

Comparison of RNA-Sequencing Methods for Degraded RNA



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Comparison between Standard, SMART-Seq, xGen, and RamDA-Seq for Expression Analysis
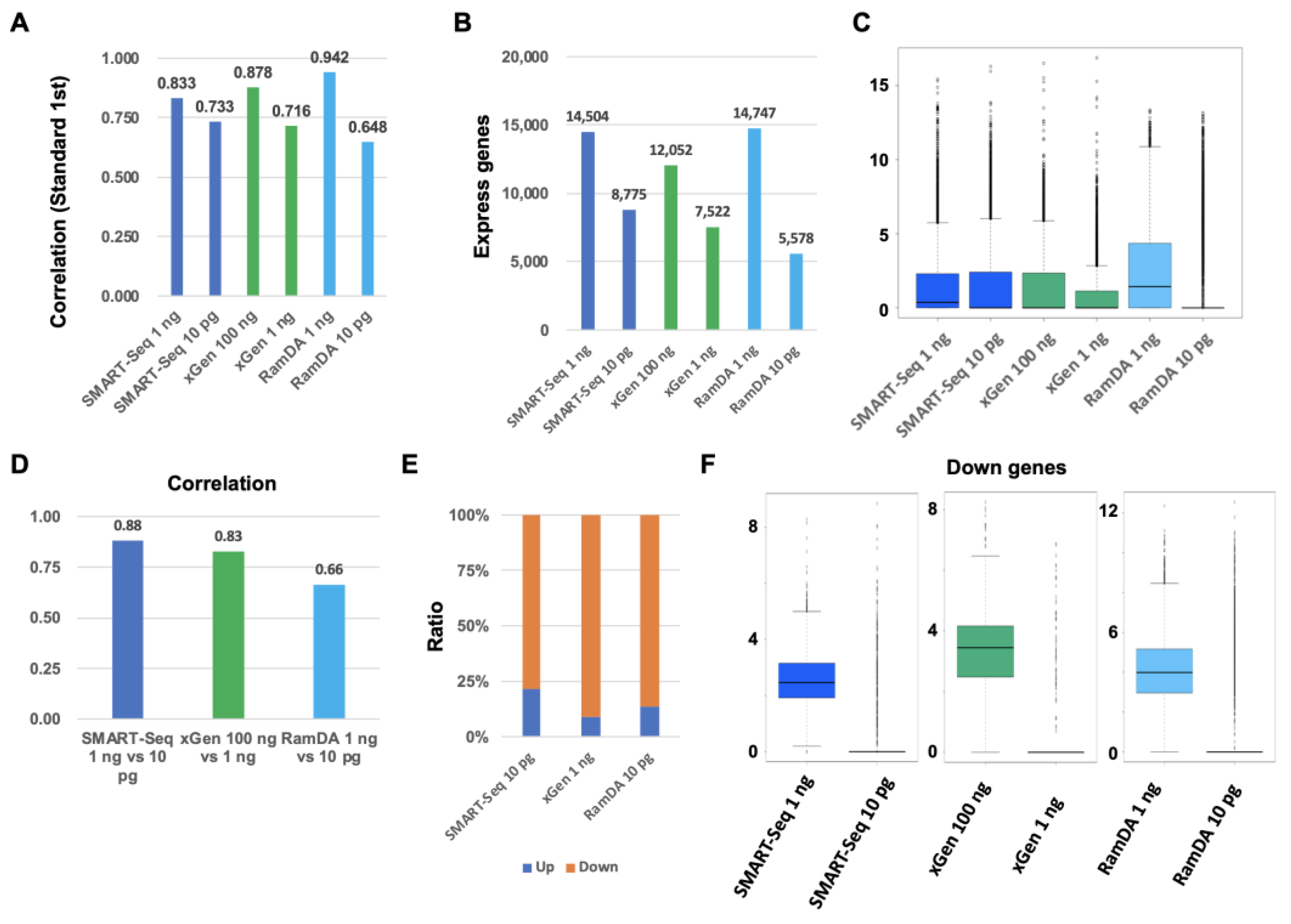
2.2. Comparison between Standard, SMART-Seq, xGen, and RamDA-Seq for Low-Input RNA
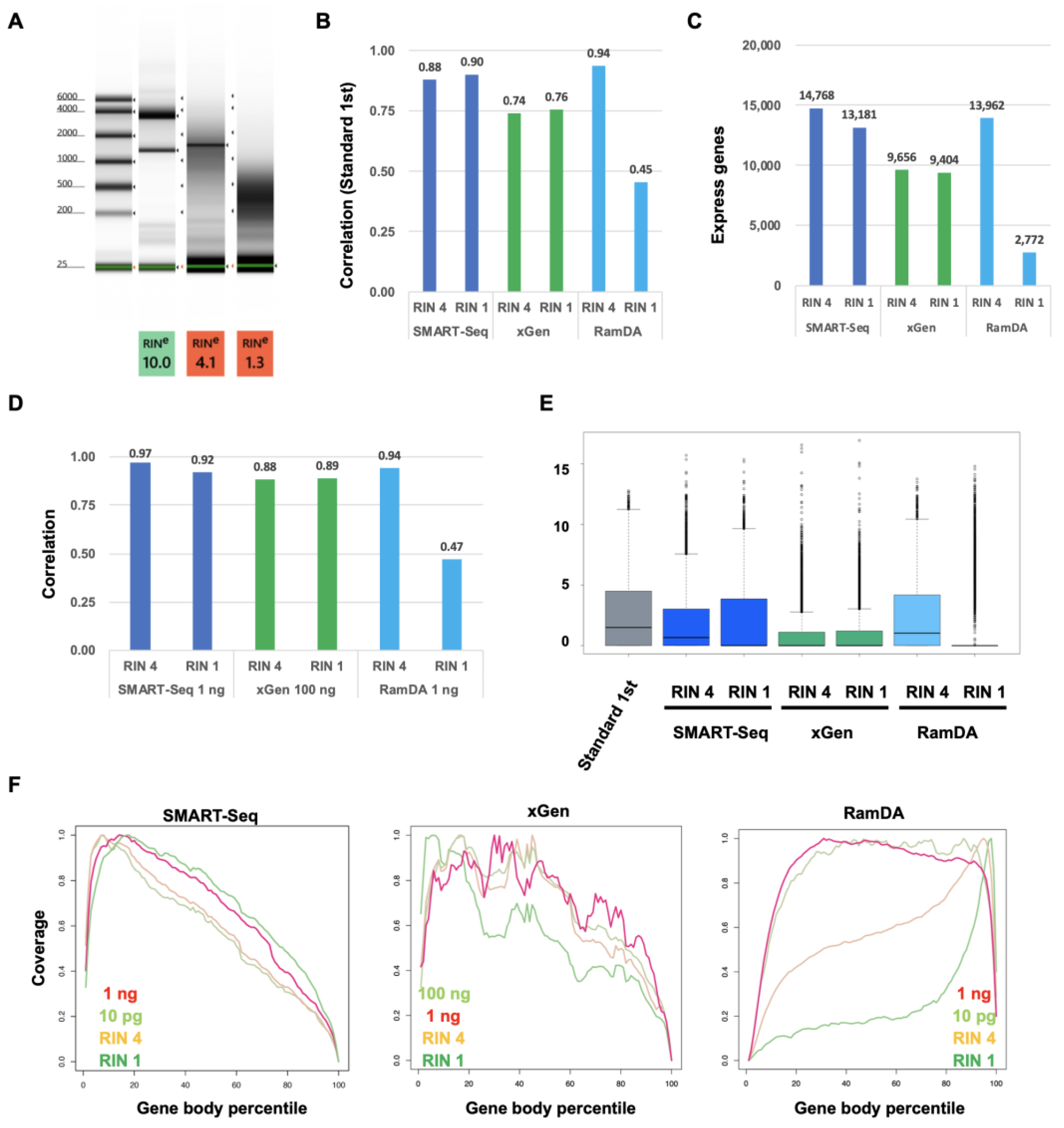
2.3. Comparison between Standard, SMART-Seq, xGen, and RamDA-Seq for Degraded RNA
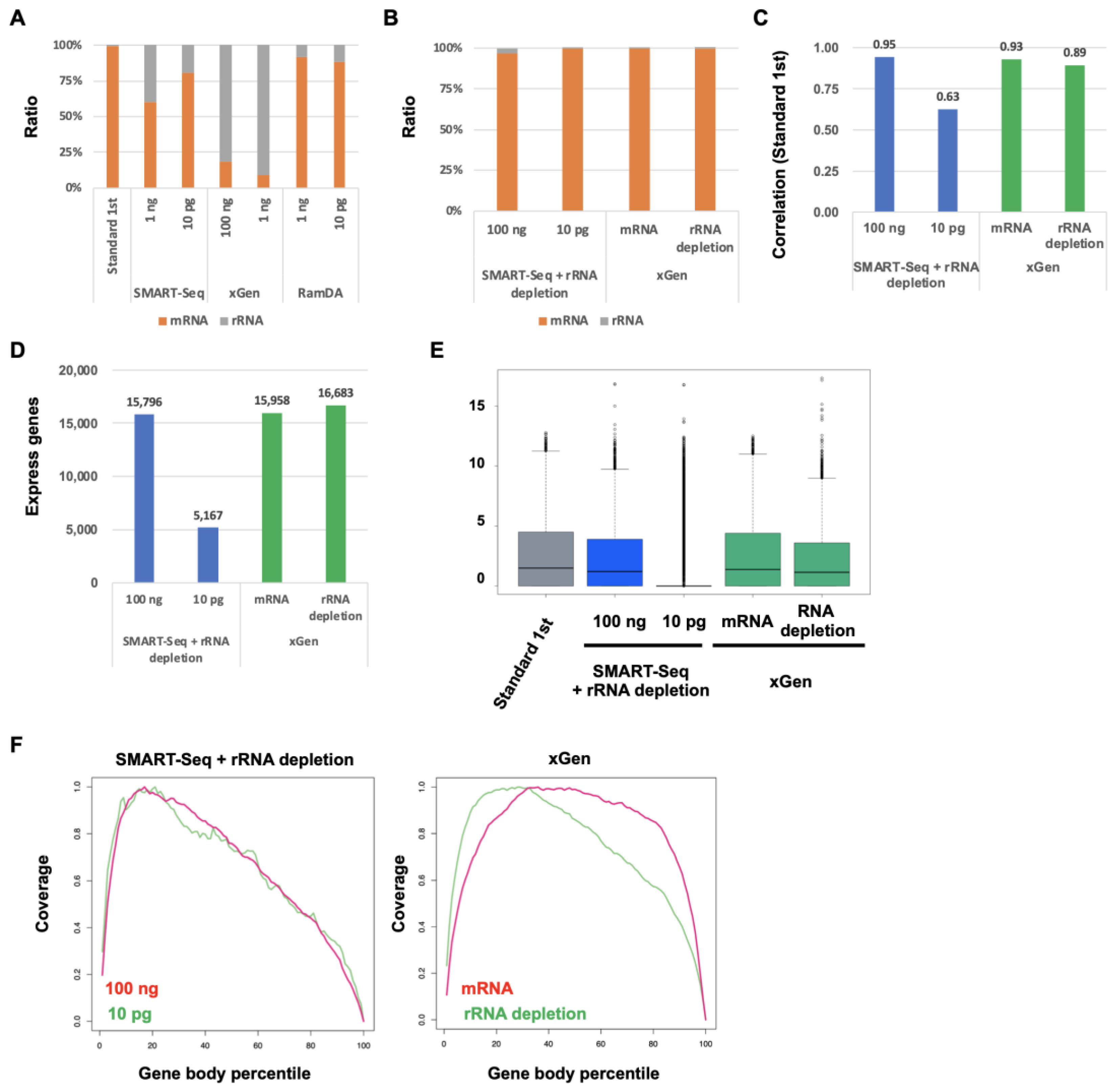
2.4. Improvement of SMART-Seq and xGen by Ribosomal RNA Depletion
2.5. Improvement of SMART-Seq and xGen for Degraded RNA by rRNA Depletion
3. Discussion
4. Materials and Methods
4.1. Total RNA Extraction and RNA Degradation by Heat-Treatment
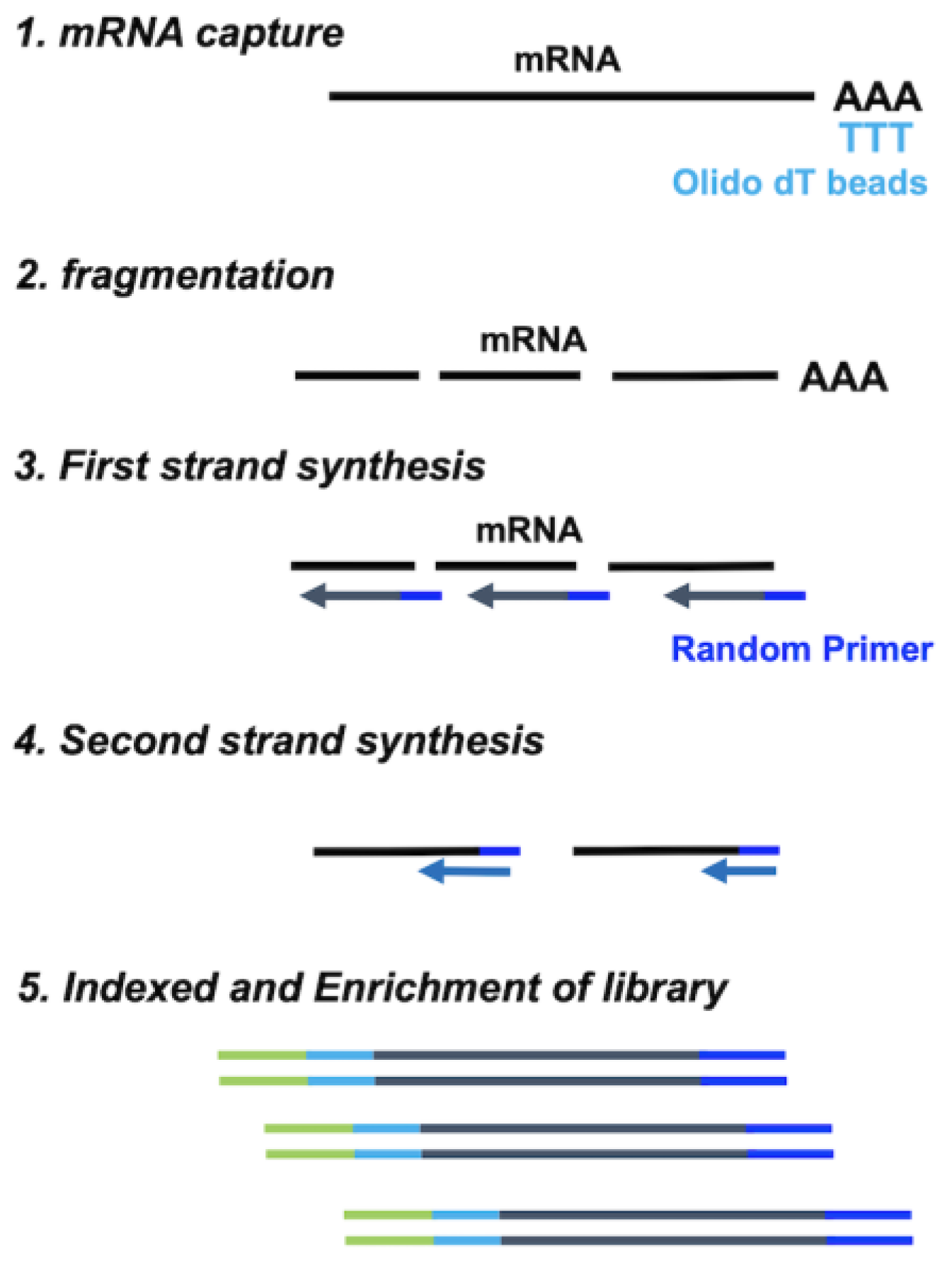
4.2. Standard RNA-Seq Library Preparation
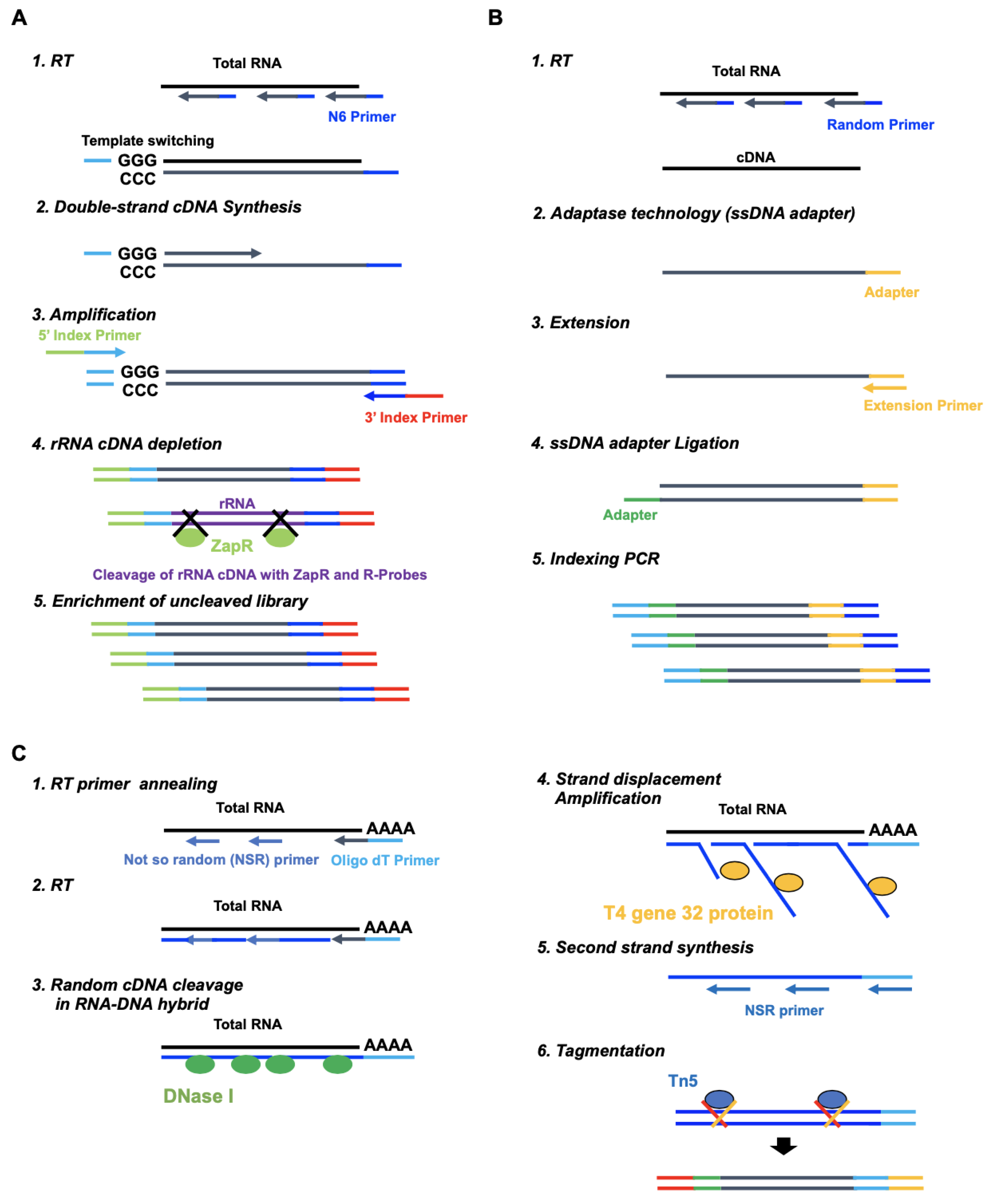
4.3. SMART-Seq Library Preparation
4.4. xGen Library Preparation
4.5. RamDA-Seq Library Preparation
4.6. rRNA Depletion
4.7. Sequencing and Generation of FASTQ Files
4.8. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Sultan, M.; Schulz, M.H.; Richard, H.; Magen, A.; Klingenhoff, A.; Scherf, M.; Seifert, M.; Borodina, T.; Soldatov, A.; Parkhomchuk, D.; et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 2008, 321, 956–960. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Melé, M.; Ferreira, P.G.; Reverter, F.; DeLuca, D.S.; Monlong, J.; Sammeth, M.; Young, T.R.; Goldmann, J.M.; Pervouchine, D.D.; Sullivan, T.J.; et al. Human genomics. The human transcriptome across tissues and individuals. Science 2015, 348, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Byron, S.A.; Van Keuren-Jensen, K.R.; Engelthaler, D.M.; Carpten, J.D.; Craig, D.W. Translating RNA sequencing into clinical diagnostics: Opportunities and challenges. Nat. Rev. Genet. 2016, 17, 257–271. [Google Scholar] [CrossRef]
- Hong, M.; Tao, S.; Zhang, L.; Diao, L.T.; Huang, X.; Huang, S.; Xie, S.J.; Xiao, Z.D.; Zhang, H. RNA sequencing: New technologies and applications in cancer research. J. Hematol. Oncol. 2020, 13, 166. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Ning, B.; Fang, H.; Hong, H.; Perkins, R.; Tong, W.; Shi, L. Next-generation sequencing and its applications in molecular diagnostics. Expert Rev. Mol. Diagn. 2011, 11, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Gallego Romero, I.; Pai, A.A.; Tung, J.; Gilad, Y. RNA-seq: Impact of RNA degradation on transcript quantification. BMC Biol. 2014, 12, 42. [Google Scholar] [CrossRef]
- Cieslik, M.; Chugh, R.; Wu, Y.M.; Wu, M.; Brennan, C.; Lonigro, R.; Su, F.; Wang, R.; Siddiqui, J.; Mehra, R.; et al. The use of exome capture RNA-seq for highly degraded RNA with application to clinical cancer sequencing. Genome Res. 2015, 25, 1372–1381. [Google Scholar] [CrossRef]
- Schuierer, S.; Carbone, W.; Knehr, J.; Petitjean, V.; Fernandez, A.; Sultan, M.; Roma, G. A comprehensive assessment of RNA-seq protocols for degraded and low-quantity samples. BMC Genom. 2017, 18, 442. [Google Scholar] [CrossRef] [PubMed]
- Jovanović, B.; Sheng, Q.; Seitz, R.S.; Lawrence, K.D.; Morris, S.W.; Thomas, L.R.; Hout, D.R.; Schweitzer, B.L.; Guo, Y.; Pietenpol, J.A.; et al. Comparison of triple-negative breast cancer molecular subtyping using RNA from matched fresh-frozen versus formalin-fixed paraffin-embedded tissue. BMC Cancer 2017, 17, 241. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Mehta, M.; Walton, A.; Talsania, K.; Levin, Y.; Shetty, J.; Gillanders, E.M.; Tran, B.; Carrick, D.M. Robustness of RNA sequencing on older formalin-fixed paraffin-embedded tissue from high-grade ovarian serous adenocarcinomas. PLoS ONE 2019, 14, e0216050. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.F.; Zumbo, P.; Fostel, J.; Gandara, J.; Hester, S.D.; Recio, L.; Williams, A.; Wood, C.E.; Yauk, C.L.; Mason, C.E. Mining the Archives: A Cross-Platform Analysis of Gene Expression Profiles in Archival Formalin-Fixed Paraffin-Embedded Tissues. Toxicol. Sci. 2015, 148, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Hedegaard, J.; Thorsen, K.; Lund, M.K.; Hein, A.M.; Hamilton-Dutoit, S.J.; Vang, S.; Nordentoft, I.; Birkenkamp-Demtröder, K.; Kruhøffer, M.; Hager, H.; et al. Next-generation sequencing of RNA and DNA isolated from paired fresh-frozen and formalin-fixed paraffin-embedded samples of human cancer and normal tissue. PLoS ONE 2014, 9, e98187. [Google Scholar] [CrossRef] [PubMed]
- Kresse, S.H.; Namløs, H.M.; Lorenz, S.; Berner, J.M.; Myklebost, O.; Bjerkehagen, B.; Meza-Zepeda, L.A. Evaluation of commercial DNA and RNA extraction methods for high-throughput sequencing of FFPE samples. PLoS ONE 2018, 13, e0197456. [Google Scholar] [CrossRef]
- Masuda, N.; Ohnishi, T.; Kawamoto, S.; Monden, M.; Okubo, K. Analysis of chemical modification of RNA from formalin-fixed samples and optimization of molecular biology applications for such samples. Nucleic Acids Res. 1999, 27, 4436–4443. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, Y.; Gamini, R.; Zhang, B.; von Schack, D. Evaluation of two main RNA-seq approaches for gene quantification in clinical RNA sequencing: PolyA+ selection versus rRNA depletion. Sci. Rep. 2018, 8, 4781. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Kankainen, M.; Parsons, A.; Kallioniemi, O.; Mattila, P.; Heckman, C.A. The impact of RNA sequence library construction protocols on transcriptomic profiling of leukemia. BMC Genom. 2017, 18, 629. [Google Scholar] [CrossRef]
- Guo, Y.; Zhao, S.; Sheng, Q.; Guo, M.; Lehmann, B.; Pietenpol, J.; Samuels, D.C.; Shyr, Y. RNAseq by Total RNA Library Identifies Additional RNAs Compared to Poly(A) RNA Library. BioMed Res. Int. 2015, 2015, 862130. [Google Scholar] [CrossRef]
- Morlan, J.D.; Qu, K.; Sinicropi, D.V. Selective depletion of rRNA enables whole transcriptome profiling of archival fixed tissue. PLoS ONE 2012, 7, e42882. [Google Scholar] [CrossRef]
- Adiconis, X.; Borges-Rivera, D.; Satija, R.; DeLuca, D.S.; Busby, M.A.; Berlin, A.M.; Sivachenko, A.; Thompson, D.A.; Wysoker, A.; Fennell, T.; et al. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat. Methods 2013, 10, 623–629. [Google Scholar] [CrossRef]
- Picelli, S.; Björklund, Å.K.; Faridani, O.R.; Sagasser, S.; Winberg, G.; Sandberg, R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 2013, 10, 1096–1098. [Google Scholar] [CrossRef]
- Hayashi, T.; Ozaki, H.; Sasagawa, Y.; Umeda, M.; Danno, H.; Nikaido, I. Single-cell full-length total RNA sequencing uncovers dynamics of recursive splicing and enhancer RNAs. Nat. Commun. 2018, 9, 619. [Google Scholar] [CrossRef]
- Song, Y.; Milon, B.; Ott, S.; Zhao, X.; Sadzewicz, L.; Shetty, A.; Boger, E.T.; Tallon, L.J.; Morell, R.J.; Mahurkar, A.; et al. A comparative analysis of library prep approaches for sequencing low input translatome samples. BMC Genom. 2018, 19, 696. [Google Scholar] [CrossRef]
- Cannizzo, M.D.; Wood, C.E.; Hester, S.D.; Wehmas, L.C. Case study: Targeted RNA-sequencing of aged formalin-fixed paraffin-embedded samples for understanding chemical mode of action. Toxicol. Rep. 2022, 9, 883–894. [Google Scholar] [CrossRef]
- Liu, Y.; Bhagwate, A.; Winham, S.J.; Stephens, M.T.; Harker, B.W.; McDonough, S.J.; Stallings-Mann, M.L.; Heinzen, E.P.; Vierkant, R.A.; Hoskin, T.L.; et al. Quality control recommendations for RNASeq using FFPE samples based on pre-sequencing lab metrics and post-sequencing bioinformatics metrics. BMC Med. Genom. 2022, 15, 195. [Google Scholar] [CrossRef]
- Yeo, J.C.; Ng, H.H. Transcriptomic analysis of pluripotent stem cells: Insights into health and disease. Genome Med. 2011, 3, 68. [Google Scholar] [CrossRef]
- Sorokin, M.; Gorelyshev, A.; Efimov, V.; Zotova, E.; Zolotovskaia, M.; Rabushko, E.; Kuzmin, D.; Seryakov, A.; Kamashev, D.; Li, X.; et al. RNA Sequencing Data for FFPE Tumor Blocks Can Be Used for Robust Estimation of Tumor Mutation Burden in Individual Biosamples. Front. Oncol. 2021, 11, 732644. [Google Scholar] [CrossRef]
- Ura, H.; Togi, S.; Iwata, Y.; Ozaki, M.; Niida, Y. Establishment of a human induced pluripotent stem cell line, KMUGMCi001-A, from a patient bearing a heterozygous c.772 + 3_772 + 4dup mutation in the ACVRL1 gene leading Telangiectasia, hereditary hemorrhagic, type 2 (HHT2). Stem Cell Res. 2022, 61, 102743. [Google Scholar] [CrossRef]
- Ura, H.; Togi, S.; Niida, Y. Target-capture full-length double-strand cDNA sequencing for alternative splicing analysis. RNA RNA Biol. 2021, 18, 1600–1607. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Sun, J.; Nishiyama, T.; Shimizu, K.; Kadota, K. TCC: An R package for comparing tag count data with robust normalization strategies. BMC Bioinform. 2013, 14, 219. [Google Scholar] [CrossRef]
- Ura, H.; Togi, S.; Niida, Y. Target-capture full-length double-stranded cDNA long-read sequencing through Nanopore revealed novel intron retention in patient with tuberous sclerosis complex. Front. Genet. 2023, 14, 1256064. [Google Scholar] [CrossRef]
- Ura, H.; Togi, S.; Niida, Y. A comparison of mRNA sequencing (RNA-Seq) library preparation methods for transcriptome analysis. BMC Genom. 2022, 23, 303. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ura, H.; Niida, Y. Comparison of RNA-Sequencing Methods for Degraded RNA. Int. J. Mol. Sci. 2024, 25, 6143. https://doi.org/10.3390/ijms25116143
Ura H, Niida Y. Comparison of RNA-Sequencing Methods for Degraded RNA. International Journal of Molecular Sciences. 2024; 25(11):6143. https://doi.org/10.3390/ijms25116143
Chicago/Turabian StyleUra, Hiroki, and Yo Niida. 2024. "Comparison of RNA-Sequencing Methods for Degraded RNA" International Journal of Molecular Sciences 25, no. 11: 6143. https://doi.org/10.3390/ijms25116143
APA StyleUra, H., & Niida, Y. (2024). Comparison of RNA-Sequencing Methods for Degraded RNA. International Journal of Molecular Sciences, 25(11), 6143. https://doi.org/10.3390/ijms25116143