
Extracellular Vesicles in Diabetic Cardiomyopathy—State of the Art and Future Perspectives

 ,
,
Abstract
1. Introduction
2. Materials and Methods
3. Extracellular Vesicles
4. Diabetic Cardiomyopathy (DCM)
5. Involvement of Extracellular Vesicles in the Pathogenesis of Diabetic Cardiomyopathy
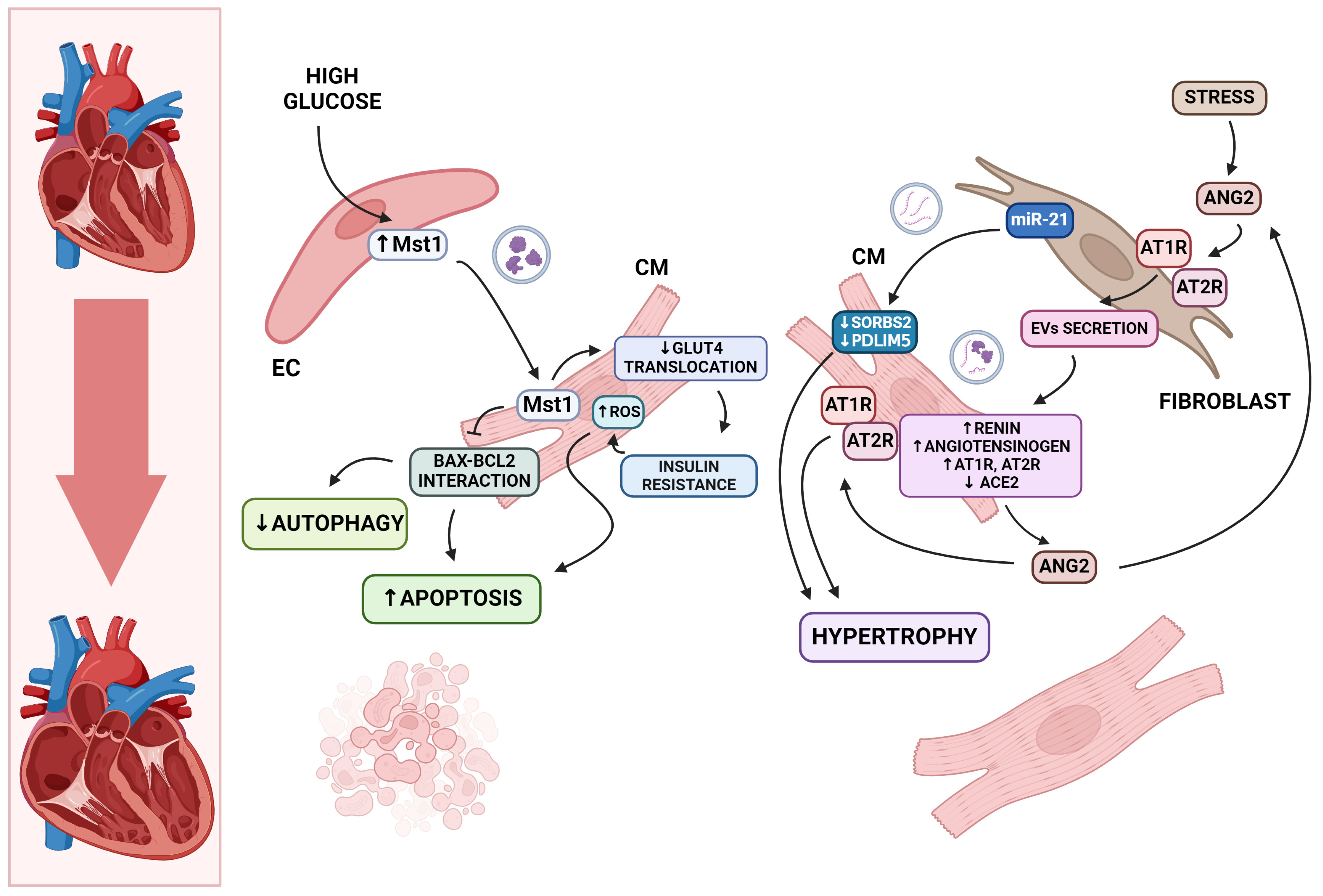
5.1. Cardiomyocyte Death and Hypertrophy
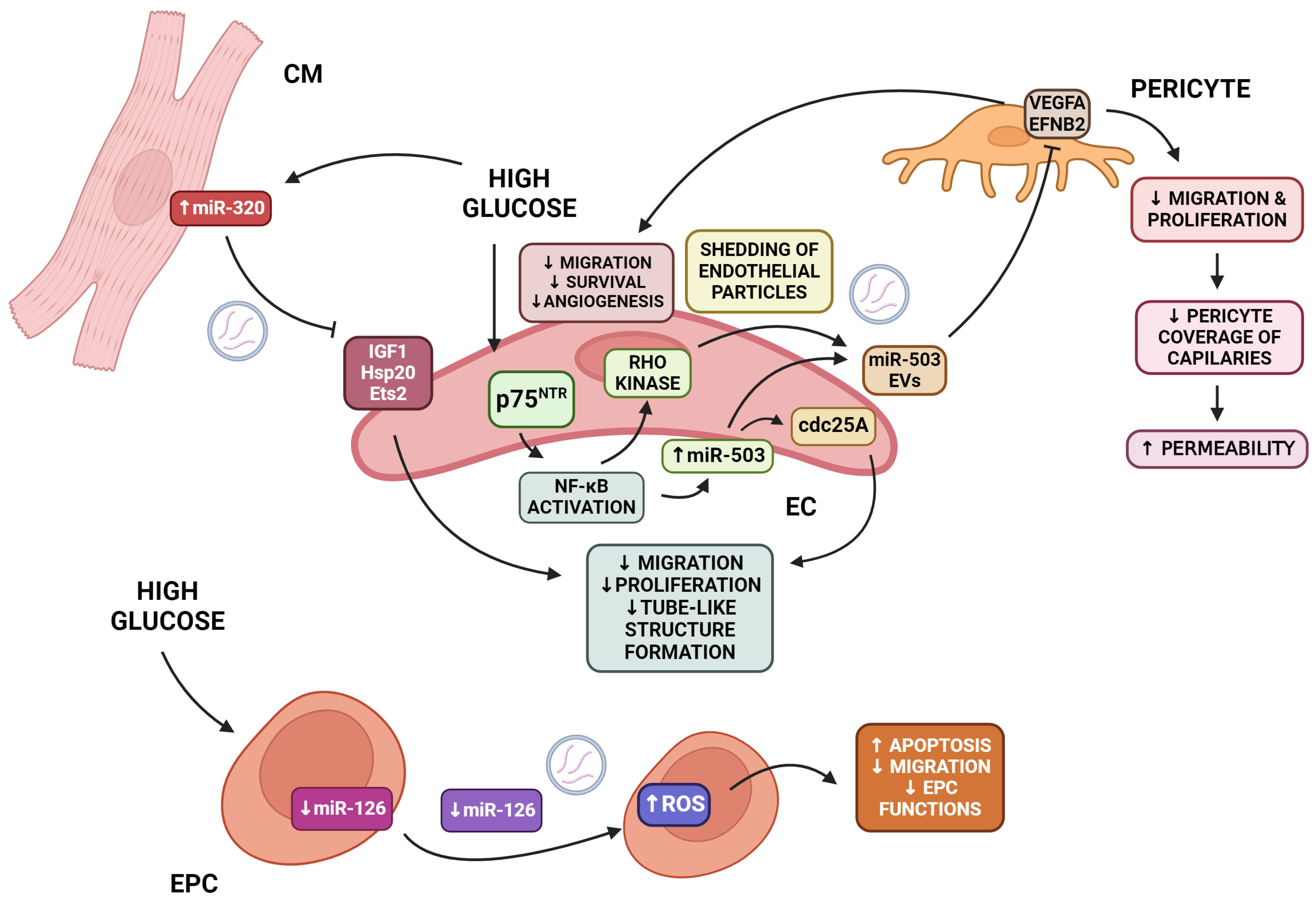
5.2. Endothelial Damage
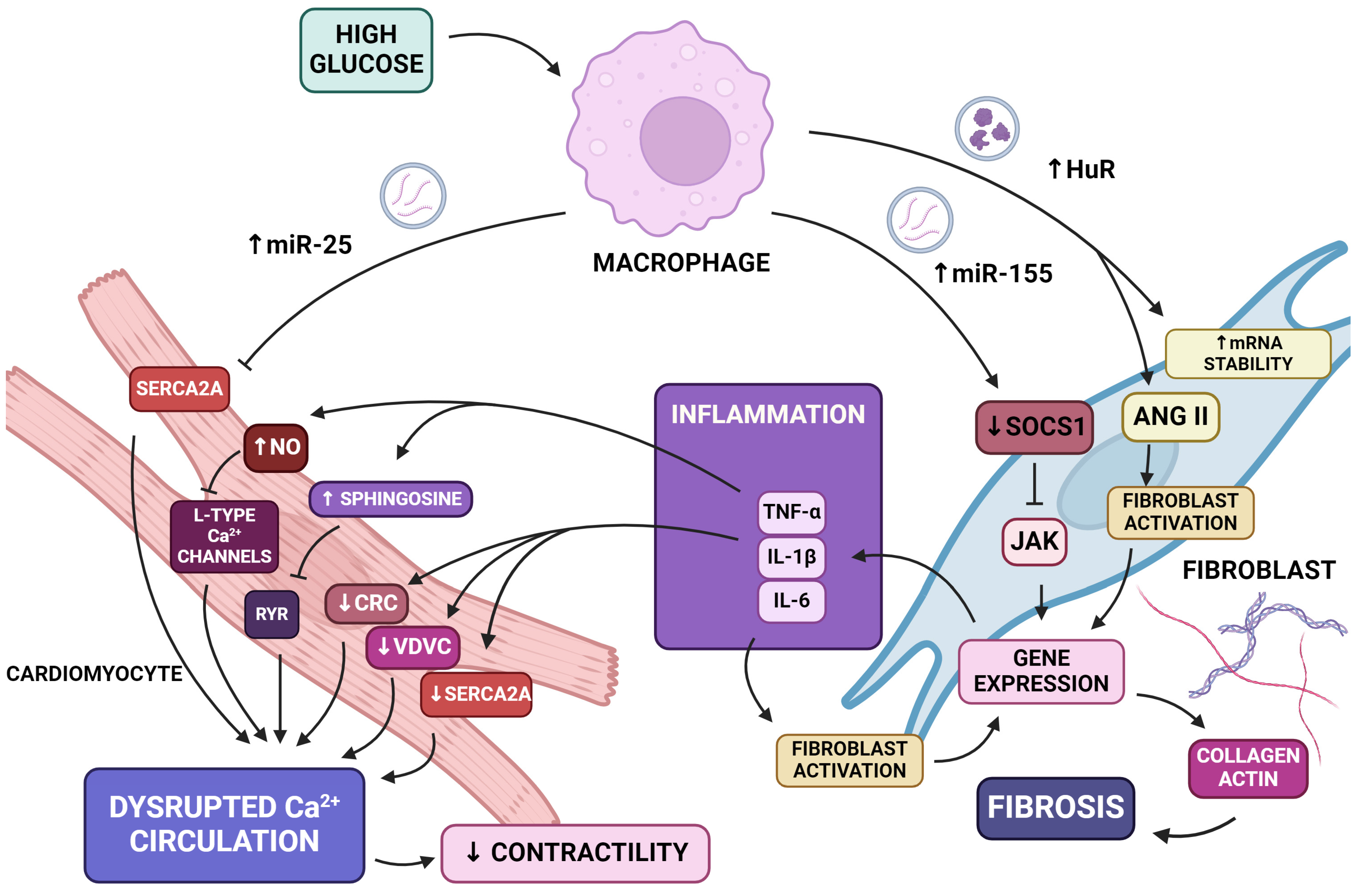
5.3. Inflammation and Fibrosis
5.4. Calcium Dyshomeostasis
5.5. Senescence
6. Beneficial Effects of Extracellular Vesicles in Diabetic Cardiomyopathy
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Cooper, M.E.; Zimmet, P. Changing epidemiology of type 2 diabetes mellitus and associated chronic kidney disease. Nat. Rev. Nephrol. 2016, 12, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Dagenais, G.; Mohan, V.; Diaz, R.; Probstfield, J.; Freeman, R.; Shaw, J.; Lanas, F.; Avezum, A.; Budaj, A.; et al. Glucose levels are associated with cardiovascular disease and death in an international cohort of normal glycaemic and dysglycaemic men and women: The EpiDREAM cohort study. Eur. J. Prev. Cardiol. 2011, 19, 755–764. [Google Scholar] [CrossRef] [PubMed]
- The Emerging Risk Factors Collaboration; Sarwar, N.; Gao, P.; Seshasai, S.R.; Gobin, R.; Kaptoge, S.; Di Angelantonio, E.; Ingelsson, E.; Lawlor, D.A.; Selvin, E.; et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: A collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375, 2215–2222. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.D.; Langenberg, C.; Rapsomaniki, E.; Denaxas, S.; Pujades-Rodriguez, M.; Gale, C.P.; Deanfield, J.; Smeeth, L.; Timmis, A.; Hemingway, H. Type 2 diabetes and incidence of a wide range of cardiovascular diseases: A cohort study in 1·9 million people. Lancet 2015, 385, S86. [Google Scholar] [CrossRef] [PubMed]
- Dal Canto, E.; Ceriello, A.; Rydén, L.; Ferrini, M.; Hansen, T.B.; Schnell, O.; Standl, E.; Beulens, J.W. Diabetes as a cardiovascular risk factor: An overview of global trends of macro and micro vascular complications. Eur. J. Prev. Cardiol. 2019, 26 (Suppl. S2), 25–32. [Google Scholar] [CrossRef] [PubMed]
- Rubler, S.; Dlugash, J.; Yuceoglu, Y.Z.; Kumral, T.; Branwood, A.W.; Grishman, A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 1972, 30, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Marx, N.; Federici, M.; Schütt, K.; Müller-Wieland, D.; A Ajjan, R.; Antunes, M.J.; Christodorescu, R.M.; Crawford, C.; Di Angelantonio, E.; Eliasson, B.; et al. 2023 ESC Guidelines for the management of cardiovascular disease in patients with diabetes. Eur. Hear. J. 2023, 44, 4043–4140. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, S.; Cai, L. Diabetic cardiomyopathy and its mechanisms: Role of oxidative stress and damage. J. Diabetes Investig. 2014, 5, 623–634. [Google Scholar] [CrossRef]
- Boudina, S.; Abel, E.D. Diabetic Cardiomyopathy Revisited. Circulation 2007, 115, 3213–3223. [Google Scholar] [CrossRef] [PubMed]
- Aneja, A.; Tang, W.W.; Bansilal, S.; Garcia, M.J.; Farkouh, M.E. Diabetic Cardiomyopathy: Insights into Pathogenesis, Diagnostic Challenges, and Therapeutic Options. Am. J. Med. 2008, 121, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Authors/Task Force Members; Rydén, L.; Grant, P.J.; Anker, S.D.; Berne, C.; Cosentino, F.; Danchin, N.; Deaton, C.; Escaned, J.; Hammes, H.-P.; et al. ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Heart J. 2013, 34, 3035–3087. [Google Scholar] [CrossRef] [PubMed]
- Diabetic cardiomyopathy: An educational review. Br. J. Cardiol. 2023. [CrossRef]
- Gottdiener, J.S.; Arnold, A.M.; Aurigemma, G.P.; Polak, J.F.; Tracy, R.P.; Kitzman, D.W.; Gardin, J.M.; E Rutledge, J.; Boineau, R.C. Predictors of congestive heart failure in the elderly: The cardiovascular health study. Circ. 2000, 35, 1628–1637. [Google Scholar] [CrossRef] [PubMed]
- From, A.M.; Leibson, C.L.; Bursi, F.; Redfield, M.M.; Weston, S.A.; Jacobsen, S.J.; Rodeheffer, R.J.; Roger, V.L. Diabetes in Heart Failure: Prevalence and Impact on Outcome in the Population. Am. J. Med. 2006, 119, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Dandamudi, S.; Slusser, J.; Mahoney, D.W.; Redfield, M.M.; Rodeheffer, R.J.; Chen, H.H. The Prevalence of Diabetic Cardiomyopathy: A Population-Based Study in Olmsted County, Minnesota. J. Card. Fail. 2014, 20, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Bouthoorn, S.; Valstar, G.B.; Gohar, A.; Ruijter, H.M.D.; Reitsma, H.B.; Hoes, A.W.; Rutten, F.H. The prevalence of left ventricular diastolic dysfunction and heart failure with preserved ejection fraction in men and women with type 2 diabetes: A systematic review and meta-analysis. Diabetes Vasc. Dis. Res. 2018, 15, 477–493. [Google Scholar] [CrossRef]
- Tan, Y.; Zhang, Z.; Zheng, C.; Wintergerst, K.A.; Keller, B.B.; Cai, L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: Preclinical and clinical evidence. Nat. Rev. Cardiol. 2020, 17, 585–607. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, S.; Wang, X.; Chen, Y.; Pang, P.; Yang, Q.; Lin, J.; Deng, S.; Wu, S.; Fan, G.; et al. Diabetic cardiomyopathy: Clinical phenotype and practice. Front. Endocrinol. 2022, 13, 1032268. [Google Scholar] [CrossRef]
- Raev, D.C. Which Left Ventricular Function Is Impaired Earlier in the Evolution of Diabetic Cardiomyopathy?: An echocardiographic study of young type I diabetic patients. Diabetes Care 1994, 17, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Tadic, M.; Cuspidi, C.; Calicchio, F.; Grassi, G.; Mancia, G. Diabetic cardiomyopathy: How can cardiac magnetic resonance help? Acta Diabetol. 2020, 57, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Dunlay, S.M.; Roger, V.L.; Weston, S.A.; Jiang, R.; Redfield, M.M. Longitudinal Changes in Ejection Fraction in Heart Failure Patients with Preserved and Reduced Ejection Fraction. Circ. Hear. Fail. 2012, 5, 720–726. [Google Scholar] [CrossRef] [PubMed]
- cMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Committees, Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Seferovic, P.M.; Paulus, W.J. Clinical diabetic cardiomyopathy: A two-faced disease with restrictive and dilated phenotypes. Eur. Hear. J. 2015, 36, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Hölscher, M.E.; Bode, C.; Bugger, H. Diabetic Cardiomyopathy: Does the Type of Diabetes Matter? Int. J. Mol. Sci. 2016, 17, 2136. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Caruso, S.; Poon, I.K.H. Apoptotic Cell-Derived Extracellular Vesicles: More Than Just Debris. Front. Immunol. 2018, 9, 1486. [Google Scholar] [CrossRef]
- Brennan, K.; Martin, K.; Fitzgerald, S.P.; O’Sullivan, J.; Wu, Y.; Blanco, A.; Richardson, C.; Mc Gee, M.M. A comparison of methods for the isolation and separation of extracellular vesicles from protein and lipid particles in human serum. Sci. Rep. 2020, 10, 1039. [Google Scholar] [CrossRef]
- Bongiovanni, L.; Andriessen, A.; Wauben, M.H.M.; Hoen, E.N.M.N.-’T.; de Bruin, A. Extracellular Vesicles: Novel Opportunities to Understand and Detect Neoplastic Diseases. Vet. Pathol. 2021, 58, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e18. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wu, H.; Geng, R.; Tang, Q. Identification of Core Gene Biomarkers in Patients with Diabetic Cardiomyopathy. Dis. Markers 2018, 2018, 1–15. [Google Scholar] [CrossRef] [PubMed]
- De Geest, B.; Mishra, M. Role of Oxidative Stress in Diabetic Cardiomyopathy. Antioxidants 2022, 11, 784. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bloem, L.J.; Yu, L.; Estridge, T.B.; Iversen, P.W.; McDonald, C.E.; Schrementi, J.P.; Wang, X.; Vlahos, C.J.; Wang, J. Protein kinase C βII activation induces angiotensin converting enzyme expression in neonatal rat cardiomyocytes. Cardiovasc. Res. 2003, 57, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. The Pathobiology of Diabetic Complications. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Wright, J.J.; Kim, J.; Buchanan, J.; Boudina, S.; Sena, S.; Bakirtzi, K.; Ilkun, O.; Theobald, H.A.; Cooksey, R.C.; Kandror, K.V.; et al. Mechanisms for increased myocardial fatty acid utilization following short-term high-fat feeding. Cardiovasc. Res. 2009, 82, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.A.; Varela-Carver, A.; Mongillo, M.; Kleinert, C.; Khan, M.T.; Leccisotti, L.; Strickland, N.; Matsui, T.; Das, S.; Rosenzweig, A.; et al. Abnormal myocardial insulin signalling in type 2 diabetes and left-ventricular dysfunction. Eur. Hear. J. 2009, 31, 100–111. [Google Scholar] [CrossRef]
- Coort, S.L.; Hasselbaink, D.M.; Koonen, D.P.; Willems, J.; Coumans, W.A.; Chabowski, A.; van der Vusse, G.J.; Bonen, A.; Glatz, J.F.; Luiken, J.J. Enhanced Sarcolemmal FAT/CD36 Content and Triacylglycerol Storage in Cardiac Myocytes From Obese Zucker Rats. Diabetes 2004, 53, 1655–1663. [Google Scholar] [CrossRef]
- Chen, S.; Zou, Y.; Song, C.; Cao, K.; Cai, K.; Wu, Y.; Zhang, Z.; Geng, D.; Sun, W.; Ouyang, N.; et al. The role of glycolytic metabolic pathways in cardiovascular disease and potential therapeutic approaches. Basic Res. Cardiol. 2023, 118, 1–27. [Google Scholar] [CrossRef]
- Du, X.; Edelstein, D.; Obici, S.; Higham, N.; Zou, M.-H.; Brownlee, M. Insulin resistance reduces arterial prostacyclin synthase and eNOS activities by increasing endothelial fatty acid oxidation. J. Clin. Investig. 2006, 116, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N. Advanced glycation endproducts—Role in pathology of diabetic complications. Diabetes Res. Clin. Pract. 2005, 67, 3–21. [Google Scholar] [CrossRef]
- Stehouwer, C.D.; Lambert, J.; Donker, A.; van Hinsbergh, V.W. Endothelial dysfunction and pathogenesis of diabetic angiopathy. Cardiovasc. Res. 1997, 34, 55–68. [Google Scholar] [CrossRef]
- I Vinik, A. Diabetic neuropathy: Pathogenesis and therapy. Am. J. Med. 1999, 107, 17–26. [Google Scholar] [CrossRef]
- Du, X.-L.; Edelstein, D.; Rossetti, L.; Fantus, I.G.; Goldberg, H.; Ziyadeh, F.; Wu, J.; Brownlee, M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc. Natl. Acad. Sci. USA 2000, 97, 12222–12226. [Google Scholar] [CrossRef]
- Musicki, B.; Kramer, M.F.; Becker, R.E.; Burnett, A.L. Inactivation of phosphorylated endothelial nitric oxide synthase (Ser-1177) by O-GlcNAc in diabetes-associated erectile dysfunction. Proc. Natl. Acad. Sci. USA 2005, 102, 11870–11875. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.J.; McDonough, P.M.; Swanson, E.; Trost, S.U.; Suzuki, M.; Fukuda, M.; Dillmann, W.H. Diabetes and the Accompanying Hyperglycemia Impairs Cardiomyocyte Calcium Cycling through Increased Nuclear O-GlcNAcylation. 2003, 278, 44230–44237. J. Biol. Chem. 2003, 278, 44230–44237. [Google Scholar] [CrossRef]
- Rajamani, U.; Essop, M.F. Hyperglycemia-mediated activation of the hexosamine biosynthetic pathway results in myocardial apoptosis. Am. J. Physiol. Physiol. 2010, 299, C139–C147. [Google Scholar] [CrossRef]
- Rajamani, U.; Joseph, D.; Roux, S.; Essop, M.F. The hexosamine biosynthetic pathway can mediate myocardial apoptosis in a rat model of diet-induced insulin resistance. Acta Physiol. 2011, 202, 151–157. [Google Scholar] [CrossRef]
- Horal, M.; Zhang, Z.; Stanton, R.; Virkamäki, A.; Loeken, M.R. Activation of the hexosamine pathway causes oxidative stress and abnormal embryo gene expression: Involvement in diabetic teratogenesis. Birth Defects Res. Part A: Clin. Mol. Teratol. 2004, 70, 519–527. [Google Scholar] [CrossRef]
- Yang, C.W.; Vlassara, H.; Peten, E.P.; He, C.J.; E Striker, G.; Striker, L.J. Advanced glycation end products up-regulate gene expression found in diabetic glomerular disease. Proc. Natl. Acad. Sci. 1994, 91, 9436–9440. [Google Scholar] [CrossRef] [PubMed]
- Strieder-Barboza, C.; Baker, N.A.; Flesher, C.G.; Karmakar, M.; Neeley, C.K.; Polsinelli, D.; Dimick, J.B.; Finks, J.F.; Ghaferi, A.A.; Varban, O.A.; et al. Advanced glycation end-products regulate extracellular matrix-adipocyte metabolic crosstalk in diabetes. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bodiga, V.L.; Eda, S.R.; Bodiga, S. Advanced glycation end products: Role in pathology of diabetic cardiomyopathy. Hear. Fail. Rev. 2013, 19, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Barlovic, D.P.; Thomas, M.C.; Jandeleit-Dahm, K. Cardiovascular Disease: What’s All the AGE/RAGE About? Cardiovasc. Hematol. Disord. Targets 2010, 10, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Deluyker, D.; Evens, L.; Haesen, S.; Driesen, R.B.; Kuster, D.; Verboven, M.; Beliën, H.; van der Velden, J.; Lambrichts, I.; Bito, V. Glycolaldehyde-Derived High-Molecular-Weight Advanced Glycation End-Products Induce Cardiac Dysfunction through Structural and Functional Remodeling of Cardiomyocytes. Cell. Physiol. Biochem. 2020, 54, 809–824. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute; Surveillance, Epidemiology, and End Result Program. Cancer Stat Facts: Myeloma. Available online: https://seer.cancer.gov/statfacts/html/mulmy.html (accessed on 13 August 2022).
- Deluyker, D.; Evens, L.; Beliën, H.; Bito, V. Acute exposure to glycated proteins reduces cardiomyocyte contractile capacity. Exp. Physiol. 2019, 104, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Levelt, E.; Mahmod, M.; Piechnik, S.K.; Ariga, R.; Francis, J.M.; Rodgers, C.T.; Clarke, W.T.; Sabharwal, N.; Schneider, J.E.; Karamitsos, T.D.; et al. Relationship Between Left Ventricular Structural and Metabolic Remodeling in Type 2 Diabetes. Diabetes 2015, 65, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.-R.; Clarke, K.; Levelt, E. Metabolic remodelling in diabetic cardiomyopathy. Cardiovasc. Res. 2017, 113, 422–430. [Google Scholar] [CrossRef]
- De Marchi, E.; Baldassari, F.; Bononi, A.; Wieckowski, M.R.; Pinton, P. Oxidative Stress in Cardiovascular Diseases and Obesity: Role of p66Shc and Protein Kinase C. Oxidative Med. Cell. Longev. 2013, 2013, 1–11. [Google Scholar] [CrossRef]
- Guo, A.; Chen, R.; Wang, Y.; Huang, C.-K.; Chen, B.; Kutschke, W.; Hong, J.; Song, L.-S. Transient activation of PKC results in long-lasting detrimental effects on systolic [Ca2+]i in cardiomyocytes by altering actin cytoskeletal dynamics and T-tubule integrity. J. Mol. Cell. Cardiol. 2018, 115, 104–114. [Google Scholar] [CrossRef]
- Marfella, R.; Di Filippo, C.; Portoghese, M.; Barbieri, M.; Ferraraccio, F.; Siniscalchi, M.; Cacciapuoti, F.; Rossi, F.; D’Amico, M.; Paolisso, G. Myocardial lipid accumulation in patients with pressure-overloaded heart and metabolic syndrome. J. Lipid Res. 2009, 50, 2314–2323. [Google Scholar] [CrossRef] [PubMed]
- Young, M.E.; Patil, S.; Ying, J.; Depre, C.; Ahuja, H.S.; Shipley, G.L.; Stepkowski, S.M.; Davies, P.J.A.; Taegtmeyer, H. Uncoupling protein 3 transcription is regulated by peroxisome proliferator-activated receptor α in the adult rodent heart. FASEB J. 2001, 15, 833–845. [Google Scholar] [CrossRef] [PubMed]
- van de Weijer, T.; Schrauwen-Hinderling, V.B.; Schrauwen, P. Lipotoxicity in type 2 diabetic cardiomyopathy. Cardiovasc. Res. 2011, 92, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.J.; Kypson, A.P.; Rodriguez, E.; Anderson, C.A.; Lehr, E.J.; Neufer, P.D. Substrate-Specific Derangements in Mitochondrial Metabolism and Redox Balance in the Atrium of the Type 2 Diabetic Human Heart. Circ. 2009, 54, 1891–1898. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.Y.; Prins, J.B.; Marwick, T.H. Diabetic Cardiomyopathy: Evidence, Mechanisms, and Therapeutic Implications. Endocr. Rev. 2004, 25, 543–567. [Google Scholar] [CrossRef] [PubMed]
- Francés, J.L.; Pagiatakis, C.; Di Mauro, V.; Climent, M. Therapeutic Potential of EVs: Targeting Cardiovascular Diseases. Biomedicines 2023, 11, 1907. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, S.; Xiong, Z.; Cheng, Z.; Yang, Z.; Lin, J.; Wang, T.; Feng, X.; Gao, E.; Wang, H.; et al. Exosomal Mst1 transfer from cardiac microvascular endothelial cells to cardiomyocytes deteriorates diabetic cardiomyopathy. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2018, 1864, 3639–3649. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Chapman, S.; Pham, D.L.; Ko, M.L.; Zhou, B.; Ko, G.Y.-P. Decreased miR-150 in obesity-associated type 2 diabetic mice increases intraocular inflammation and exacerbates retinal dysfunction. BMJ Open Diabetes Res. Care 2020, 8, e001446. [Google Scholar] [CrossRef] [PubMed]
- Diniz, G.P.; Takano, A.P.; Barreto-Chaves, M.L.M. miRNA-208a and miRNA-208b are triggered in thyroid hormone-induced cardiac hypertrophy – Role of type 1 Angiotensin II receptor (AT1R) on miRNA-208a/α-MHC modulation. Mol. Cell. Endocrinol. 2013, 374, 117–124. [Google Scholar] [CrossRef]
- Lyu, L.; Wang, H.; Li, B.; Qin, Q.; Qi, L.; Nagarkatti, M.; Nagarkatti, P.; Janicki, J.S.; Wang, X.L.; Cui, T. A critical role of cardiac fibroblast-derived exosomes in activating renin angiotensin system in cardiomyocytes. J. Mol. Cell. Cardiol. 2015, 89, 268–279. [Google Scholar] [CrossRef]
- Aoki, H.; Richmond, M.; Izumo, S.; Sadoshima, J. Specific role of the extracellular signal-regulated kinase pathway in angio-tensin II-induced cardiac hypertrophy in vitro. Biochem J 2000, 347 Pt 1, 275–284. [Google Scholar] [CrossRef]
- Gul, R.; Shawl, A.I.; Kim, S.-H.; Kim, U.-H. Cooperative interaction between reactive oxygen species and Ca2+ signals contributes to angiotensin II-induced hypertrophy in adult rat cardiomyocytes. Am. J. Physiol. Circ. Physiol. 2012, 302, H901–H909. [Google Scholar] [CrossRef] [PubMed]
- Bang, C.; Batkai, S.; Dangwal, S.; Gupta, S.K.; Foinquinos, A.; Holzmann, A.; Just, A.; Remke, J.; Zimmer, K.; Zeug, A.; et al. Cardiac fibroblast–derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J. Clin. Investig. 2014, 124, 2136–2146. [Google Scholar] [CrossRef]
- LaFramboise, W.A.; Scalise, D.; Stoodley, P.; Graner, S.R.; Guthrie, R.D.; Magovern, J.A.; Becich, M.J. Cardiac fibroblasts influence cardiomyocyte phenotype in vitro. Am. J. Physiol. Physiol. 2007, 292, C1799–C1808. [Google Scholar] [CrossRef] [PubMed]
- Rezazadeh-Gavgani, E.; Oladghaffari, M.; Bahramian, S.; Majidazar, R.; Dolati, S. MicroRNA-21: A critical underestimated molecule in diabetic retinopathy. Gene 2023, 859, 147212. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.C.; Davis, M.E.; Lisowski, L.K.; Lee, R.T. Endothelial-cardiomyocyte interactions in cardiac development and repair. Annu. Rev. Physiol. 2006, 68, 51–66. [Google Scholar] [CrossRef]
- Gustafson, D.; Veitch, S.; Fish, J.E. Extracellular Vesicles as Protagonists of Diabetic Cardiovascular Pathology. Front. Cardiovasc. Med. 2017, 4, 71. [Google Scholar] [CrossRef]
- Zeng, H.; Chen, J.-X. Microvascular Rarefaction and Heart Failure with Preserved Ejection Fraction. Front. Cardiovasc. Med. 2019, 6, 15. [Google Scholar] [CrossRef]
- Wang, X.; Qian, R.; Zhang, W.; Chen, S.; Jin, H.; Hu, R. MicroRNA-320 Expression in Myocardial Microvascular Endothelial Cells and Its Relationship with Insulin-Like Growth Factor-1 in Type 2 Diabetic Rats. Clin. Exp. Pharmacol. Physiol. 2009, 36, 181–188. [Google Scholar] [CrossRef]
- Caporali, A.; Meloni, M.; Nailor, A.; Mitić, T.; Shantikumar, S.; Riu, F.; Sala-Newby, G.B.; Rose, L.; Besnier, M.; Katare, R.; et al. p75NTR-dependent activation of NF-κB regulates microRNA-503 transcription and pericyte–endothelial crosstalk in diabetes after limb ischaemia. Nat. Commun. 2015, 6, 8024. [Google Scholar] [CrossRef]
- Gu, S.; Zhang, W.; Chen, J.; Ma, R.; Xiao, X.; Ma, X.; Yao, Z.; Chen, Y. EPC-Derived Microvesicles Protect Cardiomyocytes from Ang II-Induced Hypertrophy and Apoptosis. PLOS ONE 2014, 9, e85396. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Yang, Y.; Zhong, Y.; Ammar, H.M.; Zhang, P.; Guo, R.; Liu, H.; Cheng, C.; Koroscil, T.M.; Chen, Y.; et al. The effects of microvesicles on endothelial progenitor cells are compromised in type 2 diabetic patients via downregulation of the miR-126/VEGFR2 pathway. Am. J. Physiol. Metab. 2016, 310, E828–E837. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Dong, Y.; Yan, C.; Yu, T.; Cao, H. The role of exosomes and exosomal microRNA in diabetic cardiomyopathy. Front. Endocrinol. 2024, 14, 1327495. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Frangogiannis, N.G. Diabetes-associated cardiac fibrosis: Cellular effectors, molecular mechanisms and therapeutic opportunities. J. Mol. Cell. Cardiol. 2015, 90, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Regan, T.J.; Lyons, M.M.; Ahmed, S.S.; Levinson, G.E.; Oldewurtel, H.A.; Ahmad, M.R.; Haider, B. Evidence for Cardiomyopathy in Familial Diabetes Mellitus. J. Clin. Investig. 1977, 60, 885–899. [Google Scholar] [CrossRef]
- Li, Y.; Duan, J.-Z.; He, Q.; Wang, C.-Q. miR-155 modulates high glucose-induced cardiac fibrosis via the Nrf2/HO-1 signaling pathway. Mol. Med. Rep. 2020, 22, 4003–4016. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, C.; Liu, L.; A, X.; Chen, B.; Li, Y.; Du, J. Macrophage-Derived mir-155-Containing Exosomes Suppress Fibroblast Proliferation and Promote Fibroblast Inflammation during Cardiac Injury. Mol. Ther. 2016, 25, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Huffaker, T.B.; O’connell, R.M. miR-155-SOCS1 as a Functional Axis: Satisfying the Burden of Proof. Immunity 2015, 43, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Naka, T.; Kubo, M. SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 2007, 7, 454–465. [Google Scholar] [CrossRef]
- Alexander, W.S. Suppressors of cytokine signalling (SOCS) in the immune system. Nat. Rev. Immunol. 2002, 2, 410–416. [Google Scholar] [CrossRef]
- Wu, Y.; Li, Y.; Zhang, C.; A, X.; Wang, Y.; Cui, W.; Li, H.; Du, J. S100a8/a9 Released by CD11b+ Gr1+ Neutrophils Activates Cardiac Fibroblasts to Initiate Angiotensin II–Induced Cardiac Inflammation and Injury. Hypertension 2014, 63, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Kolb, M.; Margetts, P.J.; Anthony, D.C.; Pitossi, F.; Gauldie, J. Transient expression of IL-1β induces acute lung injury and chronic repair leading to pulmonary fibrosis. J. Clin. Investig. 2001, 107, 1529–1536. [Google Scholar] [CrossRef]
- Weber, K.T. Monitoring tissue repair and fibrosis from a distance. Circulation 1997, 96, 2488–2492. [Google Scholar]
- Ma, Z.-G.; Yuan, Y.-P.; Wu, H.-M.; Zhang, X.; Tang, Q.-Z. Cardiac fibrosis: New insights into the pathogenesis. Int. J. Biol. Sci. 2018, 14, 1645–1657. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; McTiernan, C.F.; Frye, C.S.; Slawson, S.E.; Lemster, B.H.; Koretsky, A.P.; Demetris, A.J.; Feldman, A.M. Dilated Cardiomyopathy in Transgenic Mice with Cardiac-Specific Overexpression of Tumor Necrosis Factor-α. Circ. Res. 1997, 81, 627–635. [Google Scholar] [CrossRef]
- Govindappa, P.K.; Patil, M.; Garikipati, V.N.S.; Verma, S.K.; Saheera, S.; Narasimhan, G.; Zhu, W.; Kishore, R.; Zhang, J.; Krishnamurthy, P. Targeting exosome-associated human antigen R attenuates fibrosis and inflammation in diabetic heart. FASEB J. 2020, 34, 2238–2251. [Google Scholar] [CrossRef]
- Schnee, J.M.; Hsueh, W.A. Angiotensin II, adhesion, and cardiac fibrosis. Cardiovasc. Res. 2000, 46, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Luo, L.; Wei, X.; Gong, D.; Li, Z.; Li, S.; Tang, W.; Jin, L. M1 Bone Marrow-Derived Macrophage-Derived Extracellular Vesicles Inhibit Angiogenesis and Myocardial Regeneration Following Myocardial Infarction via the MALAT1/MicroRNA-25-3p/CDC42 Axis. Oxidative Med. Cell. Longev. 2021, 2021, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Wahlquist, C.; Jeong, D.; Rojas-Muñoz, A.; Kho, C.; Lee, A.; Mitsuyama, S.; van Mil, A.; Park, W.J.; Sluijter, J.P.G.; Doevendans, P.A.F.; et al. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature 2014, 508, 531–535. [Google Scholar] [CrossRef]
- Thaik, C.M.; Calderone, A.; Takahashi, N.; Colucci, W.S. Interleukin-1 beta modulates the growth and phenotype of neonatal rat cardiac myocytes. J. Clin. Investig. 1995, 96, 1093–1099. [Google Scholar] [CrossRef]
- Stangl, V.; Baumann, G.; Stangl, K.; Felix, S.B. Negative inotropic mediators released from the heart after myocardial ischaemia–reperfusion. Cardiovasc. Res. 2002, 53, 12–30. [Google Scholar] [CrossRef] [PubMed]
- Finkel, M.S.; Oddis, C.V.; Jacob, T.D.; Watkins, S.C.; Hattler, B.G.; Simmons, R.L. Negative Inotropic Effects of Cytokines on the Heart Mediated by Nitric Oxide. Science 1992, 257, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Goldhaber, J.I.; Kim, K.H.; Natterson, P.D.; Lawrence, T.; Yang, P.; Weiss, J.N.; Petkova-Kirova, P.S.; Gursoy, E.; Mehdi, H.; McTiernan, C.F.; et al. Effects of TNF-alpha on [Ca2+]i and contractility in isolated adult rabbit ventricular myocytes. Am. J. Physiol. Circ. Physiol. 1996, 271, H1449–H1455. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, C.; Salerno, L.; Marino, F.; Scalise, M.; Salerno, N.; Pagano, L.; De Angelis, A.; Cianflone, E.; Torella, D.; Urbanek, K. Unraveling and Targeting Myocardial Regeneration Deficit in Diabetes. Antioxidants 2022, 11, 208. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Ros, J.; Mas-Bargues, C.; Romero-García, N.; Huete-Acevedo, J.; Dromant, M.; Borrás, C. Therapeutic Potential of Extracellular Vesicles in Aging and Age-Related Diseases. Int. J. Mol. Sci. 2022, 23, 14632. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Inagaki, N.; Kondoh, H. Cellular Senescence in Diabetes Mellitus: Distinct Senotherapeutic Strategies for Adipose Tissue and Pancreatic β Cells. Front. Endocrinol. 2022, 13, 869414. [Google Scholar] [CrossRef] [PubMed]
- Fafián-Labora, J.A.; O’loghlen, A. NF-κB/IKK activation by small extracellular vesicles within the SASP. Aging Cell 2021, 20, e13426. [Google Scholar] [CrossRef] [PubMed]
- Shakeri, H.; Lemmens, K.; Gevaert, A.B.; De Meyer, G.R.Y.; Segers, V.F.M. Cellular senescence links aging and diabetes in cardiovascular disease. Am. J. Physiol. Circ. Physiol. 2018, 315, H448–H462. [Google Scholar] [CrossRef]
- Huang, C.; Neupane, Y.R.; Lim, X.C.; Shekhani, R.; Czarny, B.; Wacker, M.G.; Pastorin, G.; Wang, J.W. Extracellular vesicles in cardiovascular disease. Adv Clin Chem 2021, 103, 47–95. [Google Scholar] [CrossRef]
- Pan, L.; Huang, B.-J.; Ma, X.-E.; Wang, S.-Y.; Feng, J.; Lv, F.; Liu, Y.; Liu, Y.; Li, C.-M.; Liang, D.-D.; et al. MiR-25 Protects Cardiomyocytes against Oxidative Damage by Targeting the Mitochondrial Calcium Uniporter. Int. J. Mol. Sci. 2015, 16, 5420–5433. [Google Scholar] [CrossRef]
- Sang, H.-Q.; Jiang, Z.-M.; Zhao, Q.-P.; Xin, F. MicroRNA-133a improves the cardiac function and fibrosis through inhibiting Akt in heart failure rats. Biomed. Pharmacother. 2015, 71, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jia, Z.; Zhang, C.; Sun, M.; Wang, W.; Chen, P.; Ma, K.; Zhang, Y.; Li, X.; Zhou, C. miR-499 protects cardiomyocytes from H2O2-induced apoptosis via its effects on Pdcd4 and Pacs2. RNA Biol 2014, 11, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Zhao, J.; Tuazon, J.P.; Borlongan, C.V.; Yu, G. MicroRNA-133a and Myocardial Infarction. Cell Transplant. 2019, 28, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Bitirim, C.V.; Ozer, Z.B.; Aydos, D.; Genc, K.; Demirsoy, S.; Akcali, K.C.; Turan, B. Cardioprotective effect of extracellular vesicles derived from ticagrelor-pretreated cardiomyocyte on hyperglycemic cardiomyocytes through alleviation of oxidative and endoplasmic reticulum stress. Sci. Rep. 2022, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gu, H.; Huang, W.; Peng, J.; Li, Y.; Yang, L.; Qin, D.; Essandoh, K.; Wang, Y.; Peng, T.; et al. Hsp20-Mediated Activation of Exosome Biogenesis in Cardiomyocytes Improves Cardiac Function and Angiogenesis in Diabetic Mice. Diabetes 2016, 65, 3111–3128. [Google Scholar] [CrossRef] [PubMed]
- Castaño, C.; Mirasierra, M.; Vallejo, M.; Novials, A.; Párrizas, M. Delivery of muscle-derived exosomal miRNAs induced by HIIT improves insulin sensitivity through down-regulation of hepatic FoxO1 in mice. Proc. Natl. Acad. Sci. 2020, 117, 30335–30343. [Google Scholar] [CrossRef] [PubMed]
- Ying, W.; Gao, H.; Dos Reis, F.C.G.; Bandyopadhyay, G.; Ofrecio, J.M.; Luo, Z.; Ji, Y.; Jin, Z.; Ly, C.; Olefsky, J.M. MiR-690, an exosomal-derived miRNA from M2-polarized macrophages, improves insulin sensitivity in obese mice. Cell Metab. 2021, 33, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.-J.; Jang, J.-K.; Ham, O.; Song, B.-W.; Lee, S.-Y.; Lee, C.Y.; Park, J.-H.; Lee, J.; Seo, H.-H.; Choi, E.; et al. MicroRNA-145 suppresses ROS-induced Ca2+ overload of cardiomyocytes by targeting CaMKIIδ. Biochem. Biophys. Res. Commun. 2013, 435, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Chen, J.Y.; Peng, W.M.; Yuan, B.; Bi, Q.; Xu, Y.J. Exosomes from adipose-derived stem cells promote chondrogenesis and suppress inflammation by upregulating miR-145 and miR-221. Mol. Med. Rep. 2020, 21, 1881–1889. [Google Scholar] [CrossRef]
- Zhou, H.; Zhou, J.; Teng, H.; Yang, H.; Qiu, J.; Li, X. MiR-145 enriched exosomes derived from bone marrow-derived mesenchymal stem cells protects against cerebral ischemia-reperfusion injury through downregulation of FOXO. Biochem. Biophys. Res. Commun. 2022, 632, 92–99. [Google Scholar] [CrossRef]
- Mayourian, J.; Cashman, T.J.; Ceholski, D.K.; Johnson, B.V.; Sachs, D.; Kaji, D.A.; Sahoo, S.; Hare, J.M.; Hajjar, R.J.; Sobie, E.A.; et al. Experimental and Computational Insight Into Human Mesenchymal Stem Cell Paracrine Signaling and Heterocellular Coupling Effects on Cardiac Contractility and Arrhythmogenicity. Circ. Res. 2017, 121, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Mayourian, J.; Ceholski, D.K.; Gorski, P.A.; Mathiyalagan, P.; Murphy, J.F.; Salazar, S.I.; Stillitano, F.; Hare, J.M.; Sahoo, S.; Hajjar, R.J.; et al. Exosomal microRNA-21-5p Mediates Mesenchymal Stem Cell Paracrine Effects on Human Cardiac Tissue Contractility. Circ. Res. 2018, 122, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Jansen, F.; Yang, X.; Baumann, K.; Przybilla, D.; Schmitz, T.; Flender, A.; Paul, K.; Alhusseiny, A.; Nickenig, G.; Werner, N. Endothelial microparticles reduce ICAM-1 expression in a microRNA-222-dependent mechanism. J. Cell. Mol. Med. 2015, 19, 2202–2214. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zhang, F.; Lian, X.F.; Peng, W.Q.; Yin, C.Y. Mesenchymal stem cell-derived exosomes improve diabetes mellitus-induced myocardial injury and fibrosis via inhibition of TGF-β1/Smad2 signaling pathway. Cell Mol Biol (Noisy-le-grand) 2019, 65, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Benedikter, B.J.; Weseler, A.R.; Wouters, E.F.M.; Savelkoul, P.H.M.; Rohde, G.G.U.; Stassen, F.R.M. Redox-dependent thiol modifications: Implications for the release of extracellular vesicles. Cell. Mol. Life Sci. 2018, 75, 2321–2337. [Google Scholar] [CrossRef] [PubMed]
- Chiaradia, E.; Tancini, B.; Emiliani, C.; Delo, F.; Pellegrino, R.M.; Tognoloni, A.; Urbanelli, L.; Buratta, S. Extracellular Vesicles under Oxidative Stress Conditions: Biological Properties and Physiological Roles. Cells 2021, 10, 1763. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, D.G.; Doseff, A.; Chau, B.N.; Lim, D.-S.; de Souza-Pinto, N.C.; Hansford, R.; Kastan, M.B.; Lazebnik, Y.A.; Hardwick, J.M. Caspase-3-dependent Cleavage of Bcl-2 Promotes Release of Cytochrome c. J. Biol. Chem. 1999, 274, 21155–21161. [Google Scholar] [CrossRef]
- Yildirim, S.S.; Akman, D.; Catalucci, D.; Turan, B. Relationship Between Downregulation of miRNAs and Increase of Oxidative Stress in the Development of Diabetic Cardiac Dysfunction: Junctin as a Target Protein of miR-1. Cell Biochem. Biophys. 2013, 67, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuñiga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-C.; Zhou, L.; Wang, F.; Cheng, Z.-Q.; Rong, C. Osthole decreases collagen I/III contents and their ratio in TGF- β 1-overexpressed mouse cardiac fibroblasts through regulating the TGF- β/Smad signaling pathway. Chin. J. Nat. Med. 2018, 16, 321–329. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhu, Z.; Wang, T.; Dong, Y.; Fan, Y.; Sun, D. TGF-β1-containing exosomes from cardiac microvascular endothelial cells mediate cardiac fibroblast activation under high glucose conditions. Biochem. Cell Biol. 2021, 99, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Caby, M.-P.; Lankar, D.; Vincendeau-Scherrer, C.; Raposo, G.; Bonnerot, C. Exosomal-like vesicles are present in human blood plasma. Int. Immunol. 2005, 17, 879–887. [Google Scholar] [CrossRef]
- Palanisamy, V.; Sharma, S.; Deshpande, A.; Zhou, H.; Gimzewski, J.; Wong, D.T. Nanostructural and Transcriptomic Analyses of Human Saliva Derived Exosomes. PLOS ONE 2010, 5, e8577. [Google Scholar] [CrossRef] [PubMed]
- Erdbrügger, U.; Blijdorp, C.J.; Bijnsdorp, I.V.; Borràs, F.E.; Burger, D.; Bussolati, B.; Byrd, J.B.; Clayton, A.; Dear, J.W.; Falcón-Pérez, J.M.; et al. Urinary extracellular vesicles: A position paper by the Urine Task Force of the International Society for Extracellular Vesicles. J. Extracell. Vesicles 2021, 10, e12093. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Luo, X.; Liao, B.; Li, G.; Feng, J. Insights into SGLT2 inhibitor treatment of diabetic cardiomyopathy: Focus on the mechanisms. Cardiovasc. Diabetol. 2023, 22, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.M.; Yang, Q.Y.; Monsel, A.; Yan, J.Y.; Dai, C.X.; Zhao, J.Y.; Shi, G.C.; Zhou, M.; Zhu, X.M.; Li, S.K.; et al. Preclinical efficacy and clinical safety of clinical-grade nebulized allogenic adipose mesenchymal stromal cells-derived extracellular vesicles. J. Extracell. Vesicles 2021, 10, e12134. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.; Wang, H.; Bian, L.; Huang, J.; Wu, D.; Zhang, R.; Fei, F.; Chen, Y.; Xia, J. Nebulization Therapy with Umbilical Cord Mesenchymal Stem Cell-Derived Exosomes for COVID-19 Pneumonia. Stem Cell Rev. Rep. 2022, 18, 2152–2163. [Google Scholar] [CrossRef]
- Kwon, H.H.; Yang, S.H.; Lee, J.; Park, B.C.; Park, K.Y.; Jung, J.Y.; Bae, Y.; Park, G.-H. Combination Treatment with Human Adipose Tissue Stem Cell-derived Exosomes and Fractional CO2 Laser for Acne Scars: A 12-week Prospective, Double-blind, Randomized, Split-face Study. Acta Dermato-Venereologica 2020, 100, adv00310. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Nomenclature of Extracellular Vesicles (EVs) | References | ||
|---|---|---|---|
| Classical (based on biogenesis) * | exosomes | exocytosis of MVB | [27,28,29,30,31,32] |
| microvesicles | direct budding from PM | ||
| apoptotic EVs | apoptosis | ||
| Size | small | <200 nm | |
| medium/large | >200 nm | ||
| Density | low | each range should be defined, e.g., exosome and microvesicle density—1.08–1.19 g/mL; apoptotic EV density—1.16–1.28 g/ml | |
| medium | |||
| high | |||
| Biochemical composition | e.g., CD63, CD81, CD9-positive EVs—classical exosomes; annexin-V-positive EVs—apoptotic EVs; ARRDC1-positive EVs—ARMM | ||
| Cellular origin | e.g., myocardial EV, endothelial EV, oncosome, fibroblast-derived EV | ||
| Condition | e.g., hypoxic EV, apoptotic EV | ||
| Type of miRNA | Type of Extracellular Vesicles | Secreting Cells | Target Cells | Mechanism of Changes | Ref. |
| miR-25 | No data | No data | Cardiomyocytes | Anti-apoptotic effect | [112] |
| miR-133a/b | Extracellular Vesicles | Cardiomyocytes | Cardiomyocytes | Anti-apoptotic effect | [113,114,115,116] |
| No data | Exosomes | Cardiomyocytes | Cardiomyocytes | The anti-apoptotic effect, increased myocardial vascular density | [117] |
| miR-133b | Exosomes | Muscle | Hepatocytes | Improved insulin sensitivity | [118] |
| miR-690 | Exosomes | Macrophages | Adipocytes, hepatocytes | Improved insulin sensitivity | [119] |
| miR-145 | Exosomes | Bone marrow mesenchymal stem cells and adipose-derived stem cells | Cardiomyocytes | Regulation of intracellular calcium level | [94,120,121,122] |
| miR-21-5p | Exosomes | Human mesenchymal stem cells | Cardiomyocytes | Regulation of intracellular calcium level and contractile improvement | [123,124] |
| miR-222 | Extracellular Vesicles | Endothelial cells | Endothelial cells | Anti-inflammatory effect | [125] |
| No data | Exosomes | Mesenchymal stem cells | Cardiac fibroblasts | Inhibition of fibrosis | [126] |
| Trial Number | Status | Disease(s) | Interventional or Observational | Aim of the Study | Potential Application in DCM Research |
|---|---|---|---|---|---|
| NCT05774509 | recruiting | non-ischemic dilated cardiomyopathy | interventional | assessment of the safety and efficacy of the extracellular vesicle-enriched secretome of cardiovascular progenitor cells in severely symptomatic patients with non-ischemic dilated cardiomyopathy | therapy |
| NCT06169540 | recruiting | acute decompensated heart failure and chronic heart failure | observational | determining the relationship between the levels of RNA, including ones in extracellular vesicles from different organs in the blood and the saliva of patients with HF | diagnosis |
| NCT03268135 | recruiting | heart failure, aortic stenosis | observational | investigating global transcriptome to determine the expression profile of different RNAs in patients with heart failure and aortic stenosis | diagnosis |
| NCT05726695 | active, not recruiting | heart failure with reduced ejection fraction | observational | determining the analytical characteristics of the microRNA enzymatic immunoassay method and various relations among miRNA biomarkers and heart failure | diagnosis |
| NCT04950569 | unknown | heart failure | interventional | determining the relationships between several clinical and laboratory findings (including myocardial miRNAs) after levosimendan treatment | therapy |
| NCT02138331 | unknown | T1DM | interventional | assessing the relationship between intravenous infusion of microvesicles from mesenchymal stem cells and β-cell mass as well as the glycemic control in T1DM patients | therapy |
| NCT02459106 | active, not recruiting | T2DM | observational | investigation of effects of fat tissue-released miRNA on biology and insulin sensitivity of skeletal muscle | diagnosis/therapy |
| NCT05139914 | recruiting | T2DM | interventional | assessment of the impact of dapagliflozin on endothelial cell health (including non-coding RNA assessment) | therapy, therapy monitoring |
| NCT05259449 | recruiting | T2DM | interventional | determination of the efficacy of nutritional and physical education on health-related variables (such as exosome profile) in T2DM patients | therapy monitoring |
| NCT06401876 | not yet recruiting | T2DM, obesity | observational | profiling the cargo of extracellular vesicles of obese diabetic patients before and after bariatric surgery | therapy monitoring |
| NCT06408961 | recruiting | obesity, cardiometabolic disease | observational | researching the content and function of extracellular vesicles in obese patients receiving bariatric surgery | therapy monitoring |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zygmunciak, P.; Stróżna, K.; Błażowska, O.; Mrozikiewicz-Rakowska, B. Extracellular Vesicles in Diabetic Cardiomyopathy—State of the Art and Future Perspectives. Int. J. Mol. Sci. 2024, 25, 6117. https://doi.org/10.3390/ijms25116117
Zygmunciak P, Stróżna K, Błażowska O, Mrozikiewicz-Rakowska B. Extracellular Vesicles in Diabetic Cardiomyopathy—State of the Art and Future Perspectives. International Journal of Molecular Sciences. 2024; 25(11):6117. https://doi.org/10.3390/ijms25116117
Chicago/Turabian StyleZygmunciak, Przemysław, Katarzyna Stróżna, Olga Błażowska, and Beata Mrozikiewicz-Rakowska. 2024. "Extracellular Vesicles in Diabetic Cardiomyopathy—State of the Art and Future Perspectives" International Journal of Molecular Sciences 25, no. 11: 6117. https://doi.org/10.3390/ijms25116117
APA StyleZygmunciak, P., Stróżna, K., Błażowska, O., & Mrozikiewicz-Rakowska, B. (2024). Extracellular Vesicles in Diabetic Cardiomyopathy—State of the Art and Future Perspectives. International Journal of Molecular Sciences, 25(11), 6117. https://doi.org/10.3390/ijms25116117