
Exploring the Interplay between the Hologenome and Complex Traits in Bovine and Porcine Animals Using Genome-Wide Association Analysis

 and
and
Abstract
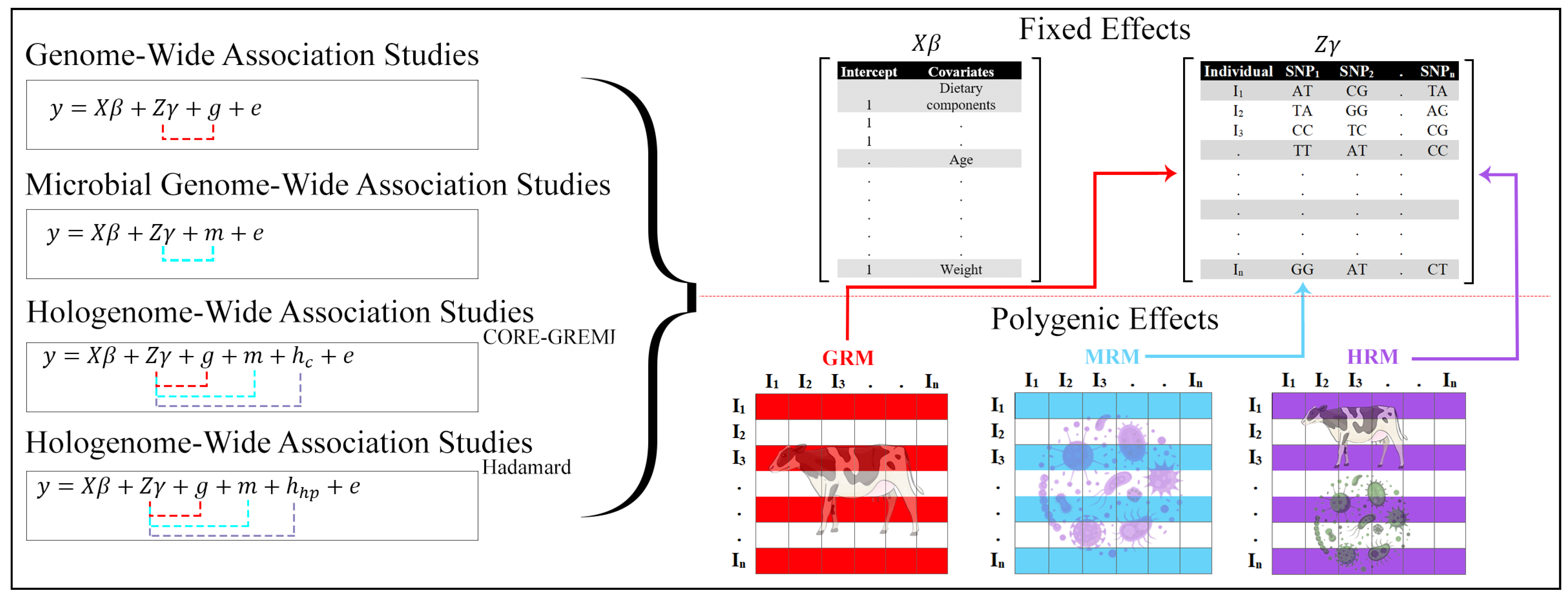
:1. Introduction
2. Results
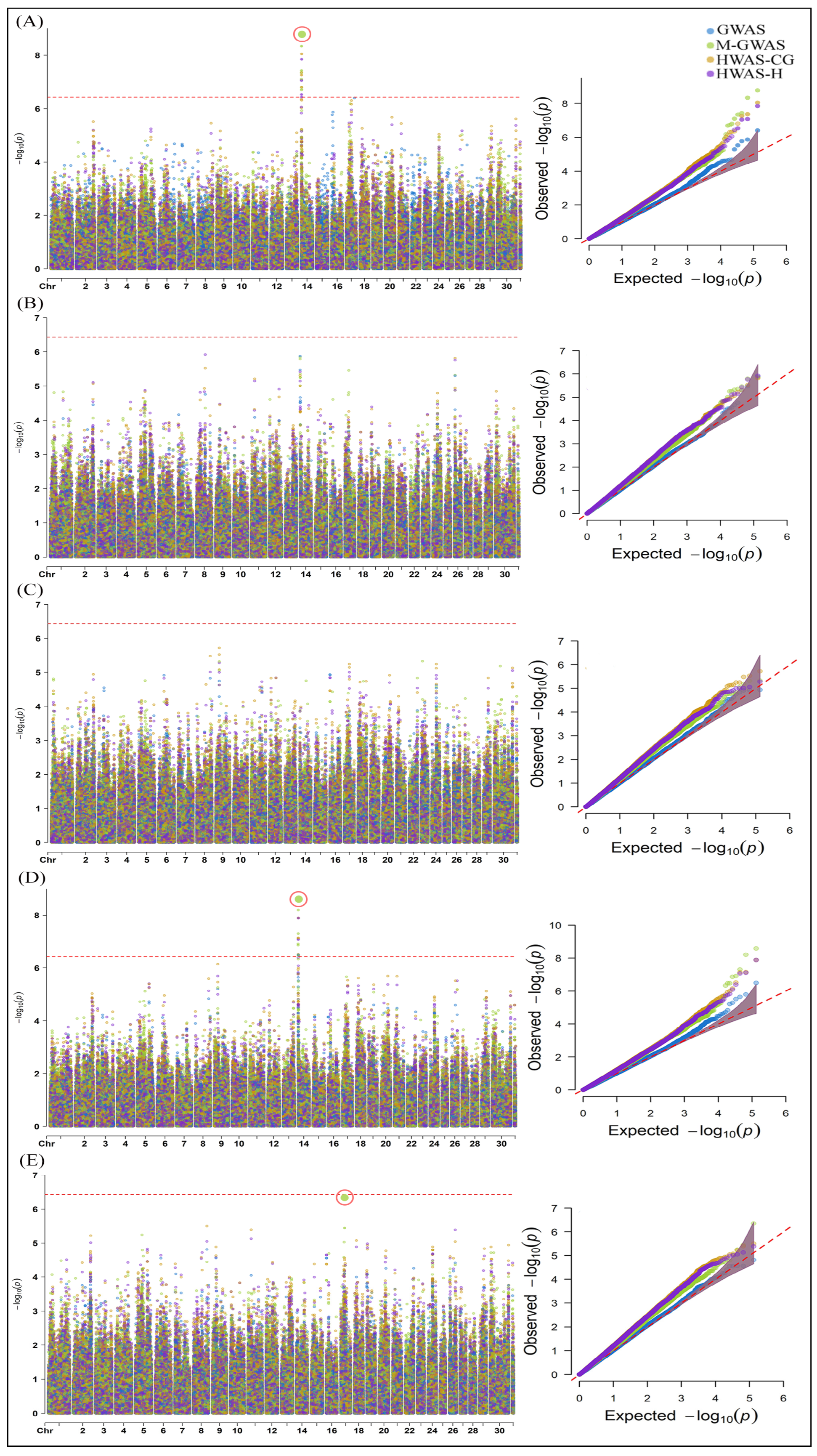
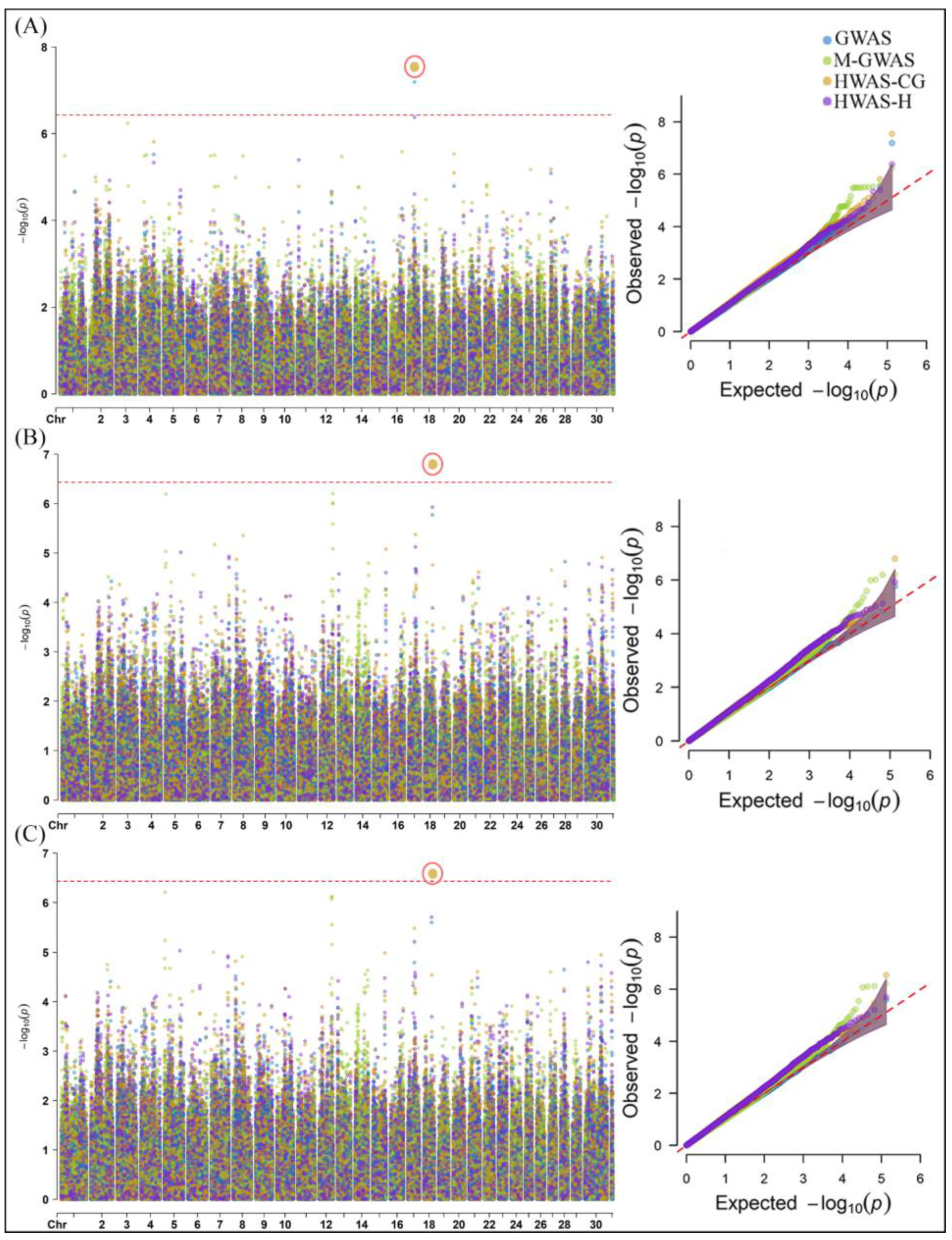
2.1. Genome-Wide Association Analysis of Milk Yield, Methane Emissions and Rumen Physiology Traits in Cattle
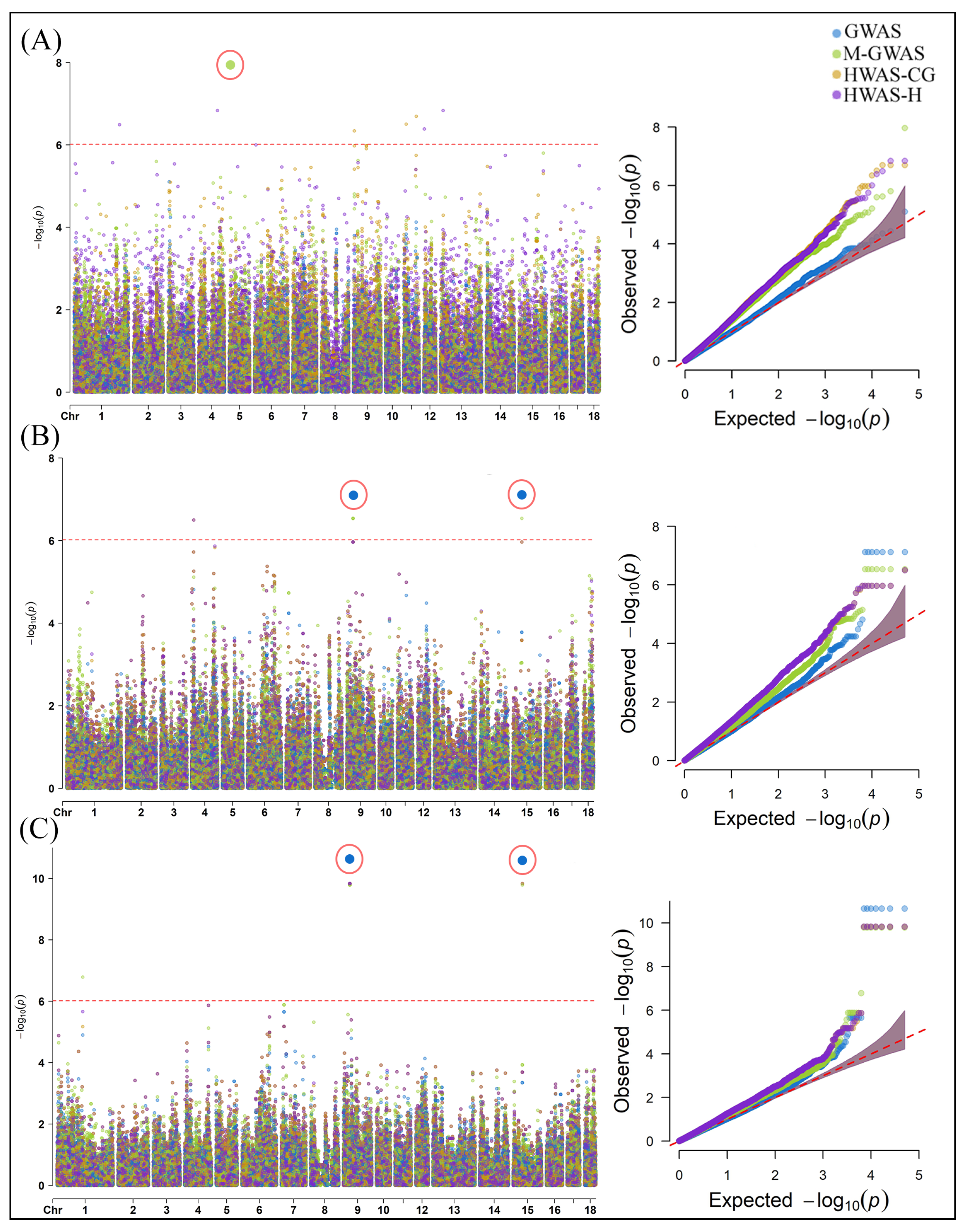
2.2. Genome-Wide Association Analysis of Gut Microbial Composition Traits in Pigs
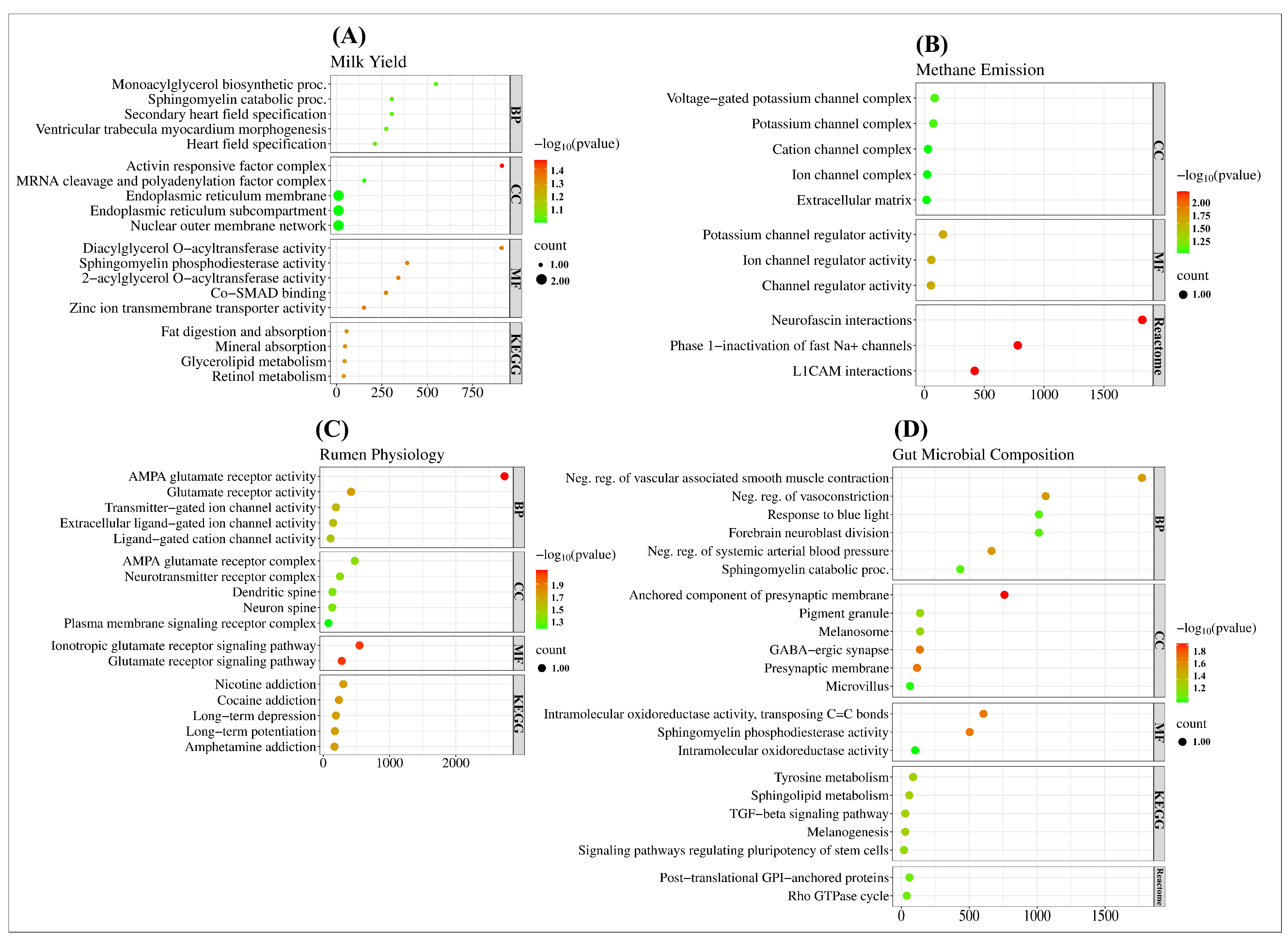
2.3. Gene Ontology and Functional Analysis of SNPs in Complex Traits
2.4. Transcriptome-Wide Association Studies of Complex Traits
2.5. Integrative Summary-Based Mendelian Randomization Analysis of Complex Traits
3. Discussion
3.1. SNPs and Candidate Genes Identified for Milk Yield, Methane Emissions and Rumen Physiology Traits in Cattle
3.2. SNPs and Candidate Genes Identified for Gut Microbial Composition Traits in Pigs
3.3. Dissecting Complex Traits—TWAS and SMR Analysis of Tissue-Specific Gene Expression
3.4. Limitations of the Analysis
4. Materials and Methods
4.1. Animal Genotype and Metagenome Data
4.2. Animal Phenotype Data
4.3. Genome-Wide Association Analysis and Linear Mixed Models
4.4. Genome-Wide Association Analysis Threshold and Functional Genomics Analysis
4.5. Transcriptome-Wide Association Analysis
4.6. Summary-Based Mendelian Randomization Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| APCDD1 | APC down-regulated 1 |
| APOBEC3H | Apolipoprotein B mRNA editing enzyme catalytic subunit 3H |
| ARHGAP39 | Rho GTPase Activating Protein 39 |
| ARHGAP42 | Rho GTPase Activating Protein 42 |
| BC | Bonferroni correction |
| BTA | Bos taurus autosome |
| C9H6orf163 | Chromosome 9 C6orf163 homolog |
| CNTN5 | Contactin 5 |
| CORE-GREML | COvariance between Random Effects Genome-based restricted maximum likelihood |
| CPSF1 | Cleavage and polyadenylation specific factor 1 |
| DCST1 | DC-STAMP Domain Containing 1 |
| DCT | Dopachrome Tautomerase |
| DCT | Dopachrome Tautomerase |
| DG | Daily gain |
| DGAT1 | Diacylglycerol O-Acyltransferase 1 |
| ENPP6 | Ectonucleotide Pyrophosphatase/Phosphodiesterase 6 |
| ENPP7 | Ectonucleotide Pyrophosphatase/Phosphodiesterase 7 |
| eQTLs | Expression quantitative trait loci |
| EXTL1 | Exostosin-like glycosyltransferase 1 |
| FBXO15 | F-Box Protein 15 |
| FC | Feed conversion |
| FCM | Fat–corrected milk |
| FDR-BH | False discovery rate control using Benjamini-Hochberg |
| FI | Feed intake |
| FOXH1 | Forkhead Box H1 |
| GO | Gene Ontology |
| GPAA1 | Glycosylphosphatidylinositol anchor attachment 1 |
| GPR180 | G Protein-Coupled Receptor 180 |
| GRIA2 | Glutamate Ionotropic Receptor AMPA Type Subunit 2 |
| GSEA | Gene set enrichment analysis |
| GWA | Genome-wide association |
| GWAS | Genome-wide association studies |
| GWST | Genome-wide significance threshold |
| HEIDI | Heterogeneity in dependent instruments |
| HRM | Hologenome relationship matrix |
| HWAS | Hologenome-wide association studies |
| HWAS-CG | Hologenome-wide association studies using CORE-GREML method |
| HWAS-H | Hologenome-wide association studies using Hadamard product method |
| IL33 | Interleukin 33 |
| IQANK1 | IQ motif and ankyrin repeat containing 1 |
| IRF2 | Interferon Regulatory Factor 2 |
| KCNIP4 | Potassium Voltage-Gated Channel Interacting Protein 4 |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LMM | Linear Mixed Model |
| lncRNA | long non-coding RNAs |
| LY6K | Lymphocyte Antigen 6 Family Member K |
| MAF | Minor allele frequency |
| MED29 | Mediator complex subunit 29 |
| MFRP | Membrane frizzled-related protein |
| M-GWAS | Microbial genome-wide association studies |
| MRM | Microbial relationship matrix |
| MWAS | Microbiome-wide association studies |
| NFASC | Neurofascin |
| NRBP2 | Nuclear receptor binding protein 2 |
| OPTC | Opticin |
| OSTF1 | Osteoclast Stimulating Factor 1 |
| OTUs | Operational taxonomic units |
| PCs | Principal components |
| PMRs | Partial-mixed rations |
| QIIME 2 | Quantitative insights into microbial ecology |
| Q-Q plot | Quantile-Quantile plot |
| RASSF3 | Ras association domain family member 3 |
| SLC39A4 | Solute carrier family 39 member 4 |
| SMAD9 | SMAD Family Member 9 |
| SMPD1 | Sphingomyelin phosphodiesterase 1 |
| SMPD5 | Sphingomyelin Phosphodiesterase 5 |
| SMR | Summary-based Mendelian randomization |
| SNP | Single nucleotide polymorphisms |
| SPOCD1 | SPOC domain containing 1 |
| SSC | Sus scrofa chromosome |
| TCF20 | Transcription Factor 20 |
| TFAP2C | Transcription factor AP-2 gamma |
| TMRs | Total-mixed rations |
| TWAS | Transcriptome-wide association studies |
References
- Raymond, B.; Yengo, L.; Costilla, R.; Schrooten, C.; Bouwman, A.C.; Hayes, B.J.; Veerkamp, R.F.; Visscher, P.M. Using prior information from humans to prioritize genes and gene-associated variants for complex traits in livestock. PLoS Genet. 2020, 16, e1008780. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Sun, H.; Wu, X.; Guan, L.L.; Liu, J. Assessment of Rumen Microbiota from a Large Dairy Cattle Cohort Reveals the Pan and Core Bacteriomes Contributing to Varied Phenotypes. Appl. Environ. Microbiol. 2018, 84, e00970-18. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.B.; Saha, S.; Do, K.-A. Analysis of microbiome data. Annu. Rev. Stat. Its Appl. 2023, 11, 483–504. [Google Scholar] [CrossRef]
- Couch, C.E.; Epps, C.W. Host, Microbiome, and Complex Space: Applying Population and Landscape Genetic Approaches to Gut Microbiome Research in Wild Populations. J. Hered. 2022, 113, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Veller, C.; Coop, G.M. Interpreting population- and family-based genome-wide association studies in the presence of confounding. PLoS Biol. 2024, 22, e3002511. [Google Scholar] [CrossRef] [PubMed]
- Surana, N.K.; Kasper, D.L. Moving beyond microbiome-wide associations to causal microbe identification. Nature 2017, 552, 244–247. [Google Scholar] [CrossRef]
- Rosenberg, E.; Zilber-Rosenberg, I. The hologenome concept of evolution after 10 years. Microbiome 2018, 6, 78. [Google Scholar] [CrossRef] [PubMed]
- Difford, G.F.; Plichta, D.R.; Løvendahl, P.; Lassen, J.; Noel, S.J.; Højberg, O.; Wright, A.-D.G.; Zhu, Z.; Kristensen, L.; Nielsen, H.B.; et al. Host genetics and the rumen microbiome jointly associate with methane emissions in dairy cows. PLoS Genet. 2018, 14, e1007580. [Google Scholar] [CrossRef]
- Weishaar, R.; Wellmann, R.; Camarinha-Silva, A.; Rodehutscord, M.; Bennewitz, J. Selecting the hologenome to breed for an improved feed efficiency in pigs—A novel selection index. J. Anim. Breed. Genet. 2019, 137, 14–22. [Google Scholar] [CrossRef]
- Qadri, Q.R.; Zhao, Q.; Lai, X.; Zhang, Z.; Zhao, W.; Pan, Y.; Wang, Q. Estimation of Complex-Trait Prediction Accuracy from the Different Holo-Omics Interaction Models. Genes 2022, 13, 1580. [Google Scholar] [CrossRef]
- Saborío-Montero, A.; Gutiérrez-Rivas, M.; López-García, A.; García-Rodríguez, A.; Atxaerandio, R.; Goiri, I.; Jiménez-Montero, J.A.; González-Recio, O. Holobiont effect accounts for more methane emission variance than the additive and microbiome effects on dairy cattle. Livest. Sci. 2021, 250, 104538. [Google Scholar] [CrossRef]
- Awany, D.; Allali, I.; Dalvie, S.; Hemmings, S.; Mwaikono, K.S.; Thomford, N.E.; Gomez, A.; Mulder, N.; Chimusa, E.R. Host and Microbiome Genome-Wide Association Studies: Current State and Challenges. Front. Genet. 2019, 9, 637. [Google Scholar] [CrossRef]
- Hua, X.; Song, L.; Yu, G.; Vogtmann, E.; Goedert, J.J.; Abnet, C.C.; Landi, M.T.; Shi, J. MicrobiomeGWAS: A Tool for Identifying Host Genetic Variants Associated with Microbiome Composition. Genes 2022, 13, 1224. [Google Scholar] [CrossRef]
- Nicolae, D.L.; Gamazon, E.; Zhang, W.; Duan, S.; Dolan, M.E.; Cox, N.J. Trait-Associated SNPs Are More Likely to Be eQTLs: Annotation to Enhance Discovery from GWAS. PLoS Genet. 2010, 6, e1000888. [Google Scholar] [CrossRef]
- Wen, X.; Luca, F.; Pique-Regi, R. Cross-Population Joint Analysis of eQTLs: Fine Mapping and Functional Annotation. PLoS Genet. 2015, 11, e1005176. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef]
- Shi, Y.; Di Giammartino, D.C.; Taylor, D.; Sarkeshik, A.; Rice, W.J.; Yates, J.R.; Frank, J.; Manley, J.L. Molecular architecture of the human pre-mRNA 3′ processing complex. Mol. Cell 2009, 33, 365–376. [Google Scholar] [CrossRef]
- Geiser, J.; Venken, K.J.T.; De Lisle, R.C.; Andrews, G.K. A Mouse Model of Acrodermatitis Enteropathica: Loss of Intestine Zinc Transporter ZIP4 (Slc39a4) Disrupts the Stem Cell Niche and Intestine Integrity. PLoS Genet. 2012, 8, e1002766. [Google Scholar] [CrossRef]
- Cochran, S.D.; Cole, J.B.; Null, D.J.; Hansen, P.J. Discovery of single nucleotide polymorphisms in candidate genes associated with fertility and production traits in Holstein cattle. BMC Genet. 2013, 14, 49. [Google Scholar] [CrossRef]
- Pedrosa, V.B.; Schenkel, F.S.; Chen, S.-Y.; Oliveira, H.R.; Casey, T.M.; Melka, M.G.; Brito, L.F. Genomewide Association Analyses of Lactation Persistency and Milk Production Traits in Holstein Cattle Based on Imputed Whole-Genome Sequence Data. Genes 2021, 12, 1830. [Google Scholar] [CrossRef]
- Atashi, H.; Salavati, M.; De Koster, J.; Ehrlich, J.; Crowe, M.; Opsomer, G.; Hostens, M.; the GplusE consortium. Genome-wide association for milk production and lactation curve parameters in Holstein dairy cows. J. Anim. Breed. Genet. 2019, 137, 292–304. [Google Scholar] [CrossRef]
- Manzanilla-Pech, C.I.V.; De Haas, Y.; Hayes, B.J.; Veerkamp, R.F.; Khansefid, M.; Donoghue, K.A.; Arthur, P.F.; Pryce, J.E. Genomewide association study of methane emissions in Angus beef cattle with validation in dairy cattle1. J. Anim. Sci. 2016, 94, 4151–4166. [Google Scholar] [CrossRef]
- Nayak, S.S.; Panigrahi, M.; Rajawat, D.; Ghildiyal, K.; Sharma, A.; Parida, S.; Bhushan, B.; Mishra, B.P.; Dutt, T. Comprehensive selection signature analyses in dairy cattle exploiting purebred and crossbred genomic data. Mamm. Genome 2023, 34, 615–631. [Google Scholar] [CrossRef]
- Weber, K.L.; Welly, B.T.; Van Eenennaam, A.L.; Young, A.E.; Porto-Neto, L.R.; Reverter, A.; Rincon, G. Identification of Gene Networks for Residual Feed Intake in Angus Cattle Using Genomic Prediction and RNA-seq. PLoS ONE 2016, 11, e0152274. [Google Scholar] [CrossRef]
- Diao, S.; Huang, S.; Xu, Z.; Ye, S.; Yuan, X.; Chen, Z.; Zhang, H.; Zhang, Z.; Li, J. Genetic Diversity of Indigenous Pigs from South China Area Revealed by SNP Array. Animals 2019, 9, 361. [Google Scholar] [CrossRef]
- Jöhnk, B.; Bayram, Ö.; Abelmann, A.; Heinekamp, T.; Mattern, D.J.; Brakhage, A.A.; Jacobsen, I.D.; Valerius, O.; Braus, G.H. SCF Ubiquitin Ligase F-box Protein Fbx15 Controls Nuclear Co-repressor Localization, Stress Response and Virulence of the Human Pathogen Aspergillus fumigatus. PLoS Pathog. 2016, 12, e1005899. [Google Scholar] [CrossRef]
- Oommen, S.; Vasu, V.T.; Leonard, S.W.; Traber, M.G.; Cross, C.E.; Gohil, K. Genome wide responses of murine lungs to dietary α-tocopherol. Free Radic. Res. 2007, 41, 98–133. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhu, Z.; Sun, L.; Yin, W.; Liang, Y.; Chen, H.; Bi, Y.; Zhai, W.; Yin, Y.; Zhang, W. Hepatic G Protein-Coupled Receptor 180 Deficiency Ameliorates High Fat Diet-Induced Lipid Accumulation via the Gi-PKA-SREBP Pathway. Nutrients 2023, 15, 1838. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, T.; Cao, M.; Yuan, C.; Reiter, R.J.; Zhao, Z.; Zhao, Y.; Chen, L.; Fan, W.; Wang, X.; et al. Gut Microbiota Dysbiosis Induced by Decreasing Endogenous Melatonin Mediates the Pathogenesis of Alzheimer’s Disease and Obesity. Front. Immunol. 2022, 13, 900132. [Google Scholar] [CrossRef]
- Abdellatif, A.M.; Smith, H.J.; Harms, R.Z.; Sarvetnick, N.E. Human Islet Response to Selected Type 1 Diabetes-Associated Bacteria: A Transcriptome-Based Study. Front. Immunol. 2019, 10, 2623. [Google Scholar] [CrossRef]
- Wu, F.; Chen, Z.; Zhang, Z.; Wang, Z.; Zhang, Z.; Wang, Q.; Pan, Y. The Role of SOCS3 in Regulating Meat Quality in Jinhua Pigs. Int. J. Mol. Sci. 2023, 24, 10593. [Google Scholar] [CrossRef]
- Huang, H.; Hu, Y.; Guo, L.; Wen, Z. Integrated bioinformatics analyses of key genes involved in hepatocellular carcinoma immunosuppression. Oncol. Lett. 2021, 22, 830. [Google Scholar] [CrossRef]
- Lynegaard, J.C.; Kjeldsen, N.J.; Hansen, C.F.; Williams, A.R.; Nielsen, J.P.; Amdi, C. Reduction in Diarrhoea and Modulation of Intestinal Gene Expression in Pigs Allocated a Low Protein Diet without Medicinal Zinc Oxide Post-Weaning. Animals 2022, 12, 989. [Google Scholar] [CrossRef]
- Zhang, H.; Xia, P.; Feng, L.; Jia, M.; Su, Y. Feeding Frequency Modulates the Intestinal Transcriptome Without Affecting the Gut Microbiota in Pigs With the Same Daily Feed Intake. Front. Nutr. 2021, 8, 743343. [Google Scholar] [CrossRef]
- Dionissopoulos, L. Understanding the Molecular Mechanisms Involved in Subacute Ruminal Acidosis And Rumenitis. Ph.D. Thesis, University of Guelph, Guelph, ON, Canada, 2013. [Google Scholar]
- Cheng, Z.; Little, M.; Ferris, C.; Takeda, H.; Ingvartsen, K.; Crowe, M.; Wathes, D. Influence of the concentrate inclusion level in a grass silage–based diet on hepatic transcriptomic profiles in Holstein-Friesian dairy cows in early lactation. J. Dairy Sci. 2023, 106, 5805–5824. [Google Scholar] [CrossRef]
- Khanal, P.; Sanglard, L.P.; Mayberry, K.; Sommer, J.; Poore, M.H.; Poole, D.H.; Serão, N.V.L. PSIII-3 Genes and functions associated with tolerance to fescue toxicosis in Angus cows. J. Anim. Sci. 2019, 97, 167. [Google Scholar] [CrossRef]
- Browne, N.; Daly, D.; Horgan, K. Differential impact of yeast cell wall products in recovery of porcine intestinal epithelial cell barrier function following Lipopolysaccharide challenge. Porc. Health Manag. 2023, 9, 18. [Google Scholar] [CrossRef]
- Imaizumi, T.; Ando, M.; Nakatochi, M.; Maruyama, S.; Yasuda, Y.; Honda, H.; Kuwatsuka, Y.; Kato, S.; Kondo, T.; Iwata, M.; et al. Association of interactions between dietary salt consumption and hypertension-susceptibility genetic polymorphisms with blood pressure among Japanese male workers. Clin. Exp. Nephrol. 2016, 21, 457–464. [Google Scholar] [CrossRef]
- Reverter, A.; Ballester, M.; Alexandre, P.A.; Marmol-Sanchez, E.; Dalmau, A.; Quintanilla, R.; Ramayo-Caldas, Y. A gene co-association network regulating gut microbial communities in a Duroc pig population. Microbiome 2021, 9, 52. [Google Scholar] [CrossRef]
- Liu, S.; Gao, Y.; Canela-Xandri, O.; Wang, S.; Yu, Y.; Cai, W.; Li, B.; Xiang, R.; Chamberlain, A.J.; Pairo-Castineira, E.; et al. A multi-tissue atlas of regulatory variants in cattle. Nat. Genet. 2022, 54, 1438–1447. [Google Scholar] [CrossRef]
- Buaban, S.; Lengnudum, K.; Boonkum, W.; Phakdeedindan, P. Genome-wide association study on milk production and somatic cell score for Thai dairy cattle using weighted single-step approach with random regression test-day model. J. Dairy Sci. 2021, 105, 468–494. [Google Scholar] [CrossRef]
- Tsartsianidou, V.; Sánchez-Molano, E.; Kapsona, V.V.; Basdagianni, Z.; Chatziplis, D.; Arsenos, G.; Triantafyllidis, A.; Banos, G. A comprehensive genome-wide scan detects genomic regions related to local adaptation and climate resilience in Mediterranean domestic sheep. Genet. Sel. Evol. 2021, 53, 90. [Google Scholar] [CrossRef]
- Oliveira, H.R.d. Genetic and Genomic Analyses of Longitudinal Traits in Dairy Cattle. Ph.D. Thesis, Federal University of Viçosa, Viçosa, MG, Brasil, 2018. [Google Scholar]
- Abbas, W. Host Genetics and Phenotype Associations within the Bovine Rumen Microbiome. Doctoral Dissertation, The University of Nebraska-Lincoln, Lincoln, Nebraska, 2021. [Google Scholar]
- Zhao, K.; Chen, Y.H.; Penner, G.B.; Oba, M.; Guan, L.L. Transcriptome analysis of ruminal epithelia revealed potential regulatory mechanisms involved in host adaptation to gradual high fermentable dietary transition in beef cattle. BMC Genom. 2017, 18, 976. [Google Scholar] [CrossRef]
- Brunes, L.C.; Baldi, F.; Lopes, F.B.; Lôbo, R.B.; Espigolan, R.; Costa, M.F.O.; Stafuzza, N.B.; Magnabosco, C.U. Weighted single-step genome-wide association study and pathway analyses for feed efficiency traits in Nellore cattle. J. Anim. Breed. Genet. 2020, 138, 23–44. [Google Scholar] [CrossRef]
- Wang, W.; Dong, Y.; Guo, W.; Zhang, X.; Degen, A.A.; Bi, S.; Ding, L.; Chen, X.; Long, R. Linkages between rumen microbiome, host, and environment in yaks, and their implications for understanding animal production and management. Front. Microbiol. 2024, 15, 1301258. [Google Scholar] [CrossRef]
- Choudhury, R.; Gu, Y.; Bolhuis, J.E.; Kleerebezem, M. Early feeding leads to molecular maturation of the gut mucosal immune system in suckling piglets. Front. Immunol. 2023, 14, 1208891. [Google Scholar] [CrossRef]
- Ponsuksili, S.; Murani, E.; Brand, B.; Schwerin, M.; Wimmers, K. Integrating expression profiling and whole-genome association for dissection of fat traits in a porcine model [S]. J. Lipid Res. 2011, 52, 668–678. [Google Scholar] [CrossRef]
- Fanalli, S.L. Effect of the Addition of Different Sources of Fatty Acids in The Pig Diet on The Transcriptomic Profile of Different Tissues. Ph.D. Thesis, Universidade de São Paulo, São Paulo, SP, Brasil, 2022. [Google Scholar]
- Óvilo, C.; Trakooljul, N.; Núñez, Y.; Hadlich, F.; Murani, E.; Ayuso, M.; García-Contreras, C.; Vázquez-Gómez, M.; Rey, A.I.; Garcia, F.; et al. SNP discovery and association study for growth, fatness and meat quality traits in Iberian crossbred pigs. Sci. Rep. 2022, 12, 16361. [Google Scholar] [CrossRef]
- Tan, X.; He, Z.; Fahey, A.G.; Zhao, G.; Liu, R.; Wen, J. Research progress and applications of genome-wide association study in farm animals. Anim. Res. One Health 2023, 1, 56–77. [Google Scholar] [CrossRef]
- Yoosefzadeh-Najafabadi, M.; Eskandari, M.; Belzile, F.; Torkamaneh, D. Genome-wide association study statistical models: A review. In Genome-Wide Association Studies; Springer: Berlin/Heidelberg, Germany, 2022; pp. 43–62. [Google Scholar]
- Kwak, I.-Y.; Pan, W. Gene- and pathway-based association tests for multiple traits with GWAS summary statistics. Bioinformatics 2016, 33, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Wallace, R.J.; Sasson, G.; Garnsworthy, P.C.; Tapio, I.; Gregson, E.; Bani, P.; Huhtanen, P.; Bayat, A.R.; Strozzi, F.; Biscarini, F.; et al. A heritable subset of the core rumen microbiome dictates dairy cow productivity and emissions. Sci. Adv. 2019, 5, eaav8391. [Google Scholar] [CrossRef]
- Harris, B.; Kolver, E. Review of Holsteinization on Intensive Pastoral Dairy Farming in New Zealand. J. Dairy Sci. 2001, 84, E56–E61. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Camarinha-Silva, A.; Maushammer, M.; Wellmann, R.; Vital, M.; Preuss, S.; Bennewitz, J. Host Genome Influence on Gut Microbial Composition and Microbial Prediction of Complex Traits in Pigs. Genetics 2017, 206, 1637–1644. [Google Scholar] [CrossRef] [PubMed]
- Stamer, E.; Brade, W.; Thaller, G. Actual genetic parameters for feed conversion rate of station tested progenies of Piétrain boars in Lower Saxony (brief report). Züchtungskunde 2009, 81, 97–105. [Google Scholar]
- Dandine-Roulland, C.; Perdry, H. Genome-wide data manipulation, association analysis and heritability estimates in R with Gaston 1.5. In Human Heredity; Karger: Basel, Switzerland, 2017; p. 6. [Google Scholar]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Vitezica, Z.G.; Legarra, A.; Toro, M.A.; Varona, L. Orthogonal Estimates of Variances for Additive, Dominance, and Epistatic Effects in Populations. Genetics 2017, 206, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Joyce, M.M.; Burghardt, J.R.; Burghardt, R.C.; Hooper, R.N.; Jaeger, L.A.; Spencer, T.E.; Bazer, F.W.; Johnson, G.A. Pig Conceptuses Increase Uterine Interferon-Regulatory Factor 1 (IRF1), but Restrict Expression to Stroma Through Estrogen-Induced IRF2 in Luminal Epithelium1. Biol. Reprod. 2007, 77, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xu, S. Statistical power in genome-wide association studies and quantitative trait locus mapping. Heredity 2019, 123, 287–306. [Google Scholar] [CrossRef]
- Aguiar, M.; Mahajan, S. Bimonoids for Hyperplane Arrangements; Cambridge University Press: Cambridge, UK, 2020; Volume 173. [Google Scholar]
- Korthauer, K.; Kimes, P.K.; Duvallet, C.; Reyes, A.; Subramanian, A.; Teng, M.; Shukla, C.; Alm, E.J.; Hicks, S.C. A practical guide to methods controlling false discoveries in computational biology. Genome Biol. 2019, 20, 118. [Google Scholar] [CrossRef]
- Li, M.-X.; Yeung, J.M.Y.; Cherny, S.S.; Sham, P.C. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum. Genet. 2011, 131, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Barsh, G.S.; Copenhaver, G.P.; Gibson, G.; Williams, S.M. Guidelines for Genome-Wide Association Studies. PLoS Genet. 2012, 8, e1002812. [Google Scholar] [CrossRef] [PubMed]
- Team, R.C.; Team, M.R.C.; Suggests, M.; Matrix, S. Package Stats; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Katsevich, E.; Sabatti, C.; Bogomolov, M. Filtering the Rejection Set While Preserving False Discovery Rate Control. J. Am. Stat. Assoc. 2021, 118, 165–176. [Google Scholar] [CrossRef]
- Kaler, A.S.; Purcell, L.C. Estimation of a significance threshold for genome-wide association studies. BMC Genom. 2019, 20, 618. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated Tool for Genome-wide Association Study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef]
- Martin, F.J.; Amode, M.R.; Aneja, A.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Becker, A.; Bennett, R.; Berry, A.; Bhai, J. Ensembl 2023. Nucleic Acids Res. 2023, 51, D933–D941. [Google Scholar] [CrossRef] [PubMed]
- Qanbari, S. On the Extent of Linkage Disequilibrium in the Genome of Farm Animals. Front. Genet. 2020, 10, 1304. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, Z.; Teng, J.; Liu, S.; Lin, Q.; Gao, Y.; Bai, Z.; Consortium, F.; Li, B.; Liu, G. FarmGTEx TWAS-server: An interactive web server for customized TWAS analysis in both human and farm animals. bioRxiv 2023. [Google Scholar] [CrossRef]
- Gamazon, E.R.; GTEx Consortium; Wheeler, H.E.; Shah, K.P.; Mozaffari, S.V.; Aquino-Michaels, K.; Carroll, R.J.; Eyler, A.E.; Denny, J.C.; Nicolae, D.L.; et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 2015, 47, 1091–1098. [Google Scholar] [CrossRef]
- Teng, J.; Gao, Y.; Yin, H.; Bai, Z.; Liu, S.; Zeng, H.; Bai, L.; Cai, Z.; Zhao, B.; Li, X.; et al. A compendium of genetic regulatory effects across pig tissues. Nat. Genet. 2024, 56, 112–123. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Arbitrary Threshold | FDR-BH | BC | GWST |
|---|---|---|---|---|
| HWAS-CG | 130,337 | 234 | 28 | 14 |
| HWAS-H | 124,212 | 118 | 20 | 12 |
| M-GWAS | 111,222 | 97 | 34 | 18 |
| GWAS | 84,281 | 29 | 19 | 10 |
| Trait | SNP ID | Gene | Function |
|---|---|---|---|
| Milk and Lactose | UFL-rs134432442 | CPSF1, SLC39A4 | mRNA cleavage and polyadenylation specificity factor complex, cellular zinc homeostasis as a zinc transporter * |
| BovineHD1400000206 | SMPD5 | lipid and sphingolipid metabolism | |
| ARS-BFGL-NGS-4939 | DGAT1 | Absorption of dietary fats and esterification of exogenous fatty acids to glycerol | |
| BovineHD1400000188 | ARHGAP39 | Post-synapse organization and signal transduction | |
| ARS-BFGL-NGS-57820 | FOXH1 | Development of the mammary gland and the regulation of milk protein | |
| BovineHD1400000447 | LY6K | Sperm migration and cell growth | |
| ARS-BFGL-NGS-103064 | |||
| BovineHD1400000262 | LOC100141215 | Unknown function | |
| Milk | BovineHD1400000453 | LY6D | Marking the earliest stage of B- and T-cell development * |
| FCM | BovineHD1700008152 | 7SK | Negatively regulating RNA polymerase II transcription * |
| CH4 ECM | ARS-BFGL-NGS-3398 | OPTC | Inhibiting angiogenesis and regulating collagen fibril organization * |
| ARS-BFGL-NGS-35483 | NFASC | Axon subcellular targeting and synapse formation * | |
| BTA-75940-no-rs | KCNIP4 | Regulation of potassium ion transmembrane transport * | |
| ARS-BFGL-NGS-34028 | PALM2 ** | Membrane–cytoskeleton interaction | |
| Acetate | Hapmap32352-BTA-153912 | GRIA2 | Various central nervous system functions * |
| Propionate and Wolin | ARS-BFGL-NGS-17066 | FBXO15 | Ubiquitin-mediated protein degradation * |
| DG | ASGA0023892 | TCF20 | Transcriptional regulator * |
| ASGA0051282 | GPR180 | Thermogenic adipocyte activity * | |
| INRA0037215 | DCT | Plays a role in melanin biosynthesis pathway * | |
| SIRI0001279 | |||
| DIAS0003214 | SMAD9 | Cell signaling and act as transcription factors * | |
| M1GA0025187 | ENSSSCG00000014730 | Putative odorant or sperm cell receptor | |
| M1GA0006139 | DCST1 | Egg-sperm fusion and antigen processing | |
| ASGA0056172 | ENSSSCG00000041418 | None (Long non-coding RNA) | |
| DRGA0002098 | OSTF1 | Mastitis resistance and bone resorption activity | |
| MARC0008607 | ENPP7 | Protecting intestinal mucosa from inflammation | |
| FC and FI | ASGA0069501 | ENPP6 | Synthesis of phosphatidylcholine and Choline metabolism * |
| ASGA0101971 | ENSSSCG00000053826 | None (Long non-coding RNA) | |
| DRGA0009289 | CNTN5 | Cell-cell adhesion and brain development * | |
| DRGA0009304 | ARHGAP42 | Regulating endothelial cell shape and angiogenesis * | |
| MARC0075559 | |||
| MARC0066941 | ENSSSCG00000053826 | None (Long non-coding RNA) | |
| MARC0073975 | IRF2 | Controlling the luminal epithelium of the endometrium | |
| FC | MARC0057350 | ENSSSCG00000047619 | None (Long non-coding RNA) |
| FI | MARC0087957 | ENSSSCG00000045481 | Unknown function |
| Trait | Model | Gene | Chr | Start | End | PFDR | Tissue |
|---|---|---|---|---|---|---|---|
| Milk | M-GWAS | GPAA1 | 14 | 750,608 | 753,850 | 1.1 × 10−6 | Muscle |
| Fat | HWAS-CG | NRBP2 | 14 | 961,099 | 968,482 | 0.00036 | Blood |
| Protein | HWAS-H | TFAP2C | 13 | 59,366,399 | 59,375,952 | 0.022 | Embryo |
| Lactose | M-GWAS | GPAA1 | 14 | 750,608 | 753,850 | 7.7 × 10−7 | Muscle |
| FCM | M-GWAS | RASSF3 | 5 | 49,114,394 | 49,190,776 | 0.0073 | Macrophage |
| CH4 g/d | GWAS | EXTL1 | 2 | 127,052,889 | 127,070,881 | 0.025 | Hypothalamus |
| CH4 DMI | GWAS | APOBEC3H | 5 | 110,559,226 | 110,574,354 | 0.00178 | Monocyte |
| CH4 ECM | HWAS-CG | C9H6orf163 | 9 | 62,541,669 | 62,569,565 | 0.0027 | Oviduct |
| Acetate | HWAS-CG | SPOCD1 | 2 | 121,894,026 | 121,924,935 | 0.022 | Uterus |
| Propionate | HWAS-CG | MED29 | 18 | 49,092,003 | 49,100,532 | 0.00021 | Macrophage |
| Wolin | HWAS-CG | MED29 | 18 | 49,092,003 | 49,100,532 | 0.00036 | Macrophage |
| DG | HWAS-CG | IL33 | 1 | 215,899,435 | 215,941,840 | 1 × 10−6 | Colon |
| FC | HWAS-CG | APCDD1 | 6 | 97,992,228 | 98,026,768 | 0.0009 | Hypothalamus |
| FI | HWAS-H | SMPD1 | 9 | 3,324,091 | 3,328,373 | 0.016 | Hypothalamus |
| Trait | Model | Gene | Chr | PGWAS | Pe-QTL | PSMR | FDR | Tissue | PHEIDI |
|---|---|---|---|---|---|---|---|---|---|
| Milk | M-GWAS | DGAT1 | 14 | 7.42 | 15.29 | 5.28 | 0.003 | Mammary | 0.0005 |
| Fat | HWAS-CG | DGAT1 | 14 | 4.25 | 16.60 | 3.56 | 0.626 | Blood | 6 × 10−5 |
| Protein | HWAS-H | KLF15 | 22 | 4.54 | 6.23 | 2.87 | 0.283 | Monocytes | 0.8467 |
| Lactose | M-GWAS | DGAT1 | 14 | 8.58 | 18.01 | 6.10 | 0.001 | Liver | 1.8 × 10−3 |
| FCM | M-GWAS | PLTP | 13 | 3.95 | 6.44 | 2.68 | 0.958 | Muscle | 0.00698 |
| CH4 g/d | GWAS | AOX1 | 2 | 4.16 | 15.04 | 3.44 | 0.141 | Adipose | - |
| CH4 DMI | GWAS | DDX17 | 5 | 4.73 | 5.74 | 2.84 | 0.927 | Mammary | - |
| CH4 ECM | HWAS-CG | PTBP3 | 8 | 4.75 | 6.85 | 3.05 | 0.891 | Macrophage | 1.6 × 10−5 |
| Acetate | HWAS-CG | ZBTB8B | 2 | 3.96 | 15.21 | 3.32 | 0.381 | Uterus | 0.9203 |
| Propionate | HWAS-CG | SLC20A2 | 27 | 3.09 | 28.16 | 2.87 | 0.929 | Blood | 0.1584 |
| Wolin | HWAS-CG | SLC20A2 | 27 | 3.18 | 28.16 | 2.95 | 0.948 | Blood | 0.1820 |
| DG | HWAS-CG | FADS1 | 23 | 4.55 | 25.60 | 4.01 | 0.52 | Muscle | 3 × 10−6 |
| FC | HWAS-CG | ENSSSCG00000037808 | 4 | 4.39 | 13.09 | 3.49 | 0.271 | Frontal cortex | 8 × 10−6 |
| FI | HWAS-H | QRSL1 | 1 | 2.75 | 8.56 | 2.24 | 0.737 | Hypothalamus | 7.3 × 10−5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qadri, Q.R.; Lai, X.; Zhao, W.; Zhang, Z.; Zhao, Q.; Ma, P.; Pan, Y.; Wang, Q. Exploring the Interplay between the Hologenome and Complex Traits in Bovine and Porcine Animals Using Genome-Wide Association Analysis. Int. J. Mol. Sci. 2024, 25, 6234. https://doi.org/10.3390/ijms25116234
Qadri QR, Lai X, Zhao W, Zhang Z, Zhao Q, Ma P, Pan Y, Wang Q. Exploring the Interplay between the Hologenome and Complex Traits in Bovine and Porcine Animals Using Genome-Wide Association Analysis. International Journal of Molecular Sciences. 2024; 25(11):6234. https://doi.org/10.3390/ijms25116234
Chicago/Turabian StyleQadri, Qamar Raza, Xueshuang Lai, Wei Zhao, Zhenyang Zhang, Qingbo Zhao, Peipei Ma, Yuchun Pan, and Qishan Wang. 2024. "Exploring the Interplay between the Hologenome and Complex Traits in Bovine and Porcine Animals Using Genome-Wide Association Analysis" International Journal of Molecular Sciences 25, no. 11: 6234. https://doi.org/10.3390/ijms25116234