
Exploring the Structure and Substance Metabolism of a Medicago sativa L. Stem Base


Abstract
1. Introduction
2. Results
2.1. Identification of Metabolites in Three Plant Tissues
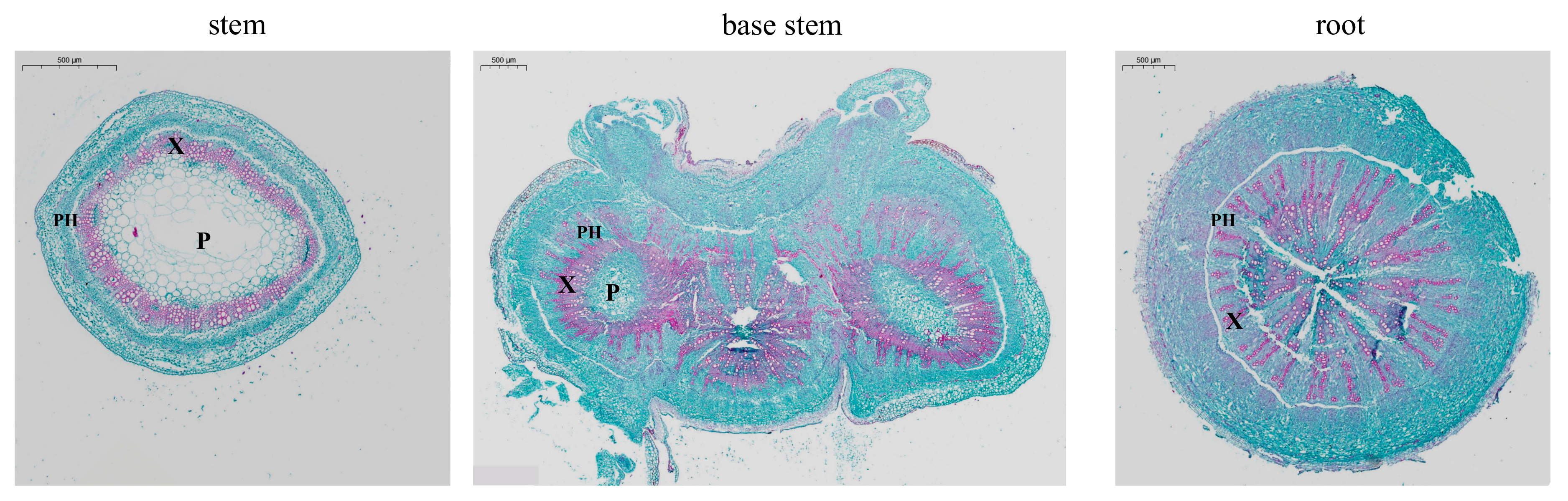
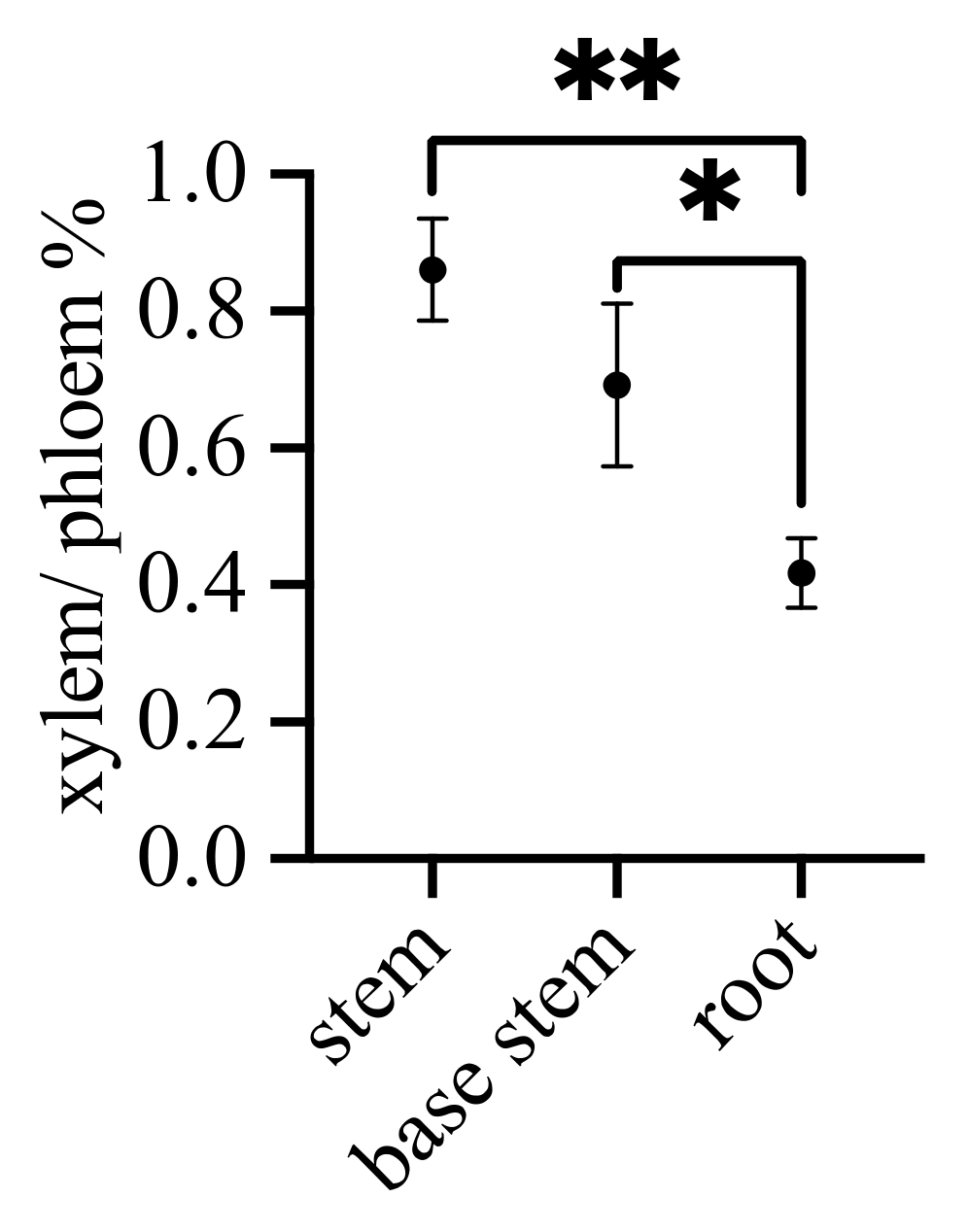
2.2. Structure Analysis
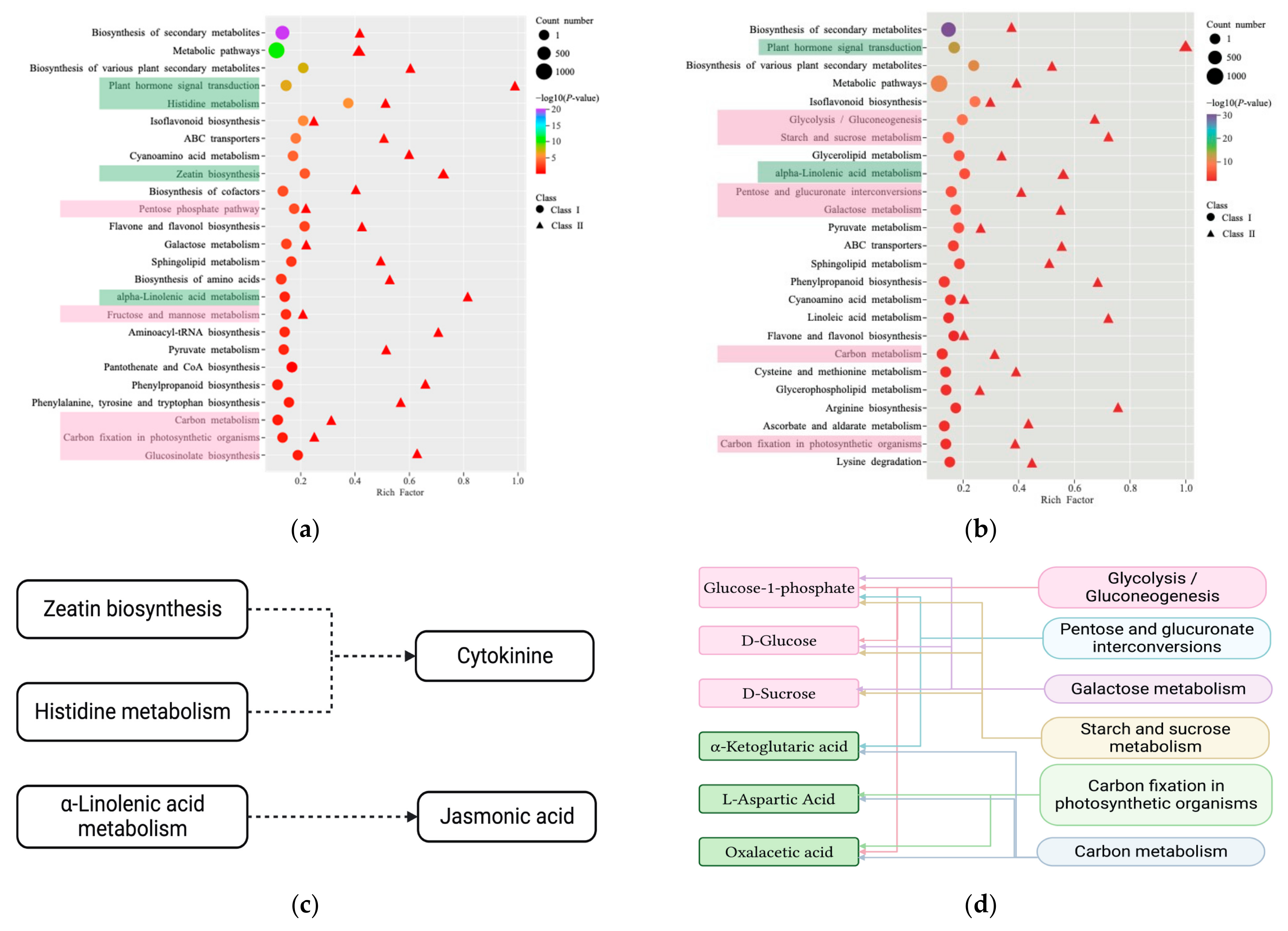
2.3. Differential Metabolites in the Stem Base
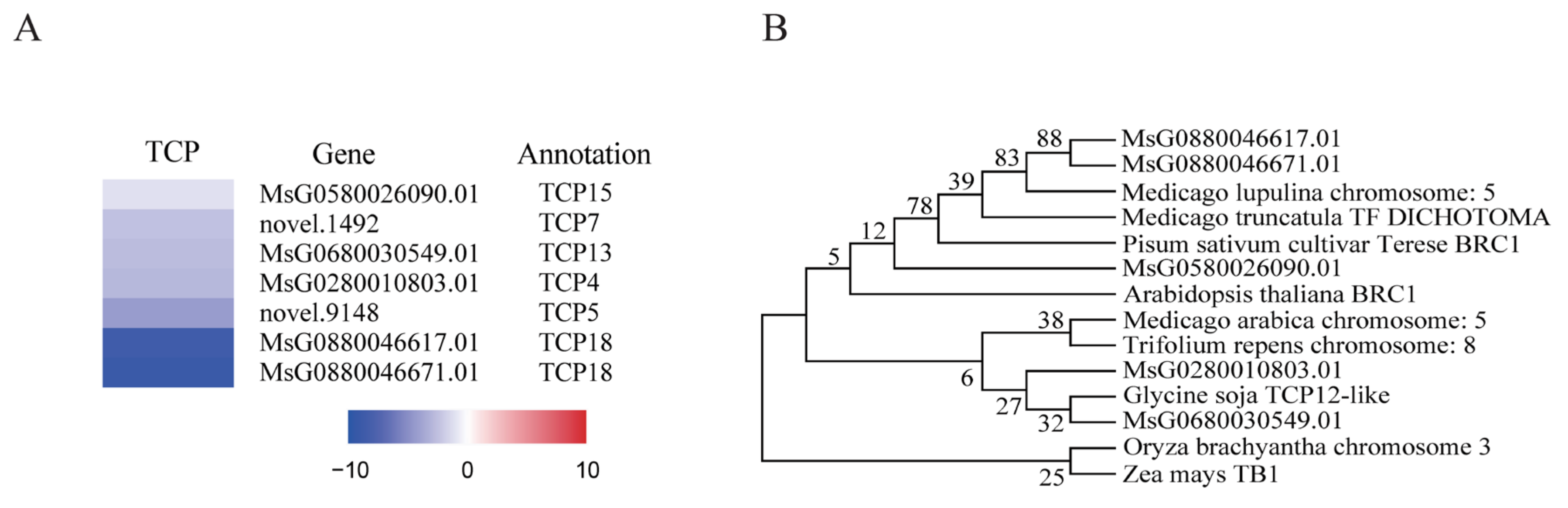
2.4. Identification of Differentially Expressed Genes
2.5. Association Analysis between DEGs and DACs
2.6. Validation of Differential Gene Expression
3. Discussion
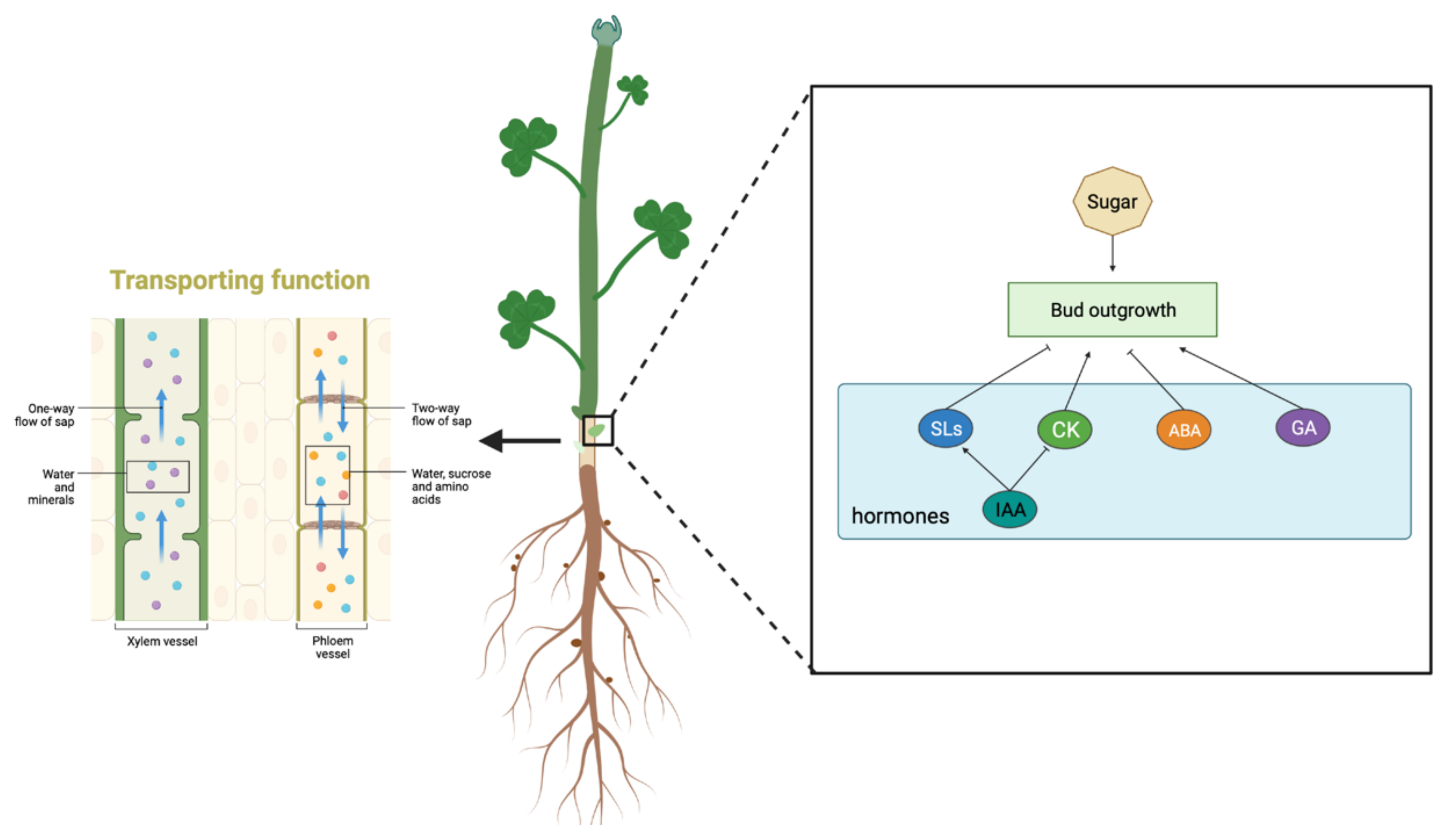
3.1. Transport Channels—Xylem and Phloem
3.2. The Regenerative Capacity of Alfalfa Stem Bases
4. Materials and Methods
4.1. Plant Materials
4.2. Histological Analysis
4.3. Extraction and Measurement of Metabolites
4.4. Transcriptome Sequencing
4.5. Real-Time RT-PCR
4.6. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, M.-X.; Zhao, L.-Y.; He, Y.-Y.; Hu, J.-P.; Hu, G.-W.; Zhu, Y.; Khan, A.; Xiong, Y.-C.; Zhang, J.-L. Potential Roles of Iron Nanomaterials in Enhancing Growth and Nitrogen Fixation and Modulating Rhizomicrobiome in Alfalfa (Medicago Sativa L.). Bioresour. Technol. 2024, 391, 129987. [Google Scholar] [CrossRef] [PubMed]
- Wolabu, T.W.; Mahmood, K.; Chen, F.; Torres-Jerez, I.; Udvardi, M.; Tadege, M.; Cong, L.; Wang, Z.; Wen, J. Mutating Alfalfa COUMARATE 3-HYDROXYLASE Using Multiplex CRISPR/Cas9 Leads to Reduced Lignin Deposition and Improved Forage Quality. Front. Plant Sci. 2024, 15, 1363182. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Liu, T.; Li, M.; Dong, X.; Han, Y.; Xu, C.; Li, S.; Zhang, J.; He, X.; Zhou, Q.; et al. MODMS: A Multi-Omics Database for Facilitating Biological Studies on Alfalfa (Medicago Sativa L.). Hortic. Res. 2024, 11, uhad245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Fan, J.; Jia, M.; Shi, C. Impacts of Long-Term Abandonment of Alfalfa Plantations on Soil Physicochemical Properties and Plant Diversity in an Agricultural Pastoral Ecotone. Plant Soil 2023, 493, 519–534. [Google Scholar] [CrossRef]
- Guerchi, A.; Mnafgui, W.; Jabri, C.; Merghni, M.; Sifaoui, K.; Mahjoub, A.; Ludidi, N.; Badri, M. Improving Productivity and Soil Fertility in Medicago Sativa and Hordeum Marinum through Intercropping under Saline Conditions. BMC Plant Biol. 2024, 24, 158. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Fan, J.; Wang, L. The Functional Division of Arbuscular Mycorrhizal Fungi and Earthworm to Efficient Cooperation on Phytoremediation in Molybdenum (Mo) Contaminated Soils. Environ. Res. 2024, 247, 118270. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Jiang, L.; Chen, Y.; Shu, Y.; Bai, Y.; Guo, C. Deep-Sequencing Transcriptome Analysis of Field-Grown Medicago Sativa L. Crown Buds Acclimated to Freezing Stress. Funct. Integr. Genom. 2016, 16, 495–511. [Google Scholar] [CrossRef] [PubMed]
- Pembleton, K.G.; Volenec, J.J.; Rawnsley, R.P.; Donaghy, D.J. Partitioning of Taproot Constituents and Crown Bud Development Are Affected by Water Deficit in Regrowing Alfalfa (Medicago Sativa L.). Crop. Sci. 2010, 50, 989–999. [Google Scholar] [CrossRef]
- Kim, H.; Kim, K.; Lee, S.J. Hydraulic Strategy of Cactus Root–Stem Junction for Effective Water Transport. Front. Plant Sci. 2018, 9, 799. [Google Scholar] [CrossRef]
- Rasl, T.; Schalk, M.; Temsch, E.; Kodym, A. Direct Cryopreservation of Winter-Acclimated Buds of Dracocephalum Austriacum (Lamiaceae) from Field Material. Plant Cell Tiss. Organ Cult. 2020, 142, 167–176. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Q.; Pang, X.P.; Xu, H.P.; Wang, J.; Zhang, W.N.; Guo, Z.G. Effect of Partial Root-Zone Drying Irrigation (PRDI) on the Biomass, Water Productivity and Carbon, Nitrogen and Phosphorus Allocations in Different Organs of Alfalfa. Agric. Water Manag. 2021, 243, 106525. [Google Scholar] [CrossRef]
- Márquez-Ortiz, J.J.; Lamb, J.F.S.; Johnson, L.D.; Barnes, D.K.; Stucker, R.E. Heritability of Crown Traits in Alfalfa. Crop Sci. 1999, 39, 38–43. [Google Scholar] [CrossRef]
- Marquez-Ortiz, J.J.; Johnson, L.D.; Barnes, D.K.; Basigalup, D.H. Crown Morphology Relationships among Alfalfa Plant Introductions and Cultivars. Crop. Sci. 1996, 36, 766–770. [Google Scholar] [CrossRef]
- Yuan, N.; Sun, L.; Du, S.; Ge, G.; Wang, Z.; Li, Y.; Bao, J.; Zhao, M.; Si, Q.; Hao, J.; et al. Effects of Harvesting Period and Storage Duration on Volatile Organic Compounds and Nutritive Qualities of Alfalfa. Agriculture 2022, 12, 1115. [Google Scholar] [CrossRef]
- Lorenzo, C.D.; García-Gagliardi, P.; Antonietti, M.S.; Sánchez-Lamas, M.; Mancini, E.; Dezar, C.A.; Vazquez, M.; Watson, G.; Yanovsky, M.J.; Cerdán, P.D. Improvement of Alfalfa Forage Quality and Management through the Down-Regulation of MsFTa1. Plant Biotechnol. J. 2020, 18, 944–954. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Zhai, C.; Li, Y.; Li, Y.; Qu, H.; Shen, Y. Effect of Nitrogen Application and Cutting Frequency on the Yield and Forage Quality of Alfalfa in Seasonal Cultivation. Agriculture 2023, 13, 1063. [Google Scholar] [CrossRef]
- Li, Z.; He, F.; Tong, Z.; Li, X.; Yang, Q.; Hannaway, D.B. Metabolomic Changes in Crown of Alfalfa (Medicago Sativa L.) during de-Acclimation. Sci. Rep. 2022, 12, 14977. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, J.; Yu, L.; Xu, Z.; Samac, D.A. Overwintering and Yield Responses of Two Late-Summer Seeded Alfalfa Cultivars to Phosphate Supply. Agronomy 2022, 12, 327. [Google Scholar] [CrossRef]
- Ventroni, L.M.; Volenec, J.J.; Cangiano, C.A. Fall Dormancy and Cutting Frequency Impact on Alfalfa Yield and Yield Components. Field Crop. Res. 2010, 119, 252–259. [Google Scholar] [CrossRef]
- Zhou, J.; Wilson, G.W.T.; Cobb, A.B.; Yang, G.; Zhang, Y. Phosphorus and Mowing Improve Native Alfalfa Establishment, Facilitating Restoration of Grassland Productivity and Diversity. Land Degrad. Dev. 2019, 30, 647–657. [Google Scholar] [CrossRef]
- Tiwari, P.; Srivastava, D.; Chauhan, A.S.; Indoliya, Y.; Singh, P.K.; Tiwari, S.; Fatima, T.; Mishra, S.K.; Dwivedi, S.; Agarwal, L.; et al. Root System Architecture, Physiological Analysis and Dynamic Transcriptomics Unravel the Drought-Responsive Traits in Rice Genotypes. Ecotoxicol. Environ. Saf. 2021, 207, 111252. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, X.; Zhang, R.; Fu, Q.; Tang, F.; Shi, F.; Temuer, B.; Zhang, Z. Comparative Transcriptome and Anatomic Characteristics of Stems in Two Alfalfa Genotypes. Plants 2022, 11, 2601. [Google Scholar] [CrossRef] [PubMed]
- Clément, C.; Schneider, H.M.; Dresbøll, D.B.; Lynch, J.P.; Thorup-Kristensen, K. Root and Xylem Anatomy Varies with Root Length, Root Order, Soil Depth and Environment in Intermediate Wheatgrass (Kernza®) and Alfalfa. Ann. Bot. 2022, 130, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Jaeger, M.; Wang, M.; Li, B.; Zhang, B.G. Three-Dimensional Distribution of Vessels, Passage Cells and Lateral Roots along the Root Axis of Winter Wheat (Triticum Aestivum). Ann. Bot. 2011, 107, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Dória, L.C.; Meijs, C.; Podadera, D.S.; Del Arco, M.; Smets, E.; Delzon, S.; Lens, F. Embolism Resistance in Stems of Herbaceous Brassicaceae and Asteraceae Is Linked to Differences in Woodiness and Precipitation. Ann. Bot. 2019, 124, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nie, W.; Dong, Y.; Liu, Y.; Tan, C.; Wang, Y.; Yuan, Y.; Ma, J.; An, S.; Liu, J.; Xiao, W.; et al. Climatic Responses and Variability in Bark Anatomical Traits of 23 Picea Species. Front. Plant Sci. 2023, 14, 1201553. [Google Scholar] [CrossRef] [PubMed]
- Min, Z.; Zhao, X.; Li, R.; Yang, B.; Liu, M.; Fang, Y. Comparative Transcriptome Analysis Provides Insight into Differentially Expressed Genes Related to Bud Dormancy in Grapevine (Vitis Vinifera). Sci. Hortic. 2017, 225, 213–220. [Google Scholar] [CrossRef]
- Simó, C.; Ibáez, C.; Valdés, A.; Cifuentes, A.; García-Cañas, V. Metabolomics of Genetically Modified Crops. Int. J. Mol. Sci. 2014, 15, 18941–18966. [Google Scholar] [CrossRef]
- Duan, S.; Wu, Y.; Fu, R.; Wang, L.; Chen, Y.; Xu, W.; Zhang, C.; Ma, C.; Shi, J.; Wang, S. Comparative Metabolic Profiling of Grape Skin Tissue along Grapevine Berry Developmental Stages Reveals Systematic Influences of Root Restriction on Skin Metabolome. Int. J. Mol. Sci. 2019, 20, 534. [Google Scholar] [CrossRef]
- De Schepper, V.; De Swaef, T.; Bauweraerts, I.; Steppe, K. Phloem Transport: A Review of Mechanisms and Controls. J. Exp. Bot. 2013, 64, 4839–4850. [Google Scholar] [CrossRef]
- Mencuccini, M.; Hölttä, T.; Sevanto, S.; Nikinmaa, E. Concurrent Measurements of Change in the Bark and Xylem Diameters of Trees Reveal a Phloem-generated Turgor Signal. New Phytol. 2013, 198, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Helfter, C.; Shephard, J.D.; Martinez-Vilalta, J.; Mencuccini, M.; Hand, D.P. A Noninvasive Optical System for the Measurement of Xylem and Phloem Sap Flow in Woody Plants of Small Stem Size. Tree Physiol. 2007, 27, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, W.; Abou-Elwafa, S.F.; Shabala, S.; Xu, L. Understanding a Mechanistic Basis of ABA Involvement in Plant Adaptation to Soil Flooding: The Current Standing. Plants 2021, 10, 1982. [Google Scholar] [CrossRef] [PubMed]
- Jaillais, Y.; Chory, J. Unraveling the Paradoxes of Plant Hormone Signaling Integration. Nat. Struct. Mol. Biol. 2010, 17, 642–645. [Google Scholar] [CrossRef] [PubMed]
- Brian, P.W. Role of Gibberellin-Like Hormones in Regulation of Plant Growth and Flowering. Nature 1958, 181, 1122–1123. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.J.; Cheng, Z.J.; Sang, Y.L.; Zhang, M.M.; Rong, X.F.; Wang, Z.W.; Tang, Y.Y.; Zhang, X.S. Type-B ARABIDOPSIS RESPONSE REGULATORs Specify the Shoot Stem Cell Niche by Dual Regulation of WUSCHEL. Plant Cell 2017, 29, 1357–1372. [Google Scholar] [CrossRef] [PubMed]
- Cancé, C.; Martin-Arevalillo, R.; Boubekeur, K.; Dumas, R. Auxin Response Factors Are Keys to the Many Auxin Doors. New Phytol. 2022, 235, 402–419. [Google Scholar] [CrossRef] [PubMed]
- Camut, L.; Gallova, B.; Jilli, L.; Sirlin-Josserand, M.; Carrera, E.; Sakvarelidze-Achard, L.; Ruffel, S.; Krouk, G.; Thomas, S.G.; Hedden, P.; et al. Nitrate Signaling Promotes Plant Growth by Upregulating Gibberellin Biosynthesis and Destabilization of DELLA Proteins. Curr. Biol. 2021, 31, 4971–4982.e4. [Google Scholar] [CrossRef] [PubMed]
- van Es, S.W.; van der Auweraert, E.B.; Silveira, S.R.; Angenent, G.C.; van Dijk, A.D.J.; Immink, R.G.H. Comprehensive Phenotyping Reveals Interactions and Functions of Arabidopsis thaliana TCP Genes in Yield Determination. Plant J. 2019, 99, 316–328. [Google Scholar] [CrossRef]
- Braun, N.; De Saint Germain, A.; Pillot, J.-P.; Boutet-Mercey, S.; Dalmais, M.; Antoniadi, I.; Li, X.; Maia-Grondard, A.; Le Signor, C.; Bouteiller, N.; et al. The Pea TCP Transcription Factor PsBRC1 Acts Downstream of Strigolactones to Control Shoot Branching. Plant Physiol. 2012, 158, 225–238. [Google Scholar] [CrossRef]
- Nicolas, M.; Cubas, P. TCP Factors: New Kids on the Signaling Block. Curr. Opin. Plant Biol. 2016, 33, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Cubas, P.; Lauter, N.; Doebley, J.; Coen, E. The TCP Domain: A Motif Found in Proteins Regulating Plant Growth and Development. Plant J. 1999, 18, 215–222. [Google Scholar] [CrossRef]
- Yuan, Y.; Khourchi, S.; Li, S.; Du, Y.; Delaplace, P. Unlocking the Multifaceted Mechanisms of Bud Outgrowth: Advances in Understanding Shoot Branching. Plants 2023, 12, 3628. [Google Scholar] [CrossRef] [PubMed]
- Barbier, F.F.; Cao, D.; Fichtner, F.; Weiste, C.; Perez-Garcia, M.; Caradeuc, M.; Le Gourrierec, J.; Sakr, S.; Beveridge, C.A. HEXOKINASE1 Signalling Promotes Shoot Branching and Interacts with Cytokinin and Strigolactone Pathways. New Phytol. 2021, 231, 1088–1104. [Google Scholar] [CrossRef] [PubMed]
- Göbel, M.; Fichtner, F. Functions of Sucrose and Trehalose 6-Phosphate in Controlling Plant Development. J. Plant Physiol. 2023, 291, 154140. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from rna-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Varet, H.; Brillet-Guéguen, L.; Coppée, J.-Y.; Dillies, M.-A. SARTools: A DESeq2- and EdgeR-based r pipeline for comprehensive differential analysis of RNA-seq data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Replicate | Raw Reads | Clean Reads | Reads Mapped | Clean Base (G) | Error Rate (%) | Q20 (%) | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|---|---|---|---|
| stem | s-1 | 46,525,180 | 44,653,312 | 35,696,908 (79.94%) | 6.7 | 0.03 | 97.77 | 93.69 | 41.49 |
| s-2 | 49,274,168 | 47,298,816 | 36,331,319 (76.81%) | 7.09 | 0.03 | 96.84 | 91.34 | 41.05 | |
| s-3 | 44,999,472 | 43,015,244 | 33,554,924 (78.01%) | 6.45 | 0.03 | 97.01 | 91.68 | 41.33 | |
| stem base | c-1 | 47,309,760 | 45,108,452 | 34,803,807 (77.16%) | 6.77 | 0.03 | 97.02 | 91.88 | 41.43 |
| c-2 | 43,655,292 | 42,248,676 | 32,729,350 (77.47%) | 6.34 | 0.03 | 96.71 | 90.99 | 41.28 | |
| c-3 | 48,859,978 | 47,321,596 | 36,753,294 (77.67%) | 7.1 | 0.03 | 96.84 | 91.29 | 41.28 | |
| root | r-1 | 47,351,972 | 45,320,980 | 34,880,708 (76.96%) | 6.8 | 0.03 | 96.91 | 91.55 | 40.81 |
| r-2 | 44,140,624 | 42,271,582 | 32,883,539 (77.79%) | 6.34 | 0.03 | 97.6 | 93.33 | 40.82 | |
| r-3 | 52,453,492 | 50,657,998 | 39,267,720 (77.52%) | 7.6 | 0.03 | 96.88 | 91.5 | 41.08 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Q.; Wang, K.; Huang, J.; Dou, P.; Miao, Z. Exploring the Structure and Substance Metabolism of a Medicago sativa L. Stem Base. Int. J. Mol. Sci. 2024, 25, 6225. https://doi.org/10.3390/ijms25116225
Gao Q, Wang K, Huang J, Dou P, Miao Z. Exploring the Structure and Substance Metabolism of a Medicago sativa L. Stem Base. International Journal of Molecular Sciences. 2024; 25(11):6225. https://doi.org/10.3390/ijms25116225
Chicago/Turabian StyleGao, Qian, Kun Wang, Jing Huang, Pengpeng Dou, and Zhengzhou Miao. 2024. "Exploring the Structure and Substance Metabolism of a Medicago sativa L. Stem Base" International Journal of Molecular Sciences 25, no. 11: 6225. https://doi.org/10.3390/ijms25116225
APA StyleGao, Q., Wang, K., Huang, J., Dou, P., & Miao, Z. (2024). Exploring the Structure and Substance Metabolism of a Medicago sativa L. Stem Base. International Journal of Molecular Sciences, 25(11), 6225. https://doi.org/10.3390/ijms25116225