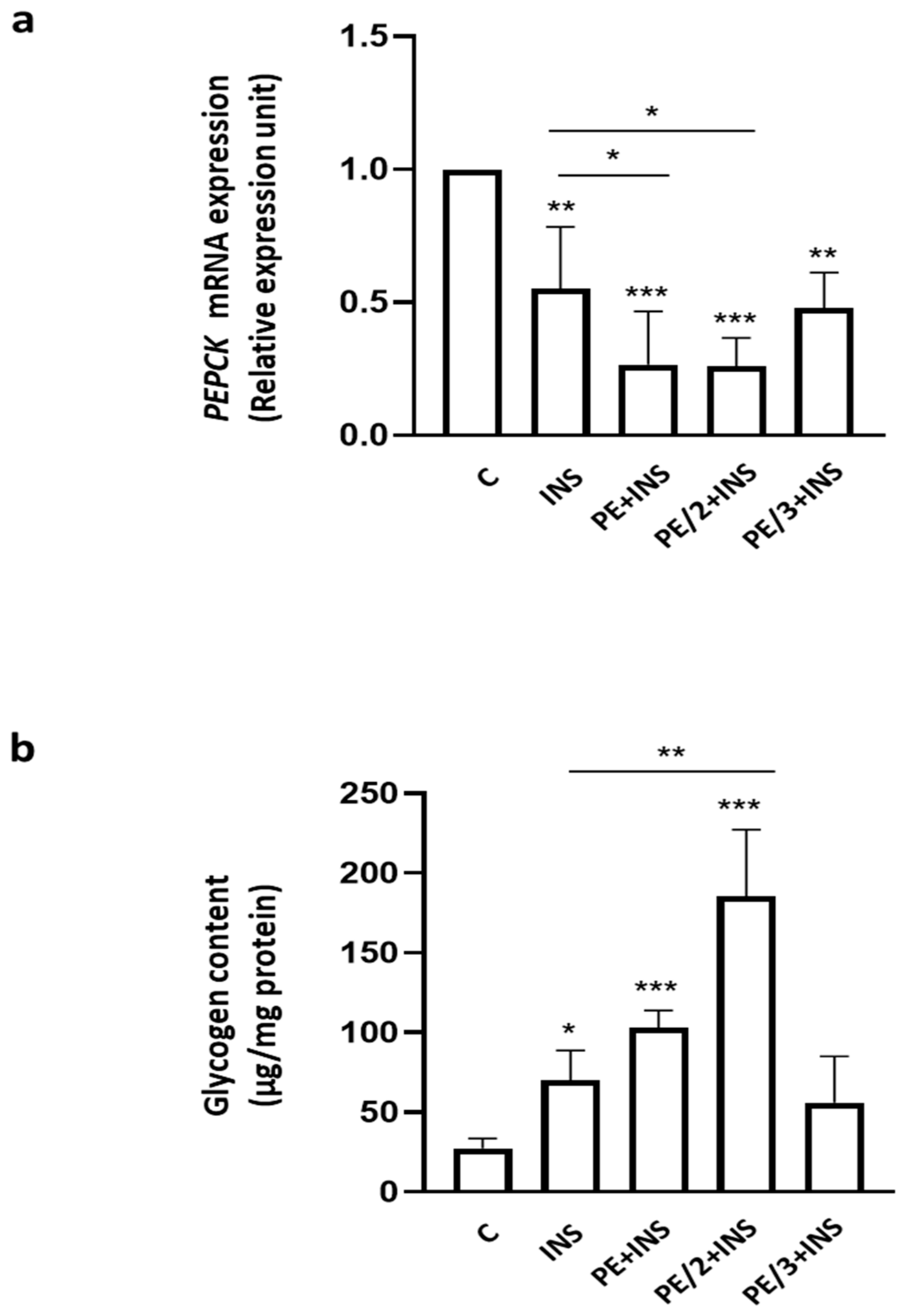
2.2. Role of Phosphoeleganin and Its Derivatives in Insulin Signaling
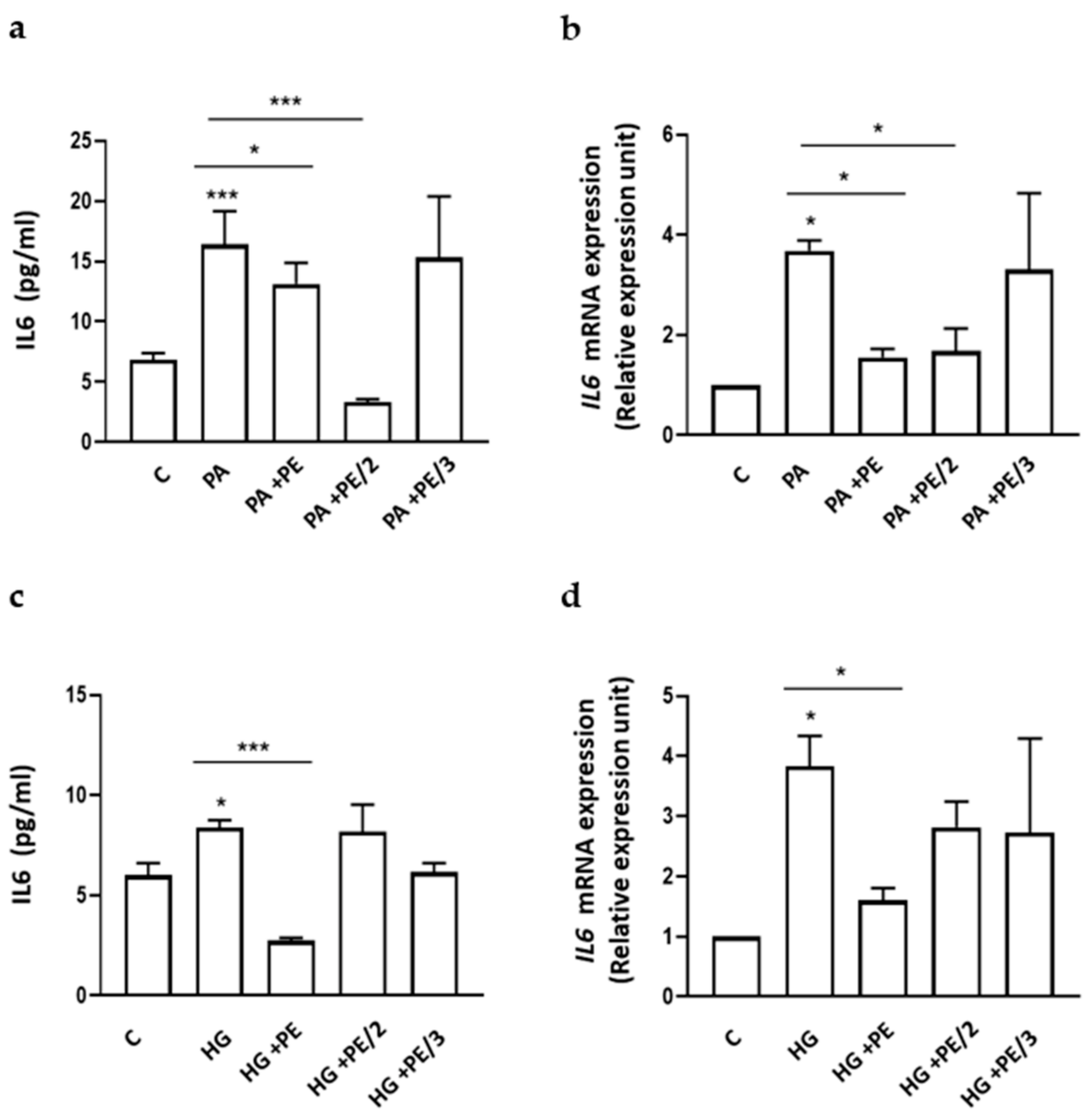
2.3. Role of Phosphoeleganin and Its Derivatives on Cytokine Secretion and Expression
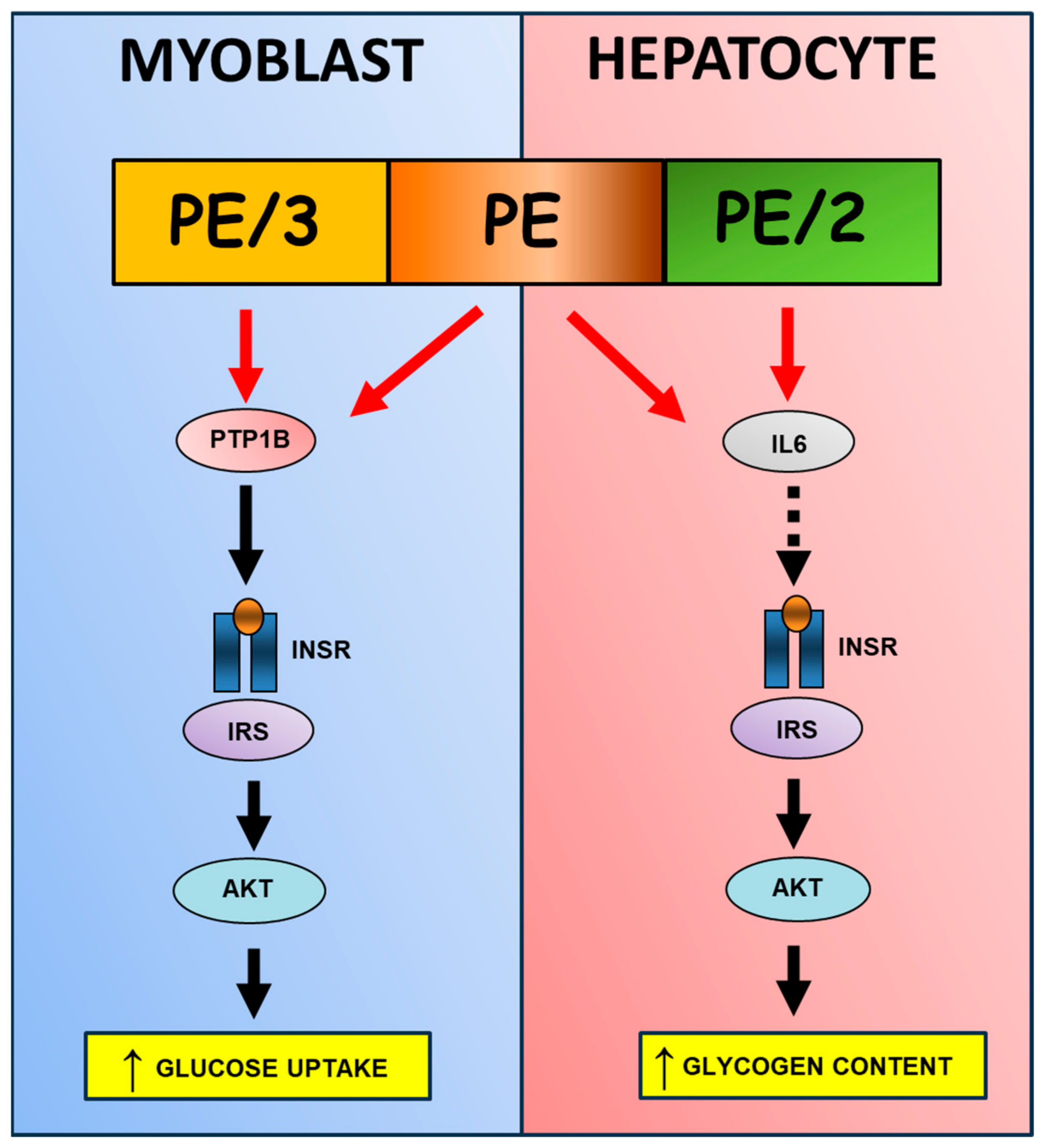
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Collection, Extraction, and Fraction of the Ascidian S. elegans
4.3. Synthesis of PE/2 and PE/3 by Oxidative Cleavage of the Natural Metabolite
4.4. Cell Culture Procedures and Cell Proliferation
4.5. Western Blot Analysis
4.6. Luminex Assay
4.7. Real-Time RT-PCR Analysis
4.8. Determination of Glycogen Content
4.9. Statistical Procedures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leclercq, I.A.; Da Silva Morais, A.; Schroyen, B.; Van Hul, N.; Geerts, A. Insulin Resistance in Hepatocytes and Sinusoidal Liver Cells: Mechanisms and Consequences. J. Hepatol. 2007, 47, 142–156. [Google Scholar] [CrossRef]
- Li, M.; Chi, X.; Wang, Y.; Setrerrahmane, S.; Xie, W.; Xu, H. Trends in Insulin Resistance: Insights into Mechanisms and Therapeutic Strategy. Signal Transduct. Target. Ther. 2022, 7, 216. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab. 2018, 27, 22–41. [Google Scholar] [CrossRef] [PubMed]
- Titchenell, P.M.; Lazar, M.A.; Birnbaum, M.J. Unraveling the Regulation of Hepatic Metabolism by Insulin. Trends Endocrinol. Metab. 2017, 28, 497–505. [Google Scholar] [CrossRef]
- Senn, J.J.; Klover, P.J.; Nowak, I.A.; Mooney, R.A. Interleukin-6 Induces Cellular Insulin Resistance in Hepatocytes. Diabetes 2002, 51, 3391–3399. [Google Scholar] [CrossRef] [PubMed]
- Kranendonk, M.E.G.; Visseren, F.L.J.; van Herwaarden, J.A.; Nolte-’t Hoen, E.N.M.; de Jager, W.; Wauben, M.H.M.; Kalkhoven, E. Effect of Extracellular Vesicles of Human Adipose Tissue on Insulin Signaling in Liver and Muscle Cells. Obesity 2014, 22, 2216–2223. [Google Scholar] [CrossRef] [PubMed]
- Nov, O.; Kohl, A.; Lewis, E.C.; Bashan, N.; Dvir, I.; Ben-Shlomo, S.; Fishman, S.; Wueest, S.; Konrad, D.; Rudich, A. Interleukin-1β May Mediate Insulin Resistance in Liver-Derived Cells in Response to Adipocyte Inflammation. Endocrinology 2010, 151, 4247–4256. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.D.; Kim, Y.H.; Cho, Y.M.; Kim, D.K.; Ahn, S.W.; Lee, J.M.; Chanda, D.; Shong, M.; Lee, C.H.; Choi, H.S. Metformin Ameliorates IL-6-Induced Hepatic Insulin Resistance via Induction of Orphan Nuclear Receptor Small Heterodimer Partner (SHP) in Mouse Models. Diabetologia 2012, 55, 1482–1494. [Google Scholar] [CrossRef]
- Kuai, Z.; Chao, X.; He, Y.; Ren, W. Metformin Attenuates Inflammation and Boosts Autophagy in the Liver and Intestine of Chronologically Aged Rats. Exp. Gerontol. 2023, 184, 112331. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Li, C.; Gao, X.; Liu, Z.; Chen, C.; Luo, D. Metformin Inhibits Tumor Growth and Affects Intestinal Flora in Diabetic Tumor-Bearing Mice. Eur. J. Pharmacol. 2021, 912, 174605. [Google Scholar] [CrossRef]
- Ghareeb, M.A.; Tammam, M.A.; El-Demerdash, A.; Atanasov, A.G. Insights about Clinically Approved and Preclinically Investigated Marine Natural Products. Curr. Res. Biotechnol. 2020, 2, 88–102. [Google Scholar] [CrossRef]
- Liang, X.; Luo, D.; Luesch, H. Advances in Exploring the Therapeutic Potential of Marine Natural Products. Pharmacol. Res. 2019, 147, 104373. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural Products in Drug Discovery: Advances and Opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.-Y.; Li, H.-J.; Li, Q.-Y.; Wu, Y.-C. Application of Marine Natural Products in Drug Research. Bioorg. Med. Chem. 2021, 35, 116058. [Google Scholar] [CrossRef] [PubMed]
- Senthilkumar, K.; Kim, S.-K. Marine Invertebrate Natural Products for Anti-Inflammatory and Chronic Diseases. Evid.-Based Complement. Altern. Med. 2013, 2013, 572859. [Google Scholar] [CrossRef]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine Natural Products. Nat. Prod. Rep. 2023, 40, 275–325. [Google Scholar] [CrossRef] [PubMed]
- Imperatore, C.; Luciano, P.; Aiello, A.; Vitalone, R.; Irace, C.; Santamaria, R.; Li, J.; Guo, Y.-W.; Menna, M. Structure and Configuration of Phosphoeleganin, a Protein Tyrosine Phosphatase 1B Inhibitor from the Mediterranean Ascidian Sidnyum elegans. J. Nat. Prod. 2016, 79, 1144–1148. [Google Scholar] [CrossRef]
- Luciano, P.; Imperatore, C.; Senese, M.; Aiello, A.; Casertano, M.; Guo, Y.-W.; Menna, M. Assignment of the Absolute Configuration of Phosphoeleganin via Synthesis of Model Compounds. J. Nat. Prod. 2017, 80, 2118–2123. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.; Imperatore, C.; Casertano, M.; Aiello, A.; Balestri, F.; Piazza, L.; Menna, M.; Del Corso, A.; Paoli, P. Dual Targeting of PTP1B and Aldose Reductase with Marine Drug Phosphoeleganin: A Promising Strategy for Treatment of Type 2 Diabetes. Mar. Drugs 2021, 19, 535. [Google Scholar] [CrossRef] [PubMed]
- Ottanà, R.; Paoli, P.; Cappiello, M.; Nguyen, T.N.; Adornato, I.; Del Corso, A.; Genovese, M.; Nesi, I.; Moschini, R.; Naß, A.; et al. In Search for Multi-Target Ligands as Potential Agents for Diabetes Mellitus and Its Complications—A Structure-Activity Relationship Study on Inhibitors of Aldose Reductase and Protein Tyrosine Phosphatase 1B. Molecules 2021, 26, 330. [Google Scholar] [CrossRef] [PubMed]
- Kousaxidis, A.; Petrou, A.; Lavrentaki, V.; Fesatidou, M.; Nicolaou, I.; Geronikaki, A. Aldose Reductase and Protein Tyrosine Phosphatase 1B Inhibitors as a Promising Therapeutic Approach for Diabetes Mellitus. Eur. J. Med. Chem. 2020, 207, 112742. [Google Scholar] [CrossRef] [PubMed]
- Casertano, M.; Genovese, M.; Piazza, L.; Balestri, F.; Del Corso, A.; Vito, A.; Paoli, P.; Santi, A.; Imperatore, C.; Menna, M. Identifying Human PTP1B Enzyme Inhibitors from Marine Natural Products: Perspectives for Developing of Novel Insulin-Mimetic Drugs. Pharmaceuticals 2022, 15, 325. [Google Scholar] [CrossRef] [PubMed]
- Sefried, S.; Häring, H.-U.; Weigert, C.; Eckstein, S.S. Suitability of Hepatocyte Cell Lines HepG2, AML12 and THLE-2 for Investigation of Insulin Signalling and Hepatokine Gene Expression. Open Biol. 2018, 8, 180147. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-M.; Min, K.-H.; Park, S.-W.; Lee, W. Data on the Expression of PEPCK in HepG2 Hepatocytes Transfected with MiR-195. Data Brief 2017, 15, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Tshivhase, A.M.; Matsha, T.; Raghubeer, S. Resveratrol Attenuates High Glucose-Induced Inflammation and Improves Glucose Metabolism in HepG2 Cells. Sci. Rep. 2024, 14, 1106. [Google Scholar] [CrossRef] [PubMed]
- Razavi, T.; Kouhsari, S.M.; Abnous, K. Morin Exerts Anti-Diabetic Effects in Human HepG2 Cells via Down-Regulation of MiR-29a. Exp. Clin. Endocrinol. Diabetes 2019, 127, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Yudhani, R.D.; Sari, Y.; Nugrahaningsih, D.A.A.; Sholikhah, E.N.; Rochmanti, M.; Purba, A.K.R.; Khotimah, H.; Nugrahenny, D.; Mustofa, M. In Vitro Insulin Resistance Model: A Recent Update. J. Obes. 2023, 2023, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Stonans, I.; Stonane, E.; Rußwurm, S.; Deigner, H.-P.; Böhm, K.J.; Wiederhold, M.; Jäger, L.; Reinhart, K. HepG2 HUMAN HEPATOMA CELLS EXPRESS MULTIPLE CYTOKINE GENES. Cytokine 1999, 11, 151–156. [Google Scholar] [CrossRef]
- Cnop, M. Fatty Acids and Glucolipotoxicity in the Pathogenesis of Type 2 Diabetes. Biochem. Soc. Trans. 2008, 36, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic Inflammation in Fat Plays a Crucial Role in the Development of Obesity-Related Insulin Resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E. Inflammation and Insulin Resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Alnahdi, A.; John, A.; Raza, H. Augmentation of Glucotoxicity, Oxidative Stress, Apoptosis and Mitochondrial Dysfunction in HepG2 Cells by Palmitic Acid. Nutrients 2019, 11, 1979. [Google Scholar] [CrossRef] [PubMed]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular Mechanisms of Lipotoxicity and Glucotoxicity in Nonalcoholic Fatty Liver Disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Siddle, K. Signalling by Insulin and IGF Receptors: Supporting Acts and New Players. J. Mol. Endocrinol. 2011, 47, R1–R10. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kim, J.-W.; Targher, G. Links between Metabolic Syndrome and Metabolic Dysfunction-Associated Fatty Liver Disease. Trends Endocrinol. Metab. 2021, 32, 500–514. [Google Scholar] [CrossRef] [PubMed]
- Androutsakos, T.; Nasiri-Ansari, N.; Bakasis, A.-D.; Kyrou, I.; Efstathopoulos, E.; Randeva, H.S.; Kassi, E. SGLT-2 Inhibitors in NAFLD: Expanding Their Role beyond Diabetes and Cardioprotection. Int. J. Mol. Sci. 2022, 23, 3107. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.S.; Chua, D.; Lim, C.O.; Ho, W.X.; Tan, N.S. Lessons on Drug Development: A Literature Review of Challenges Faced in Nonalcoholic Fatty Liver Disease (NAFLD) Clinical Trials. Int. J. Mol. Sci. 2022, 24, 158. [Google Scholar] [CrossRef] [PubMed]
- Skat-Rørdam, J.; Højland Ipsen, D.; Lykkesfeldt, J.; Tveden-Nyborg, P. A Role of Peroxisome Proliferator-activated Receptor γ in Non-alcoholic Fatty Liver Disease. Basic. Clin. Pharmacol. Toxicol. 2019, 124, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Casertano, M.; Genovese, M.; Santi, A.; Pranzini, E.; Balestri, F.; Piazza, L.; Del Corso, A.; Avunduk, S.; Imperatore, C.; Menna, M.; et al. Evidence of Insulin-Sensitizing and Mimetic Activity of the Sesquiterpene Quinone Avarone, a Protein Tyrosine Phosphatase 1B and Aldose Reductase Dual Targeting Agent from the Marine Sponge Dysidea Avara. Pharmaceutics 2023, 15, 528. [Google Scholar] [CrossRef] [PubMed]
- Izbicka, E.; Streeper, R.T. Mitigation of Insulin Resistance by Natural Products from a New Class of Molecules, Membrane-Active Immunomodulators. Pharmaceuticals 2023, 16, 913. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Fan, J.; Su, Q.; Yang, Z. Cytokines and Abnormal Glucose and Lipid Metabolism. Front. Endocrinol. 2019, 10, 703. [Google Scholar] [CrossRef] [PubMed]
- Vesković, M.; Šutulović, N.; Hrnčić, D.; Stanojlović, O.; Macut, D.; Mladenović, D. The Interconnection between Hepatic Insulin Resistance and Metabolic Dysfunction-Associated Steatotic Liver Disease—The Transition from an Adipocentric to Liver-Centric Approach. Curr. Issues Mol. Biol. 2023, 45, 9084–9102. [Google Scholar] [CrossRef] [PubMed]
- Nerstedt, A.; Cansby, E.; Amrutkar, M.; Smith, U.; Mahlapuu, M. Pharmacological Activation of AMPK Suppresses Inflammatory Response Evoked by IL-6 Signalling in Mouse Liver and in Human Hepatocytes. Mol. Cell. Endocrinol. 2013, 375, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, I.; Lorenzo, V.; Fiory, F.; Doti, N.; Ricci, S.; Cabaro, S.; Liotti, A.; Vitagliano, L.; Longo, M.; Miele, C.; et al. A Peptide Antagonist of Prep1-P160 Interaction Improves Ceramide-Induced Insulin Resistance in Skeletal Muscle Cells. Oncotarget 2017, 8, 71845–71858. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Torres-Gonzalez, M.; Tripathy, S.; Botolin, D.; Christian, B.; Jump, D.B. Elevated Hepatic Fatty Acid Elongase-5 Activity Affects Multiple Pathways Controlling Hepatic Lipid and Carbohydrate Composition. J. Lipid Res. 2008, 49, 1538–1552. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CTRL | PE | PE/2 | PE/3 | |
|---|---|---|---|---|
| IL1β | 1.02 ± 0.708 | 0.940 ± 0.589 | 1.23 ± 0.89 | 0.94 ± 0.59 |
| IL1RA | 16.0 ± 13.7 | 12.1 ± 6.78 | 11.0 ± 7.49 | 11.8 ± 11.5 |
| IL6 | 6.55 ± 0.39 | 4.82 ± 0.40 * | 4.90 ± 1.11 * | 6.67 ± 0.34 |
| IL8 | 76.7 ± 17.5 | 55.6 ± 5.64 | 48.7 ± 17.4 * | 62.4 ± 1.43 |
| IL6R | 33.3 ± 22.7 | 34.1 ± 19.2 | 16.3 ± 2.64 | 35.0 ± 23.1 |
| CCL2 | 444 ± 126 | 376 ± 15.7 | 374 ± 320 | 611 ± 46.0 |
| CCL3 | 6.24 ± 1.81 | 7.28 ± 1.80 | 11.0 ± 7.49 | 9.36 ± 1.78 |
| INHIBIN | 71.3 ± 13.9 | 65.7 ± 25.8 | 73.3 ± 26.6 | 67.7 ± 15.5 |
| VEGF | 178 ± 81.4 | 188 ± 94.4 | 161 ± 81.7 | 188 ± 88.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agognon, A.L.; Casertano, M.; Vito, A.; Orso, S.; Cabaro, S.; Mormone, F.; Morelli, C.; Perruolo, G.; Formisano, P.; Menna, M.; et al. Marine-Derived Phosphoeleganin and Its Semisynthetic Derivative Decrease IL6 Levels and Improve Insulin Signaling in Human Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2024, 25, 6039. https://doi.org/10.3390/ijms25116039
Agognon AL, Casertano M, Vito A, Orso S, Cabaro S, Mormone F, Morelli C, Perruolo G, Formisano P, Menna M, et al. Marine-Derived Phosphoeleganin and Its Semisynthetic Derivative Decrease IL6 Levels and Improve Insulin Signaling in Human Hepatocellular Carcinoma Cells. International Journal of Molecular Sciences. 2024; 25(11):6039. https://doi.org/10.3390/ijms25116039
Chicago/Turabian StyleAgognon, Ayewa L., Marcello Casertano, Alessio Vito, Sonia Orso, Serena Cabaro, Federica Mormone, Cristina Morelli, Giuseppe Perruolo, Pietro Formisano, Marialuisa Menna, and et al. 2024. "Marine-Derived Phosphoeleganin and Its Semisynthetic Derivative Decrease IL6 Levels and Improve Insulin Signaling in Human Hepatocellular Carcinoma Cells" International Journal of Molecular Sciences 25, no. 11: 6039. https://doi.org/10.3390/ijms25116039
APA StyleAgognon, A. L., Casertano, M., Vito, A., Orso, S., Cabaro, S., Mormone, F., Morelli, C., Perruolo, G., Formisano, P., Menna, M., Imperatore, C., & Oriente, F. (2024). Marine-Derived Phosphoeleganin and Its Semisynthetic Derivative Decrease IL6 Levels and Improve Insulin Signaling in Human Hepatocellular Carcinoma Cells. International Journal of Molecular Sciences, 25(11), 6039. https://doi.org/10.3390/ijms25116039