
Regulatory Role of Fatty Acid Metabolism on Glucose-Induced Changes in Insulin and Glucagon Secretion by Pancreatic Islet Cells
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Lipid Metabolism in β-Cells
2.1. Islet FFA Oxidation
2.2. Islet Lipid Biosynthesis
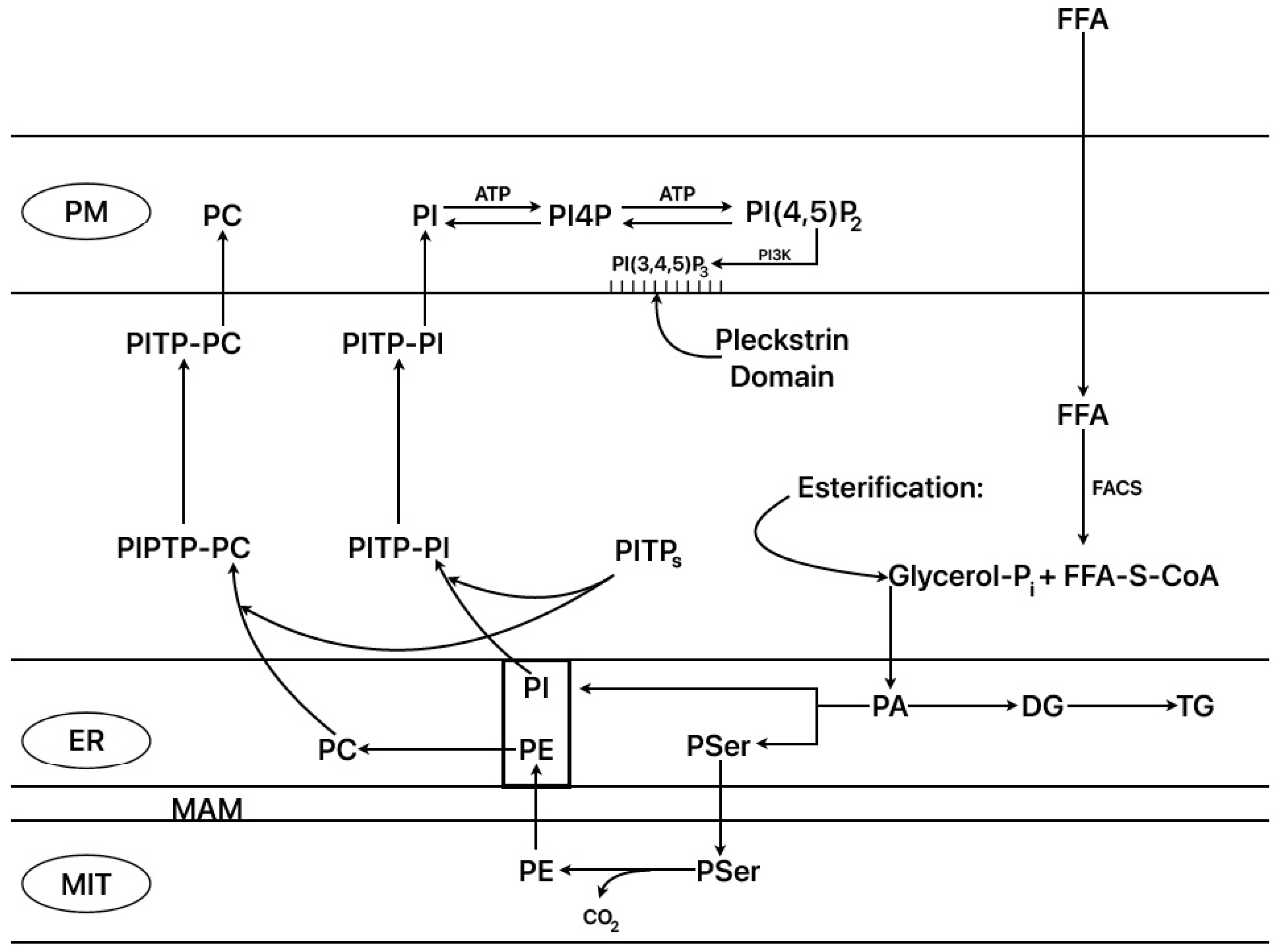
2.3. Islet 45Ca2+-Turnover and Phospholipid Biosynthesis
2.4. Stimulation of Islet Protein Kinase C Translocation through Palmitate Metabolism
2.5. Modulation of Glucose-Induced Insulin Secretion via FFA Metabolism
2.6. Norepinephrine Inhibition of Insulin Secretion
2.7. α2-Adrenergic Signaling Responsible for Norepinephrine Effects on Islet FFA Metabolism and Their Consequences for Insulin Secretion
- The effects of both messengers were assayed in incubated rat islets in a range of five concentrations (0 to 1 mM) on the insulin secretory response to 20 mM glucose at three palmitate concentrations (0, 0.25, and 1.0 mM). cAMP significantly increased the insulin response to glucose at 0.25, 0.50, and 1.0 mM in the absence and presence of 0.25 mM palmitate, but not of 1 mM. cGMP significantly inhibited insulin secretion from 0.25 onwards at the three palmitate concentrations. The simultaneous addition of 0.25 mM cGMP and 1 mM cAMP did not reverse cGMP-induced inhibition with either 0 or 1.0 mM palmitate [31].
- D-[U-14C] glucose incorporation into islet lipids (DG, TG, PA, PI, PPI) in the absence of palmitate was not modified by the presence of 1 mM 8-Br-cGMP with either 3 or 20 mM labeled glucose. In the presence of 1 mM palmitate, 1 mM 8-Br-cGMP significantly suppressed 20 mM labeled glucose incorporation into all the lipid fractions analyzed; the decrease in labeled incorporation was not recovered by the simultaneous addition of 1 mM 8-Br-cAMP [31].
- No effect was appreciated by either of the two messengers on the initial (15 min) 45Ca2+_uptake for 3 mM glucose. In addition, 0.25 mM 8-Br-cGMP did not modify the initial uptake of calcium stimulated by 20 mM glucose alone. The addition of 1 mM palmitate to 20 mM glucose potentiated around 2-fold glucose stimulation, and in the presence of 0.25 mM cGMP, the potentiation capacity of palmitate was almost completely suppressed. 1 mM 8-Br-cAMP was unable to recover 0.25 mM 8-Br-cGMP inhibition of palmitate potentiation at 20 mM glucose [31].
2.8. Norepinephrine Stimulation of Islet Adenylyl and Guanylyl Cyclase’s Activities [31]
2.9. ER–Mitochondrial Interactions (ER–Mito)
2.10. Summary and Physio-Pathological Implications
3. Implication of FFA Metabolism in the Glucagon Secretory Function of Pancreatic α-Cells
3.1. Introduction
3.2. α-Cell Glucose Metabolism and Its Implication in the Regulation of Glucagon Secretion
3.3. Effect of Starvation on Glucose Regulation of Glucagon Secretion
3.4. Precedents of the Effects of FFA Metabolism on the Regulation of Glucagon Secretion
3.5. Experimental Evidence in Favor of the Predominance of FFA Anabolism over Catabolism on the Stimulation of Islet Insulin and Glucagon Secretion
- 0.25 mM 2-bromostearate (BrS) has no effect on basal and glucose-stimulated insulin secretion of “fed” islets [81].
- 0.25 mM 2-bromostearate (BrS) blocks glucose-induced inhibition of glucagon release in “fed” islets [81].
- 0.25 mM BrS recovers the complete suppression of glucose-induced insulin secretion of “starved” islets [81].
- 0.1 mM 2-bromopalmitate (BP, homologous of BrS) stimulates glucagon secretion with 1 mM glucose around 5-fold in mouse islets [84].
- Deletion of the acetyl-CoA carboxylase gene codifying the enzyme responsible for malonyl-CoA synthesis significantly lowers fasting plasma glucagon levels [84].
- 0.25 mM BrS, in the presence of 6 mM glucose, converts a transient stimulation into a sustained stimulation of palmitate-induced insulin secretion [81].
- Under the same conditions as above, basal glucagon secretion was stimulated within approximately 50%, and it did not return to basal values after withdrawing the stimulus [81].
3.6. Possible Metabolic Mechanisms Associated with the Regulation of Glucagon Secretion
- FFA oxidation by β- and α-cells does not contribute to the stimulation of insulin and glucagon secretion in the presence of glucose. FFA anabolism to complex lipids, rather than its catabolism, is responsible for the potentiation of glucose activation of hormone secretion.
- β-cell secretion is more sensitive to the stimulation of FFA anabolism than α-cell secretion. This probably depends on a higher flux of β-cell lipogenesis than α-cell lipogenesis within the range of glucose concentrations that stimulate insulin release and inhibit FFA oxidation (from 6 to 20 mM glucose). This is concordant with the lack of BrS effect on glucose-stimulated insulin secretion in “fed” islets and its capacity to restore insulin secretion in “starved” islets to the level of their “fed” controls with further stimulation. In other words, BrS partially plays the role exerted by high glucose. In support of this view, palmitate stimulates a transient stimulation of insulin secretion at a low glucose concentration (6 mM) that is changed into a sustained release by BrS, probably because at this glucose level, the synthesis of malonyl-CoA is not as high as at greater sugar concentrations.
- 3.
- Blocking of the inhibitory effect of high glucose on glucagon secretion by starvation is synonymous with a stimulation of glucagon secretion at (not “by”) a high sugar concentration. Suppression of β-cell insulin secretion by glucose during starvation probably avoids the threat of hypoglycemia from elevated FFA plasma levels. More mechanistically, it suggests that the failure of an intra-islet hormonal action of insulin on glucagon secretion might be responsible for the restoration of glucagon release to fed control rates. We have no expert opinion on the open discussion about the mechanism responsible for the interaction between β- and α-cells, which is mediated by either a vascular mediation of secreted insulin from the β-cell core to the α-cells in the mantle (“core–mantle view”) to inhibit them or by a mantle-to-core diffusion of secreted glucagon to stimulate insulin secretion (paracrine view) [87,88]. However, we are prone to the “core–mantle” hypothesis, given that in isolated islets of starved animals, the “only” iatrogenic intervention was islet isolation, and the cause of islet functional changes was a physiological regulation of FFA body metabolism that resulted in the failure of glucose to decrease basal glucagon secretion in the absence of an insulin response to glucose. In a more naïve view, if the respective rates of secretion of insulin (ng/islet/min) and glucagon (pg/islet/min) secretion are expressed in molar units/islet/minute in one of our islet perifusion experiments, the insulin rate exceeds that of glucagon by around 6-fold, and the core-to-mantle gradient of insulin is higher than that of glucagon in the opposite direction, from the mantle to the β-cell core.
3.7. Mechanism of Inhibition of Glucagon Secretion by Co-Secreted Insulin (Core-to-Mantle View)
3.8. Hypothetical Mechanisms of Inhibition of Glucagon Secretion via Insulin-Mediated Stimulation of Somatostatin Secretion or a Direct Inhibitory Action of Secreted Insulin
3.9. Potentiation of Insulin Secretion by Glucagon or Incretin Peptides (Paracrine or Mantle-to-Core View)
Funding
Acknowledgments
Conflicts of Interest
References
- Newsholme, E.A.; Leech, A.R. Biochemistry for the Medical Sciences; Chapter 14; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 1983. [Google Scholar]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 281, 785–789. [Google Scholar] [CrossRef]
- Berne, C. The metabolism of lipids in mouse pancreatic islets. The oxidation of fatty acids and ketone bodies. Biochem. J. 1975, 152, 661–666. [Google Scholar] [CrossRef]
- Tamarit-Rodriguez, J.; Vara, E.; Tamarit, J. Starvation-induced changes of palmitate metabolism and insulin secretion in isolated rat islets stimulated by glucose. Biochem. J. 1984, 221, 317–324. [Google Scholar] [CrossRef]
- Resh, M.D. Use of analogs and inhibitors to study the functional significance of protein palmitoylation. Methods 2006, 40, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Vara, E.; Tamarit-Rodriguez, J. Glucose stimulation of insulin secretion in islets of fed and starved rats and its dependence on lipid metabolism. Metab. Clin. Exp. 1986, 35, 266–271. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D.; Stark, M.J.; Foster, D.W. Hepatic Malonyl-CoA Levels of Fed, Fasted and Diabetic Rats as measured using a simple radioisotopic assay. J. Biol. Chem. 1978, 253, 8291–8293. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D.; Foster, D.W. Importance of experimental conditions in evaluating the malonyl-CoA sensitivity of liver carnitine acyl transferase. Biochem. J. 1981, 200, 217–223. [Google Scholar] [CrossRef]
- Jeon, S.-M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef]
- Galic, S.; Loh, K.; Murray-Segal, L.; Steinberg, G.R.; Andrews, Z.B.; Kemp, B.E. AMPK signaling to acetyl-CoA carboxylase is required for fasting- and cold-induced appetite but not thermogenesis. eLife 2018, 7, e32656. [Google Scholar] [CrossRef]
- Welsh, M.; Andersson, A.; Brolin, S.; Hellerström, C. Effects of glucose, leucine and adenosine on insulin release, 45Ca2+ net uptake, NADH/NAD ratios and oxygen consumption of islets isolated from fed and starved mice. Mol. Cell. Endocrinol. 1983, 30, 51–62. [Google Scholar] [CrossRef]
- Stein, D.T.; Esser, V.; Stevenson, B.E.; Lane, K.E.; Whiteside, J.H.; Daniels, M.B.; Chen, S.; McGarry, J.D. Essentiality of circulating fatty acids for glucose-stimulated insulin secretion in the fasted rat. J. Clin. Investig. 1996, 97, 2728–2735. [Google Scholar] [CrossRef] [PubMed]
- Dobbins, R.L.; Chester, M.W.; Daniels, M.B.; McGarry, D.J.; Stein, D.T. Circulating fatty acids are essential for efficient glucose-stimulated insulin secretion after prolonged fasting in humans. Diabetes 1998, 47, 1613–1618. [Google Scholar] [CrossRef] [PubMed]
- Berne, C. The metabolism of lipids in mouse pancreatic islets. The biosynthesis of triacylglycerols and phospholipids. Biochem. J. 1975, 152, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Vara, E.; Fernández-Martín, O.; Tamarit-Rodriguez, J. Palmitate-dependence of insulin secretion, “de novo” phospholipid synthesis and 45Ca2+-turnover in glucose stimulated islets. Diabetologia 1988, 31, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Vara, E.; García, C.; Tamarit-Rodriguez, J. Trifluoperazine reproduces in rat islets the effects of calcium omission on insulin secretion and de novo lipid synthesis, without affecting 45Ca2+-uptake. Rev. Esp. physiol. 1990, 46, 163–170. [Google Scholar]
- Brindley, D.N. Intracellular translocation of phosphatidate phosphohydrolase and its possible role in the control of glycerolipid synthesis, PROG. Lipid. Res. 1984, 23, 115–133. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, M.; Larkins, R.G. Presence of membrane-associated phosphatidate phosphohydrolase activity in cultured islets and its stimulation by glucose. FEBNS Lett. 1985, 193, 231–235. [Google Scholar] [CrossRef]
- Alcázar, O.; Zhu, Q.-Y.; Giné, E.; Tamarit-Rodriguez, J. Stimulation of islet protein kinase C by palmitate requires metabolism of the fatty acid. Diabetes 1997, 46, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Hellman, B.; Sehlin, J.; Täljedal, I.-B. Effects of glucose on 45Ca2+ uptake by pancreatic islets as studied with the lanthanum method. J. Physiol. 1976, 254, 639–656. [Google Scholar] [CrossRef]
- Foskett, J.K.; White, C.; Cheung, K.-H.; Mak, D.-O.D. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2006, 87, 593–658. [Google Scholar] [CrossRef]
- Straub, S.G.; Sharp, G.W.G. Evolving insights regarding mechanisms for the inhibition of insulin release by norepinephrine and heterotrimeric G proteins. Am. J. Cell Physiol. 2012, 302, C1687–C1698. [Google Scholar] [CrossRef]
- Homaidan, F.R.; Sharp, G.W.G.; Novak, L.M. Galanin inhibits a dihydropyridine-sensitive Ca2+current in the RINm5f cell line. Proc. Natl. Acad. Sci. USA 1991, 88, 8744–8748. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.H.; Xiang, H.D.; Rajan, A.S.; Boyd, A.E., 3rd. Activation of α2-adrenergic receptors decreases Ca2+influx to inhibit insulin secretion in a hamster β-cell line: An action mediated by a guanosine triphosphate-binding protein. Endocrinology 1991, 128, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Yajima, H.; Komatsu, M.; Sato, Y.; Yamada, S.; Yamauchi, K.; Sharp, G.W.G.; Aizawa, T.; Hashizume, K. Norepinephrine inhibits glucose-stimulated, Ca2+-independent insulin release Independently from its action on adenylyl cyclase. Endocrine J. 2001, 48, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Ly, S.; Kim, K.-H. Inactivation of hepatic acetyl-CoA carboxylase by catecholamine and its agonists through the α-adrenergic receptors. J. Biol. Chem. 1981, 256, 11585–11590. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.E.; Saggerson, D. Reversible inactivation by noradrenaline of long-chain fatty acyl-CoA synthetase in rat adipocytes. Biochem. J. 1985, 226, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Vara, E.; Tamarit-Rodriguez, J. Norepinephrine inhibits lipid metabolism, 45Ca2+ uptake, and insulin secretion. Am. J. Physiol. 1989, 257, E923–E929. [Google Scholar] [CrossRef] [PubMed]
- Morgan, N.G.; Montague, W. Studies on the mechanism of inhibition of glucose-stimulated insulin secretion by noradrenaline in rat islets of Langerhans. Biochem. J. 1985, 256, 571–576. [Google Scholar] [CrossRef]
- Ullrich, S.; Wollheim, C.B. Islet cyclic AMP levels are not lowered during α2-adrenergic inhibition of insulin release. Studies with epinephrine and forskolin. J. Biol. Chem. 1984, 259, 4111–4115. [Google Scholar] [CrossRef]
- Vara, E.; Tamarit-Rodriguez, J. Does cyclic guanosine monophosphate mediate noradrenaline-induced inhibition of islet insulin secretion stimulate by glucose and palmitate? Biochem. J. 1991, 278, 243–248. [Google Scholar] [CrossRef]
- Waldman, S.A.; Murad, F. Cyclic GMP synthesis and function. Pharmacol. Rev. 1987, 39, 163–196. [Google Scholar] [PubMed]
- Komatsu, M.; McDermott, A.M.; Gillison, S.L.; Sharp, G.W.G. Time course of action of pertussis toxin to block the inhibition of stimulated insulin release by norepinephrine. Endocrinology 1995, 136, 1857–1863. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Park, S.; Bajpayee, N.S.; Nagaoka, Y.; Boulay, G.; Birnbaumer, L.; Jiang, M. Augmented glucose-induced insulin release in mice lacking Go2, but not Go1 or Gi proteins. Proc. Natl. Acad. Sci. USA 2011, 108, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Russel, M.A.; Morgan, N.G. Expression and functional roles of guanylate cyclase isoforms in BRIN-BD11 β-cells. Islets 2010, 26, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Currie, M.G.; Fokt, K.F.; Kato, J.; Moore, R.J.; Hamra, F.K.; Duffin, K.L.; Smith, C.E. Guanylin: An endogenous activator of intestinal guanylate cyclase. Proc. Natl. Acad. Sci. USA 1992, 89, 947–951. [Google Scholar] [CrossRef] [PubMed]
- Otero, A.; Becerril, S.; Martín, M.; Cienfuegos, J.A.; Valentí, V.; Moncada, R.; Catalán, V.; Gómez-Ambrosi, J.; Burrell, M.A.; Frühbeck, G.; et al. Effect of guanylin peptides on pancreas steatosis and function in experimental diet-induced obesity and after bariatric surgery. Front. Endocrinol. 2023, 14, 1185456. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Theurey, P.; Pizzo, P. The endoplasmic reticulum-mitochondria coupling in health and disease: Molecules, functions and significance. Cell Calcium 2017, 62, 1–15. [Google Scholar] [CrossRef] [PubMed]
- van der Veen, J.N.; Lingrell, S.; da Silva, R.P.; Jacobs, R.L.; Vance, D.E. The concentration of phosphatidylethanolamine in mitochondria can modulate ATP production and glucose metabolism in mice. Diabetes 2014, 63, 2620–2630. [Google Scholar] [CrossRef]
- Capelo- Diz, A.; Lachiondo-Ortega, S.; Fernández-Ramos, D.; Cañas-Martín, J.; Goikoetxea-Usandizaga, N.; Serrano-Maciá, M.; González-Rellan, M.J.; Mosca, L.; Blazquez-Vicens, J.; Tinahones-Ruano, A.; et al. Hepatic levels of S-adenosylmethionine regulate the adaptive response to fasting. Cell Metab. 2023, 35, 1373–1389. [Google Scholar] [CrossRef]
- van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2017, 1859, 15581572. [Google Scholar] [CrossRef]
- Cockcroft, S. The expanding roles of PI4P and PI(4,5)P2 at the plasma membrane: Role of phosphatidylinositol transfer proteins. BBA-Mol. Cell Biol. Lipids 2024, 1869, 159394. [Google Scholar] [CrossRef]
- Katan, M.; Cockcroft, S. Phosphatidylinositol(4,5)bisphosphate: Diverse functions at the plasma membrane. Essays Biochem. 2020, 64, 513–531. [Google Scholar] [PubMed]
- Ohashi, M.; Jan de Vries, K.; Frank, R.; Snoek, G.; Bankaitis, V.; Wirtz, K.; Huttner, W.B. A role for phosphatidylinositol transfer protein in secretory vesicle formation. Nature 1995, 377, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Ammar, M.R.; Kassas, N.; Chasserot-Golaz, S.; Bader, M.-F.; Vitale, N. Lipids in regulated exocytosis: What are they doing. Front. Endocrinol. 2013, 4, 125. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, H.; Kurosaki, M.; Takata, M.; Kurosaki, T. Genetic evidence for involvement of type 1, type 2 and type 3 inositol 1,4,5-trisphosphate receptors in signal transduction through the B-cell antigen receptor. EMBO J. 1997, 16, 3078–3088. [Google Scholar] [CrossRef]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.-H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Dingreville, F.; Panthu, B.; Thivolet, C.; Ducreuxm, S.; Gouriou, Y.; Pesenti, S.; Chauvin, M.A.; Chikh, K.; Errazuriz-Cerda, E.; Van Coppenolle, F.; et al. Differential effect of glucose on ER-mitochondria Ca2+ exchange participates in insulin secretion and glucotoxicity-mediated dysfunction of β-cells. Diabetes 2019, 68, 1778–1794. [Google Scholar] [CrossRef] [PubMed]
- Thivolet, C.; Vial, G.; Cassel, R.; Rieusset, J.; Madec, A.-M. Reduction of endoplasmic reticulum mitochondria interactions in beta cells from patients with type 2 diabetes. PLoS ONE 2017, 12, e0182027. [Google Scholar] [CrossRef] [PubMed]
- Warnotte, C.; Gilon, P.; Nenquin, M.; Henquin, J.-C. Mechanisms of the Stimulation of Insulin Release by Saturated Fatty Acids. A Study of Palmitate Effects in Mouse β-Cells. Diabetes 1994, 43, 703–711. [Google Scholar] [CrossRef]
- Remizov, O.; Jakubov, R.; Düfer, M.; Krippeit Drews, P.; Drews, G.; Waring, M.; Brabant, G.; Wienbergen, A.; Rustenbeck, I.; Schöfl, C. Palmitate-induced Ca2+-signaling in pancreatic beta-cells. Mol. Cell. Endocrinol 2003, 212, 1–9. [Google Scholar] [CrossRef]
- Kuok, I.T.; Rountree, A.M.; Jung, S.-R.; Sweet, I.R. Palmitate is not an effective fuel for pancreatic islets and amplifies insulin secretion independent of calcium release from endoplasmic reticulum. Islets 2019, 11, 51–64. [Google Scholar] [CrossRef]
- O’Connor, R.S.; Guo, L.; Ghassemi, S.; Snyder, N.W.; Worth, A.J.; Weng, L.; Kam, Y.; Philipson, B.; Trefely, S.; Nunez-Cruz, S.; et al. The CPT1a inhibitor, etomoxir induces severe oxidative stress at commonly used concentrations. Sci. Rep. 2018, 8, 6289. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.H.; Liu, G.Y.; Wang, R.; Moon, S.H.; Gross, R.W.; Patti, G.J. Identifying off-target effects of etomoxir reveals that carnitine palmitoyltransferase I is essential for cancer cell proliferation independent of β-oxidation. PLoS Biol. 2018, 16, e2003782. [Google Scholar] [CrossRef]
- Hedeskov, C.J.; Capito, K. The effect of starvation on Insulin secretion and glucose metabolism in mouse pancreatic islets. Biochem. J. 1974, 140, 423–433. [Google Scholar] [CrossRef]
- Malaisee, W.J.; Sener, A.; Levy, J. The stimulus-secretion coupling of glucose-induced Insulin release. Fasting-induced adaptation of key glycolytic enzymes in isolated islets. J. Biol. Chem. 1976, 251, 1731–1737. [Google Scholar] [CrossRef]
- Burch, P.T.; Trus, M.D.; Berner, K.; Leontire, A.; Zawalich, K.C.; Matschinsky, F.M. Adaptation of glycolytic enzymes. Glucose use and insulin release in rat pancreatic islets during fasting and refeeding. Diabetes 1981, 30, 923–928. [Google Scholar] [CrossRef]
- Howell, S.I.; Green, I.C.; Montague, W. A possible role of adenylate cyclase in the long-term dietary regulation of insulin secretion from rat islets of Langerhans. Biochem. J. 1973, 136, 343–349. [Google Scholar] [CrossRef]
- Kapito, K.; Hedeskov, C.J. The effect of starvation on phosphodiesterase activity and the content of adenosine 3′:5′-cycic monophosphate in Isolated mouse ancreatic islets. Biochem. J. 1974, 142, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Wolters, G.H.J.; Konijnendijk, W.; Bouman, P.R. Effects of fasting on insulin secretion, islet glucose metabolism, and the cyclic adenosine 3′,5′-monophosphate content of rat pancreatic islets in vitro. Diabetes 1977, 26, 530–537. [Google Scholar] [CrossRef]
- Bouman, P.R.; Wolters, G.H.J.; Konijnendijk, W. Insulin secretion and cyclic adenosine 3,5′-monophosphate levels in pancreatic islets of fed and fasted rats. Time course and dose kinetics during glucose stimulation. Diabetes 1979, 28, 132–140. [Google Scholar] [CrossRef]
- McGarry, D.J. Banting Lecture 2001, Dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes 2002, 51, 7–18. [Google Scholar] [CrossRef]
- Quesada, I.; Tudurí, E.; Ripoll, C.; Nadal, A. Physiology of the pancreatic α-cell and glucagon secretion: Role in glucose homeostasis and diabetes. J. Endocrinol 2008, 199, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Briant, L.; Salehi, A.; Vergari, E.; Zhang, Q.; Rorsman, P. Glucagon secretion from pancreatic a-cells. Upsala J. Med. Sci. 2016, 121, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Armour, S.L.; Stanley, J.E.; Cantley, J.; Dean, E.D.; Knudsen, J.G. Metabolic regulation of glucagon secretion. J. Endocrinol. 2023, 259, e230081. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.J. Glucagon 100 years. Important, but still enigmatic. Peptides 2023, 161, 170942. [Google Scholar] [CrossRef] [PubMed]
- Wendt, A.; Eliasson, L. Pancreatic α-cells—The unsung heroes in islet function. Semin. Cell Dev. Biol. 2020, 103, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Dou, H.; Rorsman, P. ‘Resistance is futile?’—Paradoxical inhibitory effects of KATP channel closure in glucagon-secreting α-cells. J. Phisiol. 2020, 598, 4765–4780. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H.; Cherrington, A.D. Glucagonocentric restructuring of diabetes: A pathophysiologic and therapeutic makeover. J. Clin. Investig. 2012, 122, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Detimary, P.; Dejonghe, S.; Ling, Z.; Pipeleers, D.; Schuit, F.; Henquin, J.C. The changes in adenine nucleotides measured in glucose-stimulated rodent islets occur in βcells but not in α cells and are also observed in human islets. J. Biol. Chem. 1998, 273, 33905–33908. [Google Scholar] [CrossRef]
- Olsen, H.L.; Theander, S.; Bokvist, K.; Buschard, K.; Wollheim, C.-B.; Gromada, J. Glucose stimulates glucagon release in single rat α-cells by mechanisms that mirror the stimulus-secretion coupling in β-ells. Endocrinology 2005, 146, 4861–4870. [Google Scholar] [CrossRef]
- Vieira, E.; Salehi, A.; Gylfe, E. Glucose inhibits glucagon secretion by a direct effect on mouse pancreatic alpha cells. Diabetologia 2007, 50, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yu, Q.; Ahooghalandari, P.; Gribble, F.M.; Reimann, F.; Tengholm, A.; Gylfe, E. Submembrane ATP and Ca2+ kinetics in α-cells: Unexpected signaling for glucagon secretion. FASEB J. 2015, 29, 3379–3388. [Google Scholar] [CrossRef] [PubMed]
- Brüning, D.; Morsi, M.; Früh, E.; Schemeck, S.; Rustenbeck, I. Metabolic regulation of hormone secretion in beta-cells and alpha cells of female mice. Fundamental differences. Endocrinology 2022, 163, bqac125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ramracheya, R.; Lahmann, C.; Tarasov, A.; Bengtsson, M.; Braha, O.; Braun, M.; Brereton, M.; Collins, S.; Galvanovskis, J.; et al. Role of KATP channels in glucose-regulated glucagon secretion and impaired counter regulation in type 2 diabetes. Cell Metab. 2013, 18, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Quesada, I.; Todorova, M.G.; Soria, B. Different Metabolic Responses in α-, β-, and δ-cells of the islet of Langerhans monitored by redox confocal microscopy. Biophys. J. 2006, 90, 2641–2650. [Google Scholar] [CrossRef] [PubMed]
- Früh, E.; Elgert, C.; Eggert, F.; Schermeck, S.; Rustenbeck, I. Glucagonotropic and glucagonostatic effecta of KATP channel closure and potassium. Endocrinolgy 2021, 162, bqaa136. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Shinozaki, Y.; Ohta, T. Sodium–glucose cotransporters: Functional properties and pharmaceutical potential. J. Diabetes Investig. 2020, 11, 770–782. [Google Scholar] [CrossRef]
- Berger, C.; Zdzieblo, D. Glucose transporters in pancreatic islets. Pflugers Arch.-Eur. J. Physiol. 2020, 472, 1249–1272. [Google Scholar] [CrossRef]
- Heimberg, H.; De Vos, A.; Pipeleers, D.; Thorens, B.; Schuit, F. Differences in glucose transporter gene expression between rat pancreatic α- and β-cells are correlated to differences in glucose transport but not in glucose utilization. J. Biol. Chem. 1995, 15, 8971–8975. [Google Scholar] [CrossRef]
- Tamarit-Rodriguez, J.; Vara, E.; Tamarit, J. Starvation-induced changes of insulin, somatostatin, and glucagon secretion and their modification by 2-bromostearate. Horm. Metab. Res. 1984, 16, 115–119. [Google Scholar] [CrossRef]
- Kristinsson, H.; Sargsyan, E.; Manell, H.; Smith, D.M.; Göpel, S.O.; Bergsten, P. Basal hypersecretion of glucagon and insulin from palmitate-exposed human islets depends on FFAR1 but not decreased somatostatin secretion. Sci. Rep. 2017, 7, 4657. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, C.S.; Salehi, A.; Göpel, S.O.; Holm, C.; Rorsman, P. Palmitate stimulation of glucagon secretion in mouse pancreatic α-cells results from activation of L-type calcium channels and elevation of cytoplasmic calcium. Diabetes 2004, 53, 2836–2843. [Google Scholar] [CrossRef] [PubMed]
- Veprik, A.; Denwood, G.; Liu, D.; Bakar, R.B.; Morfin, V.; McHugh, K.; Tebeka, N.N.; Vetterli, L.; Yonova-Doing, K.; Gribble, F.; et al. Acetyl-CoA-carboxylase 1 (ACC1) plays a critical role in glucagon secretion. Commun. Biol. 2022, 5, 238. [Google Scholar] [CrossRef] [PubMed]
- Armour, S.L.; Frueh, A.; Chibalina, M.V.; Dou, H.; Argemi-Muntadas, L.; Hamilton, A.; Katzilieris-Petras, G.; Carmeliet, P.; Davies, B.; Moritz, T.; et al. Glucose Controls Glucagon Secretion by Regulating Fatty Acid Oxidation in Pancreatic a-Cells. Diabetes 2023, 72, 1446–1459. [Google Scholar] [CrossRef]
- Briant, L.J.; Dodd, M.S.; Chibalina, M.V.; Rorsman, N.J.; Johnson, P.R.; Carmeliet, P.; Rorsman, P.; Knudsen, J.G. CPT1a-Dependent Long-Chain Fatty Acid Oxidation Contributes to Maintaining Glucagon Secretion from Pancreatic Islets. Cell Rep. 2018, 23, 3300–3311. [Google Scholar] [CrossRef] [PubMed]
- Weir, G.C.; Bonner-Weir, S. Conflicting views about interactions between pancreatic α-cells and β-cells. Diabetes 2023, 72, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Caicedo, A.; Huising, M.O.; Wess, J. An intraislet paracrine signaling pathway that enables glucagon to stimulate pancreatic β-cells. Diabetes 2023, 72, 1748–1750. [Google Scholar] [CrossRef] [PubMed]
- Kisanuki, K.; Kishikawa, H.; Araki, E.; Shirotani, T.; Uehara, M.; Isami, S.; Ura, S.; Jinnouchi, H.; Miyamura, N.; Shichiri, M. Expression of insulin receptor on clonal pancreatic alpha cells and its possible role for insulin-stimulated negative regulation of glucagon secretion. Diabetologia 1995, 38, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Kawamori, D.; Kurpad, A.J.; Hu, J.; Liew, C.W.; Shih, J.L.; Ford, E.L.; Herrera, P.L.; Polonsky, K.S.; McGuinness, O.P.; Kulkarni, R.N. Insulin Signaling in a Cells Modulates Glucagon Secretion In Vivo. Cell Metab. 2009, 9, 350–361. [Google Scholar] [CrossRef]
- Diao, J.; Asghar, Z.; Chan, C.B.; Wheeler, M.B. Glucose-regulated Glucagon Secretion Requires Insulin Receptor Expression in Pancreatic a-Cells. J. Biol. Chem. 2005, 280, 33487–33496. [Google Scholar] [CrossRef]
- Gu, W.; Bundgaard-Anker, C.C.; Bodelund-Christiansen, C.; Moede, T.; Berggren, P.-O.; Hermansen, K.; Gregersen, S.; Bendix-Jeppesen, P. Pancreatic β Cells Inhibit Glucagon Secretion from α Cells: An In Vitro Demonstration of α–β Cell Interaction. Nutrients 2021, 13, 2281. [Google Scholar] [CrossRef]
- Liu, W.; Kin, T.; Ho, S.; Dorrell, C.; Campbell, S.R.; Luo, P.; Chen, X. Abnormal regulation of glucagon secretion by human islet alpha cells in the absence of beta cells. EBioMedicine 2019, 50, 30631. [Google Scholar] [CrossRef]
- Salehi, A.; Qader, S.S.; Grapengiesser, E.; Hellman, B. Pulses of somatostatin release are slightly delayed compared with insulin and antisynchronous to glucagon. Regul. Pept. 2007, 144, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Vergari, E.; Knudsen, J.G.; Ramracheya, R.; Salehi, A.; Zhang, Q.; Adam, J.; Wernstedt -Asterholm, I.; Benrick, A.; Briant, L.J.B.; Chibalina, M.V.; et al. Insulin inhibits glucagon release by SGLT2-induced stimulation of somatostatin secretion. Nat. Commun. 2019, 10, 139. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, C.; Wright, E.M. Regulation of the human Na+-dependent glucose cotransporter hSGLT2. Am. J. Physiol.-Cell Physiol. 2012, 303, C348–C354. [Google Scholar] [CrossRef] [PubMed]
- El, K.; Gray, S.M.; Capozzi, M.E.; Knuth, E.R.; Jin, E.; Svendsen, B.; Clifford, A.; Brown, J.L.; Encisco, S.E.; Chazotte, B.M.; et al. GIP mediates the incretin effect and glucose tolerance by dual actions on α cells and β cells. Sci. Adv. 2021, 7, eabf1948. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.P.; Sorrell, J.E.; Haller AKaren, A.; Roelofs, K.; Hutch, C.R.; Kim, K.-S.; Gutierrez-Aguilar, R.; Li, B.; Drucker, D.J.; AD’Alessio, D.A.; et al. The Role of Pancreatic Preproglucagon in Glucose Homeostasis in Mice. Cell Metab. 2017, 25, 927–934. [Google Scholar] [CrossRef]
- Svendsen, B.; Larsen, O.; Nordskov Gabe, M.B.; Christiansen, C.B.; Rosenkilde, M.M.; Drucker, D.J.; Holst, J.J. Insulin Secretion Depends on Intra-islet Glucagon Signaling. Cell Rep. 2018, 25, 1127–1134. [Google Scholar] [CrossRef]
- Capozzi, M.E.; Wait, J.B.; Koech, J.; Gordon, A.N.; Coch, R.W.; Svendsen, B.; Finan, B.; D’Alessio, D.A.; Campbell, J.E. Glucagon lowers glycemia when β cells are active. JCI Insight 2019, 4, e129954. [Google Scholar] [CrossRef]
- Zhu, L.; Dattaroy, D.; Pham, J.; Wang, L.; Barella, L.F.; Cui, Y.; Wilkins, K.J.; Roth, B.L.; Hochgeschwender, U.; Matschinsky, F.M.; et al. Intraislet glucagon signaling is critical for maintaining glucose homeostasis. JCI Insight 2019, 4, e127994. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamarit-Rodriguez, J. Regulatory Role of Fatty Acid Metabolism on Glucose-Induced Changes in Insulin and Glucagon Secretion by Pancreatic Islet Cells. Int. J. Mol. Sci. 2024, 25, 6052. https://doi.org/10.3390/ijms25116052
Tamarit-Rodriguez J. Regulatory Role of Fatty Acid Metabolism on Glucose-Induced Changes in Insulin and Glucagon Secretion by Pancreatic Islet Cells. International Journal of Molecular Sciences. 2024; 25(11):6052. https://doi.org/10.3390/ijms25116052
Chicago/Turabian StyleTamarit-Rodriguez, Jorge. 2024. "Regulatory Role of Fatty Acid Metabolism on Glucose-Induced Changes in Insulin and Glucagon Secretion by Pancreatic Islet Cells" International Journal of Molecular Sciences 25, no. 11: 6052. https://doi.org/10.3390/ijms25116052
APA StyleTamarit-Rodriguez, J. (2024). Regulatory Role of Fatty Acid Metabolism on Glucose-Induced Changes in Insulin and Glucagon Secretion by Pancreatic Islet Cells. International Journal of Molecular Sciences, 25(11), 6052. https://doi.org/10.3390/ijms25116052