
Natural Product-Derived Compounds Targeting Keratinocytes and Molecular Pathways in Psoriasis Therapeutics
 , ,
, ,  and
and
Abstract
1. Introduction
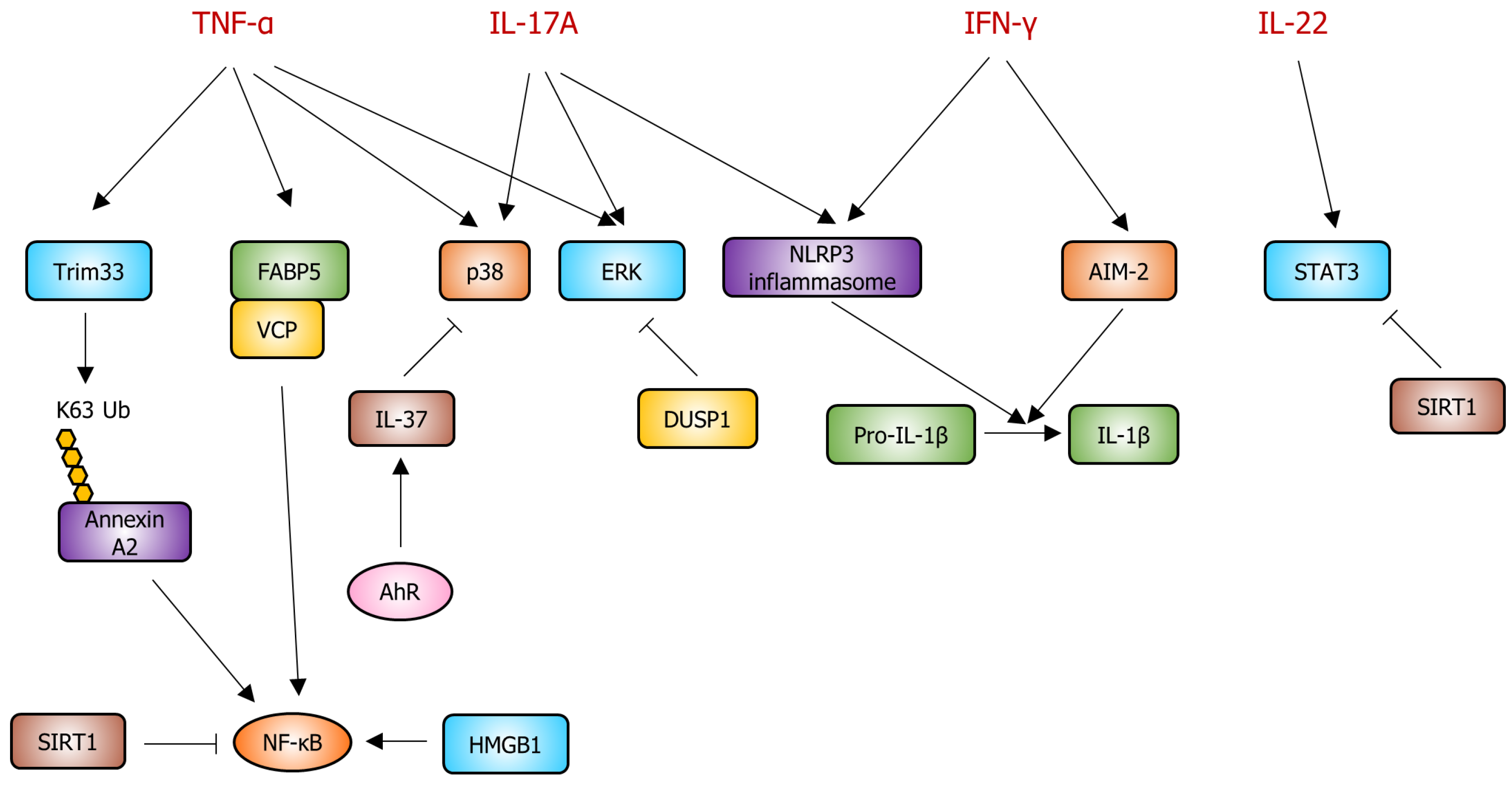
2. Pathogenesis of Psoriasis in Keratinocytes
2.1. Interleukin-17A (IL-17A)
2.2. Interleukin-22 (IL-22)
2.3. Interferon-γ (IFN-γ)
2.4. Tumor Necrosis Factor-α (TNF-α)
3. Potential Therapeutic Targets and Signaling Pathways for Psoriasis in Keratinocytes
3.1. The Janus Kinases (JAKs)
3.2. Nuclear Factor-Kappa B (NF-κB)
3.3. Tripartite Motif-Containing Protein 33 (TRIM33)
3.4. NLRP3 Inflammasome
3.5. Fatty Acid-Binding Protein (FABP)–Valosin-Containing Protein (VCP) Complex
3.6. High-Mobility Group Box-1 (HMGB1)
3.7. Sirtuins (SIRTs)
3.8. Aryl Hydrocarbon Receptor (AhR)
3.9. Mitogen-Activated Protein Kinases (MAPKs)
3.10. Dual-Specificity Phosphatase-1 (DUSP1)
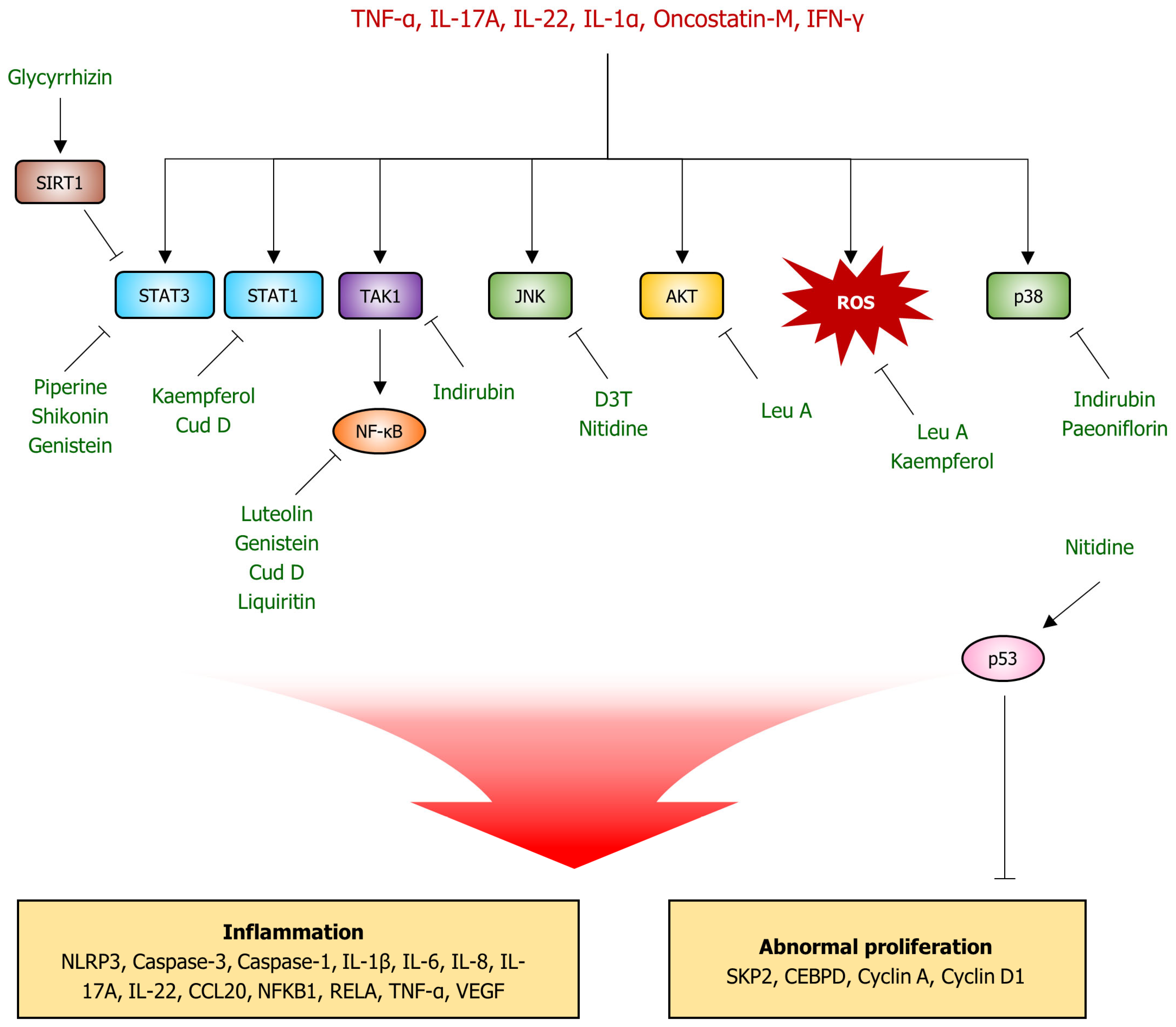
4. Natural Product-Derived Compounds for Psoriasis Therapeutics
4.1. Luteolin
4.2. Piperine
4.3. Glycyrrhizin
4.4. Kaempferol
4.5. Punicalagin
4.6. Shikonin
4.7. Genistein
4.8. Nitidine Chloride
4.9. Leucosceptoside A
4.10. Indirubin
4.11. Paeoniflorin
4.12. 3H-1,2-dithiole-3-thione (D3T)
4.13. Liquiritin
4.14. Cudraxanthone D
5. Conclusions, Perspectives, and Limitations
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Elkhawaga, O.Y.; Ellety, M.M.; Mofty, S.O.; Ghanem, M.S.; Mohamed, A.O. Review of Natural Compounds for Potential Psoriasis Treatment. Inflammopharmacolxogy 2023, 31, 1183–1198. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.P.; Weissman, F.G.; Gill, M.A. Pathophysiology and Treatment of Psoriasis. Am. J. Health Syst. Pharm. 2000, 57, 645–659; quiz 660–661. [Google Scholar] [CrossRef] [PubMed]
- Todke, P.; Shah, V.H. Psoriasis: Implication to Disease and Therapeutic Strategies, with an Emphasis on Drug Delivery Approaches. Int. J. Dermatol. 2018, 57, 1387–1402. [Google Scholar] [CrossRef] [PubMed]
- Sarac, G.; Koca, T.T.; Baglan, T. A Brief Summary of Clinical Types of Psoriasis. North. Clin. Istanb. 2016, 3, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, F.; Flores, R. Clinical Overview of Psoriasis and Psoriatic Arthritis. In Textbook of Dermatologic Ultrasound; Springer: Berlin/Heidelberg, Germany, 2022; pp. 345–365. [Google Scholar]
- Nowowiejska, J.; Baran, A.; Flisiak, I. Mutual Relationship Between Sleep Disorders, Quality of Life and Psychosocial Aspects in Patients with Psoriasis. Front. Psychiatry 2021, 12, 674460. [Google Scholar] [CrossRef] [PubMed]
- Elmets, C.A.; Korman, N.J.; Prater, E.F.; Wong, E.B.; Rupani, R.N.; Kivelevitch, D.; Armstrong, A.W.; Connor, C.; Cordoro, K.M.; Davis, D.M.R.; et al. Joint AAD-NPF Guidelines of Care for the Management and Treatment of Psoriasis with Topical Therapy and Alternative Medicine Modalities for Psoriasis Severity Measures. J. Am. Acad. Dermatol. 2021, 84, 432–470. [Google Scholar] [CrossRef] [PubMed]
- Bakker, P.; Woerdenbag, H.; Gooskens, V.; Naafs, B.; van der Kaaij, R.; Wieringa, N. Dermatological Preparations for the Tropics. A Formulary of Dermatological Preparations and Background Information on Choices, Production and Dispensing; University of Groningen: Groningen, The Netherlands, 2012. [Google Scholar]
- Committee of Psoriasis, Dermatology Branch, Chinese Medical Association. Guidelines for the Diagnosis and Treatment of Psoriasis in China: 2019 Concise Edition#. Int. J. Dermatol. Venereol. 2020, 3, 14–26. [Google Scholar] [CrossRef]
- Körver, J.E.; Van Duijnhoven, M.W.; Pasch, M.C.; Van Erp, P.E.; Van De Kerkhof, P.C. Assessment of Epidermal Subpopulations and Proliferation in Healthy Skin, Symptomless and Lesional Skin of Spreading Psoriasis. Br. J. Dermatol. 2006, 155, 688–694. [Google Scholar] [CrossRef]
- Jiang, M.; Fang, H.; Shao, S.; Dang, E.; Zhang, J.; Qiao, P.; Yang, A.; Wang, G. Keratinocyte Exosomes Activate Neutrophils and Enhance Skin Inflammation in Psoriasis. FASEB J. 2019, 33, 13241–13253. [Google Scholar] [CrossRef]
- Tsuruta, D. NF-κB Links Keratinocytes and Lymphocytes in the Pathogenesis of Psoriasis. Recent Pat. Inflamm. Allergy Drug Discov. 2009, 3, 40–48. [Google Scholar] [CrossRef]
- Hymowitz, S.G.; Filvaroff, E.H.; Yin, J.P.; Lee, J.; Cai, L.; Risser, P.; Maruoka, M.; Mao, W.; Foster, J.; Kelley, R.F.; et al. IL-17s Adopt a Cystine Knot Fold: Structure and Activity of a Novel Cytokine, IL−17F, and Implications for Receptor Binding. EMBO J. 2001, 20, 5332–5341. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, J.; Huang, A.; Stinson, J.; Heldens, S.; Foster, J.; Dowd, P.; Gurney, A.L.; Wood, W.I. Cloning and Characterization of IL-17B and IL-17C, Two New Members of the IL-17 Cytokine Family. Proc. Natl. Acad. Sci. USA 2000, 97, 773–778. [Google Scholar] [CrossRef]
- Krueger, J.G.; Wharton, K.A., Jr.; Schlitt, T.; Suprun, M.; Torene, R.I.; Jiang, X.; Wang, C.Q.; Fuentes-Duculan, J.; Hartmann, N.; Peters, T.; et al. IL-17A Inhibition by Secukinumab Induces Early Clinical, Histopathologic, and Molecular Resolution of Psoriasis. J. Allergy Clin. Immunol. 2019, 144, 750–763. [Google Scholar] [CrossRef] [PubMed]
- Hot, A.; Zrioual, S.; Toh, M.-L.; Lenief, V.; Miossec, P. IL-17A-Versus IL-17F-Induced Intracellular Signal Transduction Pathways and Modulation by IL-17RA and IL-17RC RNA interference in Rheumatoid Synoviocytes. Ann. Rheum. Dis. 2011, 70, 341–348. [Google Scholar] [CrossRef]
- Su, Y.; Huang, J.; Zhao, X.; Lu, H.; Wang, W.; Yang, X.O.; Shi, Y.; Wang, X.; Lai, Y.; Dong, C. Interleukin-17 Receptor D Constitutes an Alternative Receptor for interleukin-17A Important in Psoriasis-Like Skin Inflammation. Sci. Immunol. 2019, 4, eaau9657. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cai, G.; Liu, C.; Zhao, J.; Gu, C.; Wu, L.; Hamilton, T.A.; Zhang, C.-J.; Ko, J.; Zhu, L.; et al. IL-17R-EGFR Axis Links Wound Healing to Tumorigenesis in Lrig1+ Stem Cells. J. Exp. Med. 2019, 216, 195–214. [Google Scholar] [CrossRef]
- Miyoshi, K.; Takaishi, M.; Nakajima, K.; Ikeda, M.; Kanda, T.; Tarutani, M.; Iiyama, T.; Asao, N.; DiGiovanni, J.; Sano, S. Stat3 as a Therapeutic Target for the Treatment of Psoriasis: A Clinical Feasibility Study with STA-21, a Stat3 Inhibitor. J. Investig. Dermatol. 2011, 131, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Jin, L.; Dang, E.; Chang, T.; Feng, Z.; Liu, Y.; Wang, G. IL-17A Upregulates Keratin 17 Expression in Keratinocytes Through STAT1- and STAT3-Dependent Mechanisms. J. Investig. Dermatol. 2011, 131, 2401–2408. [Google Scholar] [CrossRef] [PubMed]
- Moos, S.; Mohebiany, A.N.; Waisman, A.; Kurschus, F.C. Imiquimod-Induced Psoriasis in Mice Depends on the IL-17 Signaling of Keratinocytes. J. Investig. Dermatol. 2019, 139, 1110–1117. [Google Scholar] [CrossRef]
- Fischer, B.; Kübelbeck, T.; Kolb, A.; Ringen, J.; Waisman, A.; Wittmann, M.; Karbach, S.; Kölsch, S.M.; Kramer, D. IL-17A-Driven Psoriasis Is Critically Dependent on IL-36 Signaling. Front. Immunol. 2023, 14, 1256133. [Google Scholar] [CrossRef]
- Dhamija, B.; Marathe, S.; Sawant, V.; Basu, M.; Attrish, D.; Mukherjee, D.; Kumar, S.; Pai, M.G.J.; Wad, S.; Sawant, A.; et al. IL-17A Orchestrates Reactive Oxygen Species/HIF1alpha-Mediated Metabolic Reprogramming in Psoriasis. J. Immunol. 2024, 212, 302–316. [Google Scholar] [CrossRef] [PubMed]
- Moran, E.M.; Mullan, R.; McCormick, J.; Connolly, M.; Sullivan, O.; Fitzgerald, O.; Bresnihan, B.; Veale, D.J.; Fearon, U. Human Rheumatoid Arthritis Tissue Production of IL-17A Drives Matrix and Cartilage Degradation: Synergy with Tumour Necrosis Factor-Alpha, Oncostatin M and Response to Biologic Therapies. Arthritis Res. Ther. 2009, 11, R113. [Google Scholar] [CrossRef] [PubMed]
- Kolbinger, F.; Huppertz, C.; Mir, A.; Padova, F.D. IL-17A and Multiple Sclerosis: Signaling Pathways, Producing Cells and Target Cells in the Central Nervous System. Curr. Drug Targets 2016, 17, 1882–1893. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Diegelmann, J.; Beigel, F.; Brand, S. IL-17A Alone Weakly Affects the Transcriptome of Intestinal Epithelial Cells but Strongly Modulates the TNF-alpha-Induced Expression of Inflammatory Mediators and Inflammatory Bowel Disease Susceptibility Genes. Inflamm. Bowel Dis. 2014, 20, 1502–1515. [Google Scholar] [CrossRef] [PubMed]
- Silfvast-Kaiser, A.; Paek, S.Y.; Menter, A. Anti-IL17 Therapies for Psoriasis. Expert Opin. Biol. Ther. 2019, 19, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.-H.; Torii, K.; Saito, C.; Furuhashi, T.; Maeda, A.; Morita, A. Serum IL-22 Correlates with Psoriatic Severity and Serum IL-6 Correlates with Susceptibility to Phototherapy. J. Dermatol. Sci. 2010, 58, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Wolk, K.; Kunz, S.; Witte, E.; Friedrich, M.; Asadullah, K.; Sabat, R. IL-22 Increases the Innate Immunity of Tissues. Immunity 2004, 21, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Ma, W.; Yan, J.; Zhong, H. Evaluation of the Effects of IL-22 on the Proliferation and Differentiation of Keratinocytes In Vitro. Mol. Med. Rep. 2020, 22, 2715–2722. [Google Scholar] [CrossRef]
- Zhang, W.; Dang, E.; Shi, X.; Jin, L.; Feng, Z.; Hu, L.; Wu, Y.; Wang, G. The Pro-inflammatory Cytokine IL-22 Up-Regulates Keratin 17 Expression in Keratinocytes via STAT3 and ERK1/2. PLoS ONE 2012, 7, e40797. [Google Scholar] [CrossRef]
- Prignano, F.; Donetti, E. Looking at Interleukin-22 from a New Dermatological Perspective: From Epidermal Homeostasis to Its Role in Chronic Skin Diseases. Dermatology 2022, 238, 829–836. [Google Scholar] [CrossRef]
- Lopez, D.V.; Kongsbak-Wismann, M. Role of IL-22 in Homeostasis and Diseases of the Skin. APMIS 2022, 130, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Adamopoulos, I.E. Psoriatic Arthritis Under the Influence of IFN-gamma. Clin. Immunol. 2020, 218, 108513. [Google Scholar] [CrossRef] [PubMed]
- Kurhade, C.; Zegenhagen, L.; Weber, E.; Nair, S.; Michaelsen-Preusse, K.; Spanier, J.; Gekara, N.O.; Kröger, A.; Överby, A.K. Type I Interferon Response in Olfactory Bulb, the Site of Tick-Borne Flavivirus Accumulation, Is Primarily Regulated by IPS-1. J. Neuroinflammation 2016, 13, 22. [Google Scholar] [CrossRef] [PubMed]
- Kambayashi, T.; Assarsson, E.; Lukacher, A.E.; Ljunggren, H.-G.; Jensen, P.E. Memory CD8+ T Cells Provide an Early Source of IFN-gamma. J. Immunol. 2003, 170, 2399–2408. [Google Scholar] [CrossRef] [PubMed]
- Gallais Sérézal, I.; Classon, C.; Cheuk, S.; Barrientos-Somarribas, M.; Wadman, E.; Martini, E.; Chang, D.; Xu Landén, N.; Ehrström, M.; Nylén, S.; et al. Resident T Cells in Resolved Psoriasis Steer Tissue Responses That Stratify Clinical Outcome. J. Investig. Dermatol. 2018, 138, 1754–1763. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.; Chu, A.; Lúdvíksson, B.R.; Berg, E.L.; Ehrhardt, R.O. IL-12, Independently of IFN-gamma, Plays a Crucial Role in the Pathogenesis of a Murine Psoriasis-Like Skin Disorder. J. Immunol. 1999, 162, 7480–7491. [Google Scholar] [CrossRef] [PubMed]
- Harden, J.L.; Johnson-Huang, L.M.; Chamian, M.F.; Lee, E.; Pearce, T.; Leonardi, C.L.; Haider, A.; Lowes, M.A.; Krueger, J.G. Humanized Anti-IFN-gamma (HuZAF) in the Treatment of Psoriasis. J. Allergy Clin. Immunol. 2015, 135, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Suh, C.-H.; Jung, J.-Y.; Ahn, M.-H.; Han, M.H.; Kwon, J.E.; Yim, H.; Kim, H.-A. Elevated Circulating Levels of the Interferon-Gamma-Induced Chemokines Are Associated with Disease Activity and Cutaneous Manifestations in Adult-Onset Still’s Disease. Sci. Rep. 2017, 7, 46652. [Google Scholar] [CrossRef] [PubMed]
- Belpaire, A.; van Geel, N.; Speeckaert, R. From IL-17 to IFN-gamma in Inflammatory Skin Disorders: Is Transdifferentiation a Potential Treatment Target? Front. Immunol. 2022, 13, 932265. [Google Scholar] [CrossRef]
- Shao, S.; Tsoi, L.C.; Sarkar, M.K.; Xing, X.; Xue, K.; Uppala, R.; Berthier, C.C.; Zeng, C.; Patrick, M.; Billi, A.C.; et al. IFN-gamma Enhances Cell-Mediated Cytotoxicity Against Keratinocytes via JAK2/STAT1 in Lichen Planus. Sci. Transl. Med. 2019, 11, eaav7561. [Google Scholar] [CrossRef]
- Hongqin, T.; Xinyu, L.; Heng, G.; Lanfang, X.; Yongfang, W.; Shasha, S. Triptolide Inhibits IFN-gamma Signaling via the Jak/STAT Pathway in HaCaT Keratinocytes. Phytother. Res. 2011, 25, 1678–1685. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Darnell, J.E., Jr. The JAK-STAT Pathway at Twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Blouin, C.M.; Lamaze, C. Interferon Gamma Receptor: The Beginning of the Journey. Front. Immunol. 2013, 4, 267. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ye, L.; Bai, Y.; Mojidi, H.; Simister, N.E.; Zhu, X. Activation of the JAK/STAT-1 Signaling Pathway by IFN-gamma Can Down-Regulate Functional Expression of the MHC Class I-Related Neonatal Fc Receptor for IgG. J. Immunol. 2008, 181, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.-F.; Teng, Y.-C.; Yoon, Y.-S.; Kim, D.-H.; Cai, D.-Q.; Lee, K.-J. Reactive Oxygen Species Are Involved in the IFN-gamma-Stimulated Production of Th2 Chemokines in HaCaT Keratinocytes. J. Cell. Physiol. 2011, 226, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, L.; Morelli, M.; Scarponi, C.; Scaglione, G.L.; Pallotta, S.; Albanesi, C.; Madonna, S. PI3Kdelta Sustains Keratinocyte Hyperproliferation and Epithelial Inflammation: Implications for a Topically Druggable Target in Psoriasis. Cells 2021, 10, 2636. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.Z.; Chaturvedi, V.; Denning, M.F.; Choubey, D.; Diaz, M.O.; Nickoloff, B.J. Role of NF-kappaB in the Apoptotic-Resistant Phenotype of Keratinocytes. J. Biol. Chem. 1999, 274, 37957–37964. [Google Scholar] [CrossRef] [PubMed]
- Mease, P. TNFalpha Therapy in Psoriatic Arthritis and Psoriasis. Ann. Rheum. Dis. 2004, 63, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Mazloom, S.E.; Yan, D.; Hu, J.Z.; Ya, J.; Husni, M.E.; Warren, C.B.; Fernandez, A.P. TNF-alpha Inhibitor-Induced Psoriasis: A Decade of Experience at the Cleveland Clinic. J. Am. Acad. Dermatol. 2020, 83, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Borghi, A.; Verstrepen, L.; Beyaert, R. TRAF2 Multitasking in TNF Receptor-Induced Signaling to NF-κB, MAP Kinases and Cell Death. Biochem. Pharmacol. 2016, 116, 1–10. [Google Scholar] [CrossRef]
- Chiricozzi, A.; Guttman-Yassky, E.; Suárez-Fariñas, M.; Nograles, K.E.; Tian, S.; Cardinale, I.; Chimenti, S.; Krueger, J.G. Integrative Responses to IL-17 and TNF-alpha in Human Keratinocytes Account for Key Inflammatory Pathogenic Circuits in Psoriasis. J. Investig. Dermatol. 2011, 131, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Bonnet, M.C.; Ulvmar, M.H.; Wolk, K.; Karagianni, N.; Witte, E.; Uthoff-Hachenberg, C.; Renauld, J.-C.; Kollias, G.; Toftgard, R.; et al. Tumor Necrosis Factor Receptor Signaling in Keratinocytes Triggers Interleukin-24-Dependent Psoriasis-Like Skin Inflammation in Mice. Immunity 2013, 39, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhang, H.; Lin, W.; Lu, L.; Su, J.; Chen, X. Signaling Pathways and Targeted Therapies for Psoriasis. Signal Transduct. Target. Ther. 2023, 8, 437. [Google Scholar] [CrossRef] [PubMed]
- Kalliolias, G.D.; Ivashkiv, L.B. TNF Biology, Pathogenic Mechanisms and Emerging Therapeutic Strategies. Nat. Rev. Rheumatol. 2016, 12, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of Tumour Necrosis Factor Signalling: Live or Let Die. Nat. Rev. Immunol. 2015, 15, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Menter, A.; Strober, B.; Langley, R.G.; Buonanno, M.; Wolk, R.; Gupta, P.; Krishnaswami, S.; Tan, H.; Harness, J.A. Efficacy and Safety of Tofacitinib, an Oral Janus Kinase Inhibitor, in the Treatment of Psoriasis: A Phase 2b Randomized Placebo-Controlled Dose-Ranging Study. Br. J. Dermatol. 2012, 167, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Ibáñez, K.; Alsina, M.M.; Muñoz-Santos, C. Tofacitinib and Other Kinase Inhibitors in the Treatment of Psoriasis. Actas Dermo Sifiliogr. 2013, 104, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yang, T.; Tang, M.; Yang, Z.; Pei, H.; Ye, H.; Tang, Y.; Cheng, Z.; Lin, P.; Chen, L. Studies on the Anti-psoriasis Effects and Its Mechanism of a Dual JAK2/FLT3 Inhibitor Flonoltinib Maleate. Biomed. Pharmacother. 2021, 137, 111373. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, V.P.; Gurevich, I.; Aneskievich, B.J. Emerging Roles for TNIP1 in Regulating Post-receptor Signaling. Cytokine Growth Factor Rev. 2012, 23, 109–118. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Morgan, M.; Kim, D.-G.; Lee, J.-Y.; Bai, L.; Lin, Y.; Liu, Z.-G.; Kim, Y.-S. TNFα Induced Noncanonical NF-κB Activation Is Attenuated by RIP1 through Stabilization of TRAF2. J. Cell Sci. 2011, 124, 647–656. [Google Scholar] [CrossRef]
- Liang, Y.; Sarkar, M.K.; Tsoi, L.C.; Gudjonsson, J.E. Psoriasis: A Mixed Autoimmune and Autoinflammatory Disease. Curr. Opin. Immunol. 2017, 49, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Ding, Z.; Liang, H.; Zhang, B.; Chen, X. The Roles of TIF1γ in Cancer. Front. Oncol. 2019, 9, 979. [Google Scholar] [CrossRef] [PubMed]
- Petit, V.; Parcelier, A.; Mathé, C.; Barroca, V.; Torres, C.; Lewandowski, D.; Ferri, F.; Gallouët, A.-S.; Dalloz, M.; Dinet, O.; et al. TRIM33 Deficiency in Monocytes and Macrophages Impairs Resolution of Colonic Inflammation. EBioMedicine 2019, 44, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhu, J.; Chen, X.; Xia, H.; Yang, L. E3 Ubiquitin Ligase Trim33 Ubiquitylates Annexin A2 to Promote NF-κB Induced Skin Inflammation in Psoriasis. J. Dermatol. Sci. 2022, 107, 160–168. [Google Scholar] [CrossRef]
- Verma, D.; Fekri, S.Z.; Sigurdardottir, G.; Bivik Eding, C.; Sandin, C.; Enerbäck, C. Enhanced Inflammasome Activity in Patients with Psoriasis Promotes Systemic Inflammation. J. Investig. Dermatol. 2021, 141, 586–595.e5. [Google Scholar] [CrossRef] [PubMed]
- Carlström, M.; Ekman, A.-K.; Petersson, S.; Söderkvist, P.; Enerbäck, C. Genetic Support for the Role of the NLRP3 Inflammasome in Psoriasis Susceptibility. Exp. Dermatol. 2012, 21, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Hao, J.; Zeng, J.; Sauter, E.R. SnapShot: FABP Functions. Cell 2020, 182, 1066–1066.e1. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Yu, J.; Yorek, M.S.; Yu, C.-L.; Pope, R.M.; Chimenti, M.S.; Xiong, Y.; Klingelhutz, A.; Jabbari, A.; Li, B. Keratinocyte FABP5-VCP Complex Mediates Recruitment of Neutrophils in Psoriasis. Cell Rep. 2023, 42, 113449. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Fu, Y.; Xiong, S. Keratinocyte Derived HMGB1 Aggravates Psoriasis Dermatitis via Facilitating Inflammatory Polarization of Macrophages and Hyperproliferation of Keratinocyte. Mol. Immunol. 2023, 163, 1–12. [Google Scholar] [CrossRef]
- Zhang, W.; Guo, S.; Li, B.; Liu, L.; Ge, R.; Cao, T.; Wang, H.; Gao, T.; Wang, G.; Li, C. Proinflammatory Effect of High-Mobility Group Protein B1 on Keratinocytes: An Autocrine Mechanism Underlying Psoriasis Development. J. Pathol. 2017, 241, 392–404. [Google Scholar] [CrossRef]
- Haigis, M.C.; Sinclair, D.A. Mammalian Sirtuins: Biological Insights and Disease Relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef]
- Fan, X.; Yan, K.; Meng, Q.; Sun, R.; Yang, X.; Yuan, D.; Li, F.; Deng, H. Abnormal Expression of SIRTs in Psoriasis: Decreased Expression of SIRT 1–5 and Increased Expression of SIRT 6 and 7. Int. J. Mol. Med. 2019, 44, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Di Meglio, P.; Duarte, J.H.; Ahlfors, H.; Owens, N.D.L.; Li, Y.; Villanova, F.; Tosi, I.; Hirota, K.; Nestle, F.O.; Mrowietz, U.; et al. Activation of the Aryl Hydrocarbon Receptor Dampens the Severity of Inflammatory Skin Conditions. Immunity 2014, 40, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Schiering, C.; Vonk, A.; Das, S.; Stockinger, B.; Wincent, E. Cytochrome P4501-Inhibiting Chemicals Amplify Aryl Hydrocarbon Receptor Activation and IL-22 Production in T Helper 17 Cells. Biochem. Pharmacol. 2018, 151, 47–58. [Google Scholar] [CrossRef]
- Smith, S.H.; Jayawickreme, C.; Rickard, D.J.; Nicodeme, E.; Bui, T.; Simmons, C.; Coquery, C.M.; Neil, J.; Pryor, W.M.; Mayhew, D.; et al. Tapinarof Is a Natural AhR Agonist That Resolves Skin Inflammation in Mice and Humans. J. Investig. Dermatol. 2017, 137, 2110–2119. [Google Scholar] [CrossRef]
- Tsuji, G.; Yamamura, K.; Kawamura, K.; Kido-Nakahara, M.; Ito, T.; Nakahara, T. Regulatory Mechanism of the IL-33-IL-37 Axis via Aryl Hydrocarbon Receptor in Atopic Dermatitis and Psoriasis. Int. J. Mol. Sci. 2023, 24, 14633. [Google Scholar] [CrossRef]
- Tsuji, G.; Hashimoto-Hachiya, A.; Matsuda-Taniguchi, T.; Takai-Yumine, A.; Takemura, M.; Yan, X.; Furue, M.; Nakahara, T. Natural Compounds Tapinarof and Galactomyces Ferment Filtrate Downregulate IL-33 Expression via the AHR/IL-37 Axis in Human Keratinocytes. Front. Immunol. 2022, 13, 745997. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-J.; Li, C.-Y.; Dai, H.-Y.; Cai, D.-X.; Wang, K.-Y.; Xu, Y.-H.; Chen, L.-M.; Zhou, C.-L. Expression and Localization of the Activated Mitogen-Activated Protein Kinase in Lesional Psoriatic Skin. Exp. Mol. Pathol. 2007, 83, 413–418. [Google Scholar] [CrossRef]
- Chen, L.; Wu, J.; Ren, W.; Yang, X.; Shen, Z. C-Jun N-Terminal Kinase (JNK)-Phospho-C-Jun (ser63/73) Pathway Is Essential for FOXP3 Nuclear Translocation in Psoriasis. J. Dermatol. Sci. 2013, 69, 114–121. [Google Scholar] [CrossRef]
- Hau, C.S.; Kanda, N.; Noda, S.; Tatsuta, A.; Kamata, M.; Shibata, S.; Asano, Y.; Sato, S.; Watanabe, S.; Tada, Y. Visfatin Enhances the Production of Cathelicidin Antimicrobial Peptide, Human Beta-defensin-2, Human Beta-defensin-3, and S100A7 in Human Keratinocytes and Their Orthologs in Murine Imiquimod-Induced Psoriatic Skin. Am. J. Pathol. 2013, 182, 1705–1717. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, J.; Zhai, T.; Li, H.; Li, H.; Huo, R.; Shen, B.; Wang, B.; Chen, X.; Li, N.; et al. CCN1 promotes IL-1beta production in keratinocytes by activating p38 MAPK signaling in psoriasis. Sci. Rep. 2017, 7, 43310. [Google Scholar] [CrossRef] [PubMed]
- Funding, A.T.; Johansen, C.; Kragballe, K.; Iversen, L. Mitogen- and Stress-Activated Protein Kinase 2 and Cyclic AMP Response Element Binding Protein Are Activated in Lesional Psoriatic Epidermis. J. Investig. Dermatol. 2007, 127, 2012–2019. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Sun, L.; Han, J.; Zheng, W.; Peng, W. DUSP1/MKP-1 Regulates Proliferation and Apoptosis in Keratinocytes Through the ERK/Elk-1/Egr-1 Signaling Pathway. Life Sci. 2019, 223, 47–53. [Google Scholar] [CrossRef]
- Hsu, L.; Armstrong, A.W. JAK Inhibitors: Treatment Efficacy and Safety Profile in Patients with Psoriasis. J. Immunol. Res. 2014, 2014, 283617. [Google Scholar] [CrossRef] [PubMed]
- Goldminz, A.M.; Au, S.C.; Kim, N.; Gottlieb, A.B.; Lizzul, P.F. NF-kappaB: An Essential Transcription Factor in Psoriasis. J. Dermatol. Sci. 2013, 69, 89–94. [Google Scholar] [CrossRef]
- Nedoszytko, B.; Szczerkowska-Dobosz, A.; Stawczyk-Macieja, M.; Owczarczyk-Saczonek, A.; Reich, A.; Bartosiñska, J.; Batycka-Baran, A.; Czajkowski, R.; Dobrucki, I.T.; Dobrucki, L.W.; et al. Pathogenesis of Psoriasis in the “omic” Era. Part II. Genetic, Genomic and Epigenetic Changes in Psoriasis. Postepy Dermatol. Alergol. 2020, 37, 283–298. [Google Scholar] [CrossRef]
- Ciążyńska, M.; Olejniczak-Staruch, I.; Sobolewska-Sztychny, D.; Narbutt, J.; Skibińska, M.; Lesiak, A. The Role of NLRP1, NLRP3, and AIM2 Inflammasomes in Psoriasis: Review. Int. J. Mol. Sci. 2021, 22, 5898. [Google Scholar] [CrossRef] [PubMed]
- Göblös, A.; Danis, J.; Vas, K.; Bata-Csörgő, Z.; Kemény, L.; Széll, M. Keratinocytes Express Functional CARD18, a Negative Regulator of Inflammasome Activation, and Its Altered Expression in Psoriasis May Contribute to Disease Pathogenesis. Mol. Immunol. 2016, 73, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Andersson, U. Targeting Inflammation Driven by HMGB1. Front. Immunol. 2020, 11, 484. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, H.; Zheng, H.; Zhou, X.; Shen, G.; Teng, X.; Liu, X.; Zhang, J.; Wei, X.; Hu, Z.; et al. Autophagy-Based Unconventional Secretion of HMGB1 by Keratinocytes Plays a Pivotal Role in Psoriatic Skin Inflammation. Autophagy 2021, 17, 529–552. [Google Scholar] [CrossRef]
- Furue, M.; Hashimoto-Hachiya, A.; Tsuji, G. Aryl Hydrocarbon Receptor in Atopic Dermatitis and Psoriasis. Int. J. Mol. Sci. 2019, 20, 5424. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, M.G.; Stein Gold, L.; Strober, B.; Papp, K.A.; Armstrong, A.W.; Bagel, J.; Kircik, L.; Ehst, B.; Hong, H.C.-H.; Soung, J.; et al. Phase 3 Trials of Tapinarof Cream for Plaque Psoriasis. N. Engl. J. Med. 2021, 385, 2219–2229. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, J.I.; Boguniewicz, M.; Quintana, F.J.; Clark, R.A.; Gross, L.; Hirano, I.; Tallman, A.M.; Brown, P.M.; Fredericks, D.; Rubenstein, D.S.; et al. Tapinarof Validates the Aryl Hydrocarbon Receptor as a Therapeutic Target: A Clinical Review. J. Allergy Clin. Immunol. 2023, in press. [Google Scholar] [CrossRef] [PubMed]
- Boutros, T.; Chevet, E.; Metrakos, P. Mitogen-Activated Protein (MAP) Kinase/MAP Kinase Phosphatase Regulation: Roles in Cell Growth, Death, and Cancer. Pharmacol. Rev. 2008, 60, 261–310. [Google Scholar] [CrossRef] [PubMed]
- Weng, Z.; Patel, A.B.; Vasiadi, M.; Therianou, A.; Theoharides, T.C. Luteolin Inhibits Human Keratinocyte Activation and Decreases NF-κB Induction That Is Increased in Psoriatic Skin. PLoS ONE 2014, 9, e90739. [Google Scholar] [CrossRef] [PubMed]
- Biswas, P.; Ghorai, M.; Mishra, T.; Gopalakrishnan, A.V.; Roy, D.; Mane, A.B.; Mundhra, A.; Das, N.; Mohture, V.M.; Patil, M.T.; et al. Piper longum L.: A Comprehensive Review on Traditional Uses, Phytochemistry, Pharmacology, and Health-Promoting Activities. Phytother. Res. 2022, 36, 4425–4476. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Gong, H.; Du, J.; Gao, W.; Xu, J.; Cai, X.; Yang, Y.; Xiao, H. Piperine Ameliorates Psoriatic Skin Inflammation by Inhibiting the Phosphorylation of STAT3. Int. Immunopharmacol. 2023, 119, 110221. [Google Scholar] [CrossRef]
- Qiong, H.; Han, L.; Zhang, N.X.; Chen, H.Y.; Yan, K.X.; Zhang, Z.H.; Ma, Y.; Xu, J.H. Glycyrrhizin Improves the Pathogenesis of Psoriasis Partially Through IL-17A and the SIRT1-STAT3 Axis. BMC Immunol. 2021, 22, 34. [Google Scholar] [CrossRef]
- Li, Y.; Cui, H.; Li, S.; Li, X.; Guo, H.; Nandakumar, K.S.; Li, Z. Kaempferol Modulates IFN-gamma Induced JAK-STAT Signaling Pathway and Ameliorates Imiquimod-Induced Psoriasis-Like Skin Lesions. Int. Immunopharmacol. 2023, 114, 109585. [Google Scholar] [CrossRef]
- Tang, L.; Zhang, B.; Li, G.; Zhu, Y.; Feng, B.; Su, Z.; Han, W.; Huang, H.; Li, Q.; Wang, M.; et al. Punicalagin Alleviates the Hyperproliferation of Keratinocytes in Psoriasis Through Inhibiting SKP2 Expression. J. Nat. Med. 2023, 77, 712–720. [Google Scholar] [CrossRef]
- Lan, X.-O.; Wang, H.-X.; Qi, R.-Q.; Xu, Y.-Y.; Yu, Y.-J.; Yang, Y.; Guo, H.; Gao, X.-H.; Geng, L. Shikonin Inhibits CEBPD Downregulation in IL-17-Treated HaCaT Cells and in an Imiquimod-Induced Psoriasis Model. Mol. Med. Rep. 2020, 22, 2263–2272. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, X.; Gao, X.; Chen, H.; Geng, L. Shikonin Suppresses IL-17-Induced VEGF Expression via Blockage of JAK2/STAT3 Pathway. Int. Immunopharmacol. 2014, 19, 327–333. [Google Scholar] [CrossRef]
- Wang, A.; Wei, J.; Lu, C.; Chen, H.; Zhong, X.; Lu, Y.; Li, L.; Huang, H.; Dai, Z.; Han, L. Genistein Suppresses Psoriasis-Related Inflammation through a STAT3-NF-κB-Dependent Mechanism in Keratinocytes. Int. Immunopharmacol. 2019, 69, 270–278. [Google Scholar] [CrossRef]
- Yang, X.-G.; Jiang, B.-W.; Jing, Q.-Q.; Li, W.-J.; Tan, L.-P.; Bao, Y.-L.; Song, Z.-B.; Yu, C.-L.; Liu, L.; Liu, Y.-C.; et al. Nitidine Chloride Induces S Phase Cell Cycle Arrest and Mitochondria-Dependent Apoptosis in HaCaT Cells and Ameliorates Skin Lesions in Psoriasis-Like Mouse Models. Eur. J. Pharmacol. 2019, 863, 172680. [Google Scholar] [CrossRef]
- Koycheva, I.K.; Mihaylova, L.V.; Todorova, M.N.; Balcheva-Sivenova, Z.P.; Alipieva, K.; Ferrante, C.; Orlando, G.; Georgiev, M.I. Leucosceptoside A from Devil’s Claw Modulates Psoriasis-Like Inflammation via Suppression of the PI3K/AKT Signaling Pathway in Keratinocytes. Molecules 2021, 26, 7014. [Google Scholar] [CrossRef]
- Zhao, J.; Xie, X.; Di, T.; Liu, Y.; Qi, C.; Chen, Z.; Li, P.; Wang, Y. Indirubin Attenuates IL-17A-Induced CCL20 Expression and Production in Keratinocytes Through Repressing TAK1 Signaling Pathway. Int. Immunopharmacol. 2021, 94, 107229. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, J.; Huo, R.; Zhai, T.; Li, H.; Wu, P.; Zhu, X.; Zhou, Z.; Shen, B.; Li, N. Paeoniflorin Inhibits Skin Lesions in Imiquimod-Induced Psoriasis-Like Mice by Downregulating Inflammation. Int. Immunopharmacol. 2015, 24, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Xiao, Z.; Zhao, R.; Lu, C.; Zhang, Y. Paeoniflorin Suppressed IL-22 via p38 MAPK Pathway and Exerts Anti-psoriatic Effect. Life Sci. 2017, 180, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Shih, M.-C.; Li, C.-L.; Liao, E.-C.; Yen, C.-Y.; Yen, L.-J.; Wang, K.-C.; Lu, L.-Y.; Chou, T.-Y.; Chen, Y.-C.; Yu, S.-J. Inhibition of NLRP3 Inflammasome Activation by 3H-1,2-Dithiole-3-Thione: A Potential Therapeutic Approach for Psoriasis Treatment. Int. J. Mol. Sci. 2023, 24, 13528. [Google Scholar] [CrossRef]
- Guo, D.; Wang, Q.; Li, A.; Li, S.; Wang, B.; Li, Y.; Yuan, J.; Guo, T.; Feng, S. Liquiritin Targeting Th17 Cells Differentiation and Abnormal Proliferation of Keratinocytes Alleviates Psoriasis via NF-κB and AP-1 Pathway. Phytother. Res. 2024, 38, 174–186. [Google Scholar] [CrossRef]
- Kim, N.; Lee, S.; Kang, J.; Choi, Y.-A.; Jang, Y.H.; Jeong, G.-S.; Kim, S.-H.; Cudraxanthone, D. Cudraxanthone D Ameliorates Psoriasis-Like Skin Inflammation in an Imiquimod-Induced Mouse Model via Inhibiting the Inflammatory Signaling Pathways. Molecules 2021, 26, 6086. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zeng, M.; Wang, Z.; Qin, F.; Chen, J.; He, Z. Dietary Luteolin: A Narrative Review Focusing on Its Pharmacokinetic Properties and Effects on Glycolipid Metabolism. J. Agric. Food Chem. 2021, 69, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Heidari, S.; Mehri, S.; Hosseinzadeh, H. The Genus Glycyrrhiza (Fabaceae Family) and Its Active Constituents as Protective Agents Against Natural or Chemical Toxicities. Phytother. Res. 2021, 35, 6552–6571. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Lo, S.-H.; Dubey, R.; Kumar, S.; Chaubey, K.K.; Kumar, S. Plant-Derived Natural Compounds as an Emerging Antiviral in Combating COVID-19. Indian J. Microbiol. 2023, 63, 429–446. [Google Scholar] [CrossRef] [PubMed]
- Periferakis, A.; Periferakis, K.; Badarau, I.A.; Petran, E.M.; Popa, D.C.; Caruntu, A.; Costache, R.S.; Scheau, C.; Caruntu, C.; Costache, D.O. Kaempferol: Antimicrobial Properties, Sources, Clinical, and Traditional Applications. Int. J. Mol. Sci. 2022, 23, 5054. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, N.; Saifi, A.; Chaudhary, A.; Tripathi, P.N.; Chaudhary, A.; Sharma, A. Multifaceted Neuroprotective Role of Punicalagin: A Review. Neurochem. Res. 2024, 49, 1427–1436. [Google Scholar] [CrossRef]
- Malik, S.; Brudzyńska, P.; Khan, M.R.; Sytar, O.; Makhzoum, A.; Sionkowska, A. Natural Plant-Derived Compounds in Food and Cosmetics: A Paradigm of Shikonin and Its Derivatives. Materials 2023, 16, 4377. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhou, M.; Mu, Z.; Guo, J.; Hou, Y.; Xu, Y.; Geng, L. Recent Advances in Shikonin for the Treatment of Immune-Related Diseases: Anti-inflammatory and Immunomodulatory Mechanisms. Biomed. Pharmacother. 2023, 165, 115138. [Google Scholar] [CrossRef]
- Nazari-Khanamiri, F.; Ghasemnejad-Berenji, M. Cellular and Molecular Mechanisms of Genistein in Prevention and Treatment of Diseases: An Overview. J. Food Biochem. 2021, 45, e13972. [Google Scholar] [CrossRef]
- Sharifi-Rad, J.; Quispe, C.; Imran, M.; Rauf, A.; Nadeem, M.; Gondal, T.A.; Ahmad, B.; Atif, M.; Mubarak, M.S.; Sytar, O.; et al. Genistein: An Integrative Overview of Its Mode of Action, Pharmacological Properties, and Health Benefits. Oxid. Med. Cell. Longev. 2021, 2021, 3268136. [Google Scholar] [CrossRef]
- Lu, Q.; Luo, S.; Shi, Z.; Yu, M.; Guo, W.; Li, C. Nitidine Chloride, a Benzophenanthridine Alkaloid from Zanthoxylum nitidum (Roxb.) DC., Exerts Multiple Beneficial Properties, Especially in Tumors and Inflammation-Related Diseases. Front. Pharmacol. 2022, 13, 1046402. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.; Hadda, T.B.; Touzani, R. Diverse Therapeutic Potential of Nitidine, A Comprehensive Review. Curr. Drug Metab. 2018, 19, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; De Vita, D.; Toniolo, C.; Sciubba, F.; Tomassini, L.; Venditti, A.; Bianco, A.; Serafini, M.; Foddai, S. Leucosceptosides A and B: Two Phenyl-Ethanoid Glycosides with Important Occurrence and Biological Activities. Biomolecules 2022, 12, 1807. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, X.; Huang, W.; Rao, X.; Lai, Y. Pharmacological Properties of Indirubin and Its Derivatives. Biomed. Pharmacother. 2022, 151, 113112. [Google Scholar] [CrossRef] [PubMed]
- Jiao, F.; Varghese, K.; Wang, S.; Liu, Y.; Yu, H.; Booz, G.W.; Roman, R.J.; Liu, R.; Fan, F. Recent Insights into the Protective Mechanisms of Paeoniflorin in Neurological, Cardiovascular, and Renal Diseases. J. Cardiovasc. Pharmacol. 2021, 77, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Chen, J.; Peng, F.; Sun, C.; Lei, Y.; Chen, G.; Li, G.; Yin, Y.; Lin, Z.; Wu, L.; et al. Pharmacological Activities and Pharmacokinetics of Liquiritin: A Review. J. Ethnopharmacol. 2022, 293, 115257. [Google Scholar] [CrossRef]
- Xin, L.-T.; Yue, S.-J.; Fan, Y.-C.; Wu, J.-S.; Yan, D.; Guan, H.-S.; Wang, C.-Y. Cudrania tricuspidata: An Updated Review on Ethnomedicine, Phytochemistry and Pharmacology. RSC Adv. 2017, 7, 31807–31832. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Expression or Activity | Signaling Pathways | Inhibitors or Agonists | References |
|---|---|---|---|---|
| JAKs (JAK1, JAK2, JAK3, TYK2) |
|
|
| [58,59,60] |
| NF-κB |
|
| Inhibitors: tumor necrosis factor-α blockers; glucocorticoids; BAY 11-7082 | [61,62,63] |
| TRIM33 | TRIM33 expression increases in the epidermis of patients with psoriasis |
| - | [64,65,66] |
| NLRP3 inflammasome |
|
| Inhibitor: BAY11-7082 (inhibiting the NLRP3 inflammasome activity and the NF-κB pathway) | [67,68] |
| FABP-VCP complex | FABP5 expression increases in the epidermal tissues of patients with psoriasis | FABP5 expression increases, interacting with VCP, ultimately leading to NF-κB pathway activation | - | [69,70] |
| HMGB1 | HMGB1 expression increases in keratinocytes of a psoriasis mouse model | HMGB1 expression increases, leading to NF-κB pathway activation and an increase in IL-18 expression | - | [71,72] |
| SIRTs |
| SIRT1 activation decreases the activity of the MAPK, NF-κB, and STAT3 pathways | SIRT1 activator: resveratrol; catapol | [73,74] |
| AhR | Activated by AhR ligands | IL-37 expression increases, decreasing p38 pathway activity and IL-33 expression | Agonists: Tapinarof, galactomyces ferment filtrate (GFF) | [75,76,77,78,79] |
| MAPKs | MAPKs (ERK, p38, and JNK) are activated in psoriatic lesions |
|
| [55,80,81,82,83,84] |
| DUSP1 | DUSP1 expression decreases in keratinocytes | ERK/ELK-1/EGR-1 pathway activity increases | - | [85] |
| Compound | Experimental Models | Inducers for Psoriasis | Concentrations of the Compounds | Effects of the Compounds | Reference |
|---|---|---|---|---|---|
| Luteolin | HaCaT and primary keratinocyte | In vitro: 50 ng/mL TNF-α | 10–100 μM in vitro | Luteolin demonstrated a reduction in proliferation and a decrease in the expressions of IL-6, IL-8, VEGF, NFKB1, and RELA mRNA levels in vitro. | [97] |
| Piperine | HaCaT in vitro; BALB/c mice (male, 8–10 weeks old) in vivo | In vitro: M5 (10 ng/mL) containing TNF-α, IL-17A, IL-22, IL-1α, and Oncostatin-M; in vivo: 62.5 mg of 5% IMQ cream | 10–40 μM in vitro; 2 mM and 4 mM in vivo (topical) | Piperine exhibited a reduction in S100A7 protein levels and mRNA expression of IL-6, IL-23, β-defensin 2, and CCL20 both in vitro and in vivo. Moreover, it decreased the levels of various cytokines and mRNAs associated with psoriasis in vivo. | [98,99] |
| Glycyrrhizin | HaCaT in vitro; BALB/c mice (male, 10 weeks old) in vivo | In vitro: 100 ng/mL IL-17A; in vivo: 62.5 mg of 5% IMQ cream | 2 μM in vitro; 20 mg/kg in vivo (topical) | Glycyrrhizin decreased the proliferation and secretion of IL-6, CCL20, and TNF-α in vitro, while increasing the expression of SIRT1 and reducing STAT3 phosphorylation. Additionally, it reduced the secretion of IL-17A and IFN-γ in the serum in vivo. | [100] |
| Kaempferol | HaCaT in vitro; BALB/c mice (female, 10 weeks old) in vivo | In vitro: IFN-γ (500 U/mL); in vivo: 62.5 mg of 5% IMQ cream | 5–40 μM in vitro; 9.01, 27.03, and 81.09 mg/kg in vivo (oral) | Kaempferol decreased JAK-STAT phosphorylation, ROS production, and IFN-γR1 expression while increasing SOCS1 expression in vitro. In vivo, it reduced the number of dendritic cells in the skin. | [101] |
| Punicalagin | HaCaT in vitro | IL-6 (50 ng/mL), IL-17A (12.5 ng/mL), and TNF-α (12.5 ng/mL) | 2.5–160 μM in vitro | Punicalagin reduced abnormal proliferation, SKP2 expression, and the cytokine-enhanced S-phase fraction in vitro. | [102] |
| Shikonin | HaCaT in vitro; BALB/c (male, 8 weeks old) in vivo | In vitro: IL-17A (40 ng/mL); in vivo: 50 mg of 5% IMQ cream | 5 μM in vitro; 1 μM in vivo (topical) | Shikonin reduced VEGF expression, JAK/STAT3 pathway activity, CEBPD expression, and keratinocyte abnormal proliferation both in vitro and in vivo. | [103,104] |
| Genistein | HaCaT in vitro; BALB/c (male, 7–8 weeks old) | In vitro: 20 ng/mL TNF-α;in vivo: 62.5 mg of 5% IMQ cream | 0.5% and 2% in vivo (topical) | Genistein decreased TNF-α-induced proliferation, inflammatory factor expression, STAT3 phosphorylation, and NF-κB signaling in vitro. It also reduced epidermal thickness and the expression of inflammatory factors in vivo. | [105] |
| Nitidine chloride | HaCaT in vitro; BALB/c mice (female, 6–8 weeks old) in vivo | In vivo: 20 μL TPA (50 μg/mL per site); 62.5 mg of 5% IMQ cream | 7.8 nM in vitro; 1.5 μg (topical) in vivo | Nitidine chloride inhibited S-phase cell cycle arrest caused by cell proliferation, decreased the expression of cyclin A and cyclin D1, increased p53 expression and apoptosis, and enhanced JNK phosphorylation both in vitro and in vivo. | [106] |
| Leucosceptoside A | HaCaT in vitro | IFN-γ, IL-17A, and IL-22 (1 ng/mL each) | 20 μM in vitro | Leucosceptoside A inhibited PI3K/AKT signaling and reduced the expression of STAT3, PI3KCA, and AKT mRNAs in vitro. | [107] |
| Indirubin | HaCaT in vitro; BALB/c mice (male, 8 weeks old) in vivo | In vitro: 100 ng/mL IL-17A;in vivo: 42 mg of 5% IMQ cream | 4–16 μM in vitro; 12.5, 25, and 50 mg/kg in vivo (topical) | Indirubin decreased CCL20 expression; TAK1-mediated NF-κB signaling; and p38 and MKK4 phosphorylation both in vitro and in vivo. Moreover, it reduced ki67-positive cells in vivo. | [108] |
| Paeoniflorin | HaCaT in vitro; BALB/c mice (female, 8–11 weeks old) or Hartley guinea pigs (male, 4 weeks old) in vivo | In vitro: 5 μg/mL LPS; in vivo: 0.2 mL of 5% propranolol cream (guinea pigs) or 60 mg of 5% IMQ cream (BALB/c mice) | 6.24–104.07 μM in vitro; 9% paeoniflorin emulsion (topical, guinea pigs), 75, 150, or 300 mg/kg/day (topical, mice) | Paeoniflorin reduced IL-6, IL-17A, and IL-22 secretion and mRNA expression in vitro. It also decreased p38 phosphorylation, propranolol chloride-induced parakeratosis, and hyperkeratinization in vivo. Moreover, it alleviated IMQ-induced psoriatic symptoms, inflammation, and cytokine production in vivo. | [109,110] |
| 3H-1,2-dithiole-3-thione (D3T) | HaCaT in vitro; BALB/c (female, 8 weeks old) in vivo | In vitro: 10 ng/mL TNF-α; in vivo: 62.5 mg of 5% IMQ cream | 50–200 μM in vitro; 10 and 30 mg/kg in vivo (intraperitoneal) | D3T reduced the expression of NLRP3, caspase-1, and IL-1β in vitro and attenuated JNK pathway activity. In vivo, it decreased ear thickness; skin redness; scaling; ki-67 levels; and NLRP3 and inflammasome levels, while cleaving caspase-1, IL-6, and IL-17A. Moreover, it inhibited Th17 differentiation. | [111] |
| Liquirtin | HaCaT in vitro; C57BL/6 (female, 8–10 weeks) in vivo | In vitro: 10 ng/mL TNF-α; in vivo: 62.5 mg of 5% IMQ cream | 5, 10, and 20 μM in vitro; 1 and 2 mg/kg in vivo (intragastric) | Liquirtin reduced TNF-α-induced proliferation and the mRNA expression of IL-6, IL-8, and IL-1β, as well as the activation of NF-κB and AP-1 pathways in vitro. In vivo, it diminished psoriasis-like phenotypes and the expression of IL-6, TNF-α, IL-23, and IL-17A, as well as the population and polarization of Th17 cells. | [112] |
| Cudraxanthone D | HaCaT in vitro; C57BL/6 (female, 8 weeks old) in vivo | In vitro: 10 ng/mL TNF-α + 10 ng/mL IFN-γ; in vivo: 62.5 mg of 5% IMQ cream | 0.01–1 μM in vitro; 0.1, 1, and 10 mg/kg in vivo (intragastric) | Cudraxanthone D reduced the expression of CCL17, IL-6, IL-8, and IL-1β in vitro and inhibited the NF-κB and STAT1 pathways. In vivo, it alleviated inflammation in psoriasis-like skin and reduced the expression of CXCL1, IL-6, and IL-4. | [113] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.G.; Jung, Y.; Choi, H.-K.; Lee, J.-I.; Lim, T.-G.; Lee, J. Natural Product-Derived Compounds Targeting Keratinocytes and Molecular Pathways in Psoriasis Therapeutics. Int. J. Mol. Sci. 2024, 25, 6068. https://doi.org/10.3390/ijms25116068
Lee YG, Jung Y, Choi H-K, Lee J-I, Lim T-G, Lee J. Natural Product-Derived Compounds Targeting Keratinocytes and Molecular Pathways in Psoriasis Therapeutics. International Journal of Molecular Sciences. 2024; 25(11):6068. https://doi.org/10.3390/ijms25116068
Chicago/Turabian StyleLee, Yu Geon, Younjung Jung, Hyo-Kyoung Choi, Jae-In Lee, Tae-Gyu Lim, and Jangho Lee. 2024. "Natural Product-Derived Compounds Targeting Keratinocytes and Molecular Pathways in Psoriasis Therapeutics" International Journal of Molecular Sciences 25, no. 11: 6068. https://doi.org/10.3390/ijms25116068
APA StyleLee, Y. G., Jung, Y., Choi, H.-K., Lee, J.-I., Lim, T.-G., & Lee, J. (2024). Natural Product-Derived Compounds Targeting Keratinocytes and Molecular Pathways in Psoriasis Therapeutics. International Journal of Molecular Sciences, 25(11), 6068. https://doi.org/10.3390/ijms25116068