
Arrestins: A Small Family of Multi-Functional Proteins


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction—Discovery of Arrestin Subtypes
2. Arrestin Structure and Modes of Self-Association
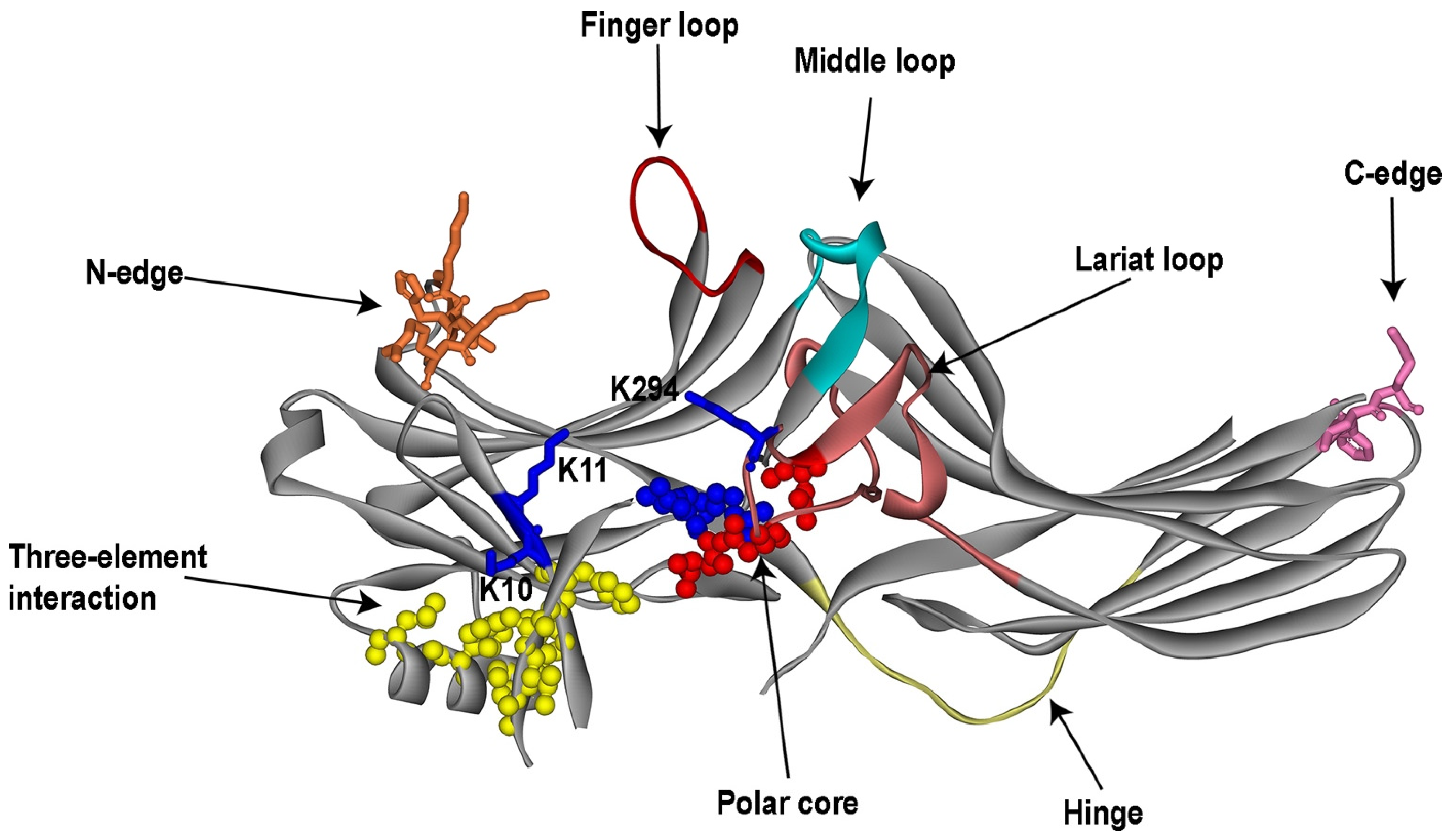
2.1. Structure of Free Arrestins
2.2. Self-Association of Arrestins
3. GPCR Binding
3.1. Receptor-Binding Elements
3.2. Arrestin Selectivity for the Active Phosphorylated GPCRs
3.2.1. “Coincidence Detector” Mechanism
3.2.2. Identification of Sensors in Arrestins
3.2.3. The Role of Receptor-Attached Phosphates
3.2.4. Conformational Rearrangements Associated with Receptor Binding
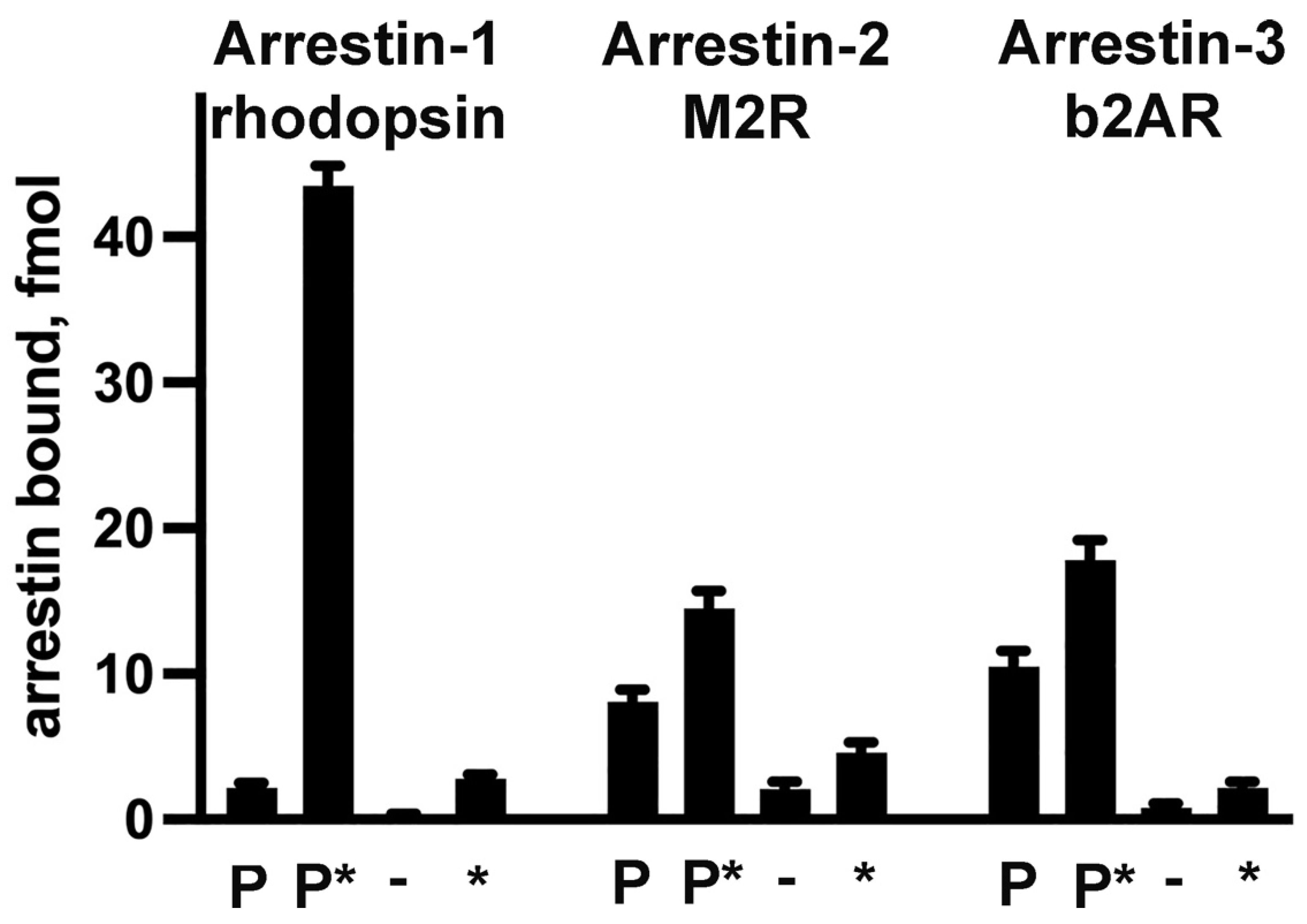
3.3. Receptor Preference of Arrestins
3.4. Structure of the Arrestin-GPCR Complexes: Possible Functional Implications
4. Arrestins in GPCR Trafficking
5. Arrestin Interactions with Non-Receptor-Binding Partners
5.1. Microtubules
5.2. MAP Kinases
5.3. Src Family Kinases
6. Ubiquitin Ligases and Deubiquitinating Enzymes
7. Calmodulin, NSF, and Other Proteins
8. Different Signaling Proteins Preferentially Bind Arrestins in All Known Conformations
9. Arrestin-Assisted Signaling: GPCR-Dependent and Independent
10. Arrestin-Based Molecular Tools
10.1. Signaling-Biased Full-Length Arrestins
10.2. Monofunctional Arrestin Elements
11. Conclusions
Funding
Conflicts of Interest
References
- Indrischek, H.; Prohaska, S.J.; Gurevich, V.V.; Gurevich, E.V.; Stadler, P.F. Uncovering missing pieces: Duplication and deletion history of arrestins in deuterostomes. BMC Evol. Biol. 2017, 17, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Carman, C.V.; Benovic, J.L. G-protein-coupled receptors: Turn-ons and turn-offs. Curr. Opin. Neurobiol. 1998, 8, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, H. Light-regulated binding of rhodopsin kinase and other proteins to cattle photoreceptor membranes. Biochemistry 1978, 17, 4389–4395. [Google Scholar] [CrossRef] [PubMed]
- Kühn, H.; Hall, S.; Wilden, U. Light-induced binding of 48-kDa protein to photoreceptor membranes is highly enhanced by phosphorylation of rhodopsin. FEBS Lett. 1984, 176, 473–478. [Google Scholar] [CrossRef]
- Hamm, H.E.; Bownds, M.D. Protein complement of rod outer segments of the frog retina. Biochemistry 1986, 25, 4512–4523. [Google Scholar] [CrossRef] [PubMed]
- Wacker, W.B.; Donoso, L.A.; Kalsow, C.M.; Yankeelov, J.J.A.; Organisciak, D.T. Experimental Allergic Uveitis. Isolation, Characterization, and Localization of a Soluble Uveitopathogenic Antigen from Bovine Retina. J. Immunol. 1977, 119, 1949–1958. [Google Scholar] [CrossRef]
- Wacker, W.B.; Lipton, M.M. Experimental Allergic Uveitis: Homologous Retina as Uveitogenic Antigen. Nature 1965, 206, 253–254. [Google Scholar] [CrossRef]
- Wilden, U.; Hall, S.W.; Kühn, H. Phosphodiesterase activation by photoexcited rhodopsin is quenched when rhodopsin is phosphorylated and binds the intrinsic 48-kDa protein of rod outer segments. Proc. Natl. Acad. Sci. USA 1986, 83, 1174–1178. [Google Scholar] [CrossRef] [PubMed]
- Pfister, C.; Chabre, M.; Plouet, J.; Tuyen, V.V.; De Kozak, Y.; Faure, J.P.; Kühn, H. Retinal S Antigen Identified as the 48K Protein Regulating Light-Dependent Phosphodiesterase in Rods. Science 1985, 228, 891–893. [Google Scholar] [CrossRef]
- Pfister, C.; Dorey, C.; Vadot, E.; Mirshahi, M.; Deterre, P.; Chabre, M.; Faure, J.P. Identification of the so-called 48 K protein that interacts with illuminated rhodopsin in retinal rods, and the retinal S antigen, inductor of experimental autoimmune uveoretinitis. C. R. Acad. Sci. Ser. III 1984, 299, 261–265. [Google Scholar]
- Zuckerman, R.; Cheasty, J.E. A 48 kDa protein arrests cGMP phosphodiesterase activation in retinal rod disk membranes. FEBS Lett. 1986, 207, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, R.; Cheasty, J.E. Sites of arrestin action during the quench phenomenon in retinal rods. FEBS Lett. 1988, 238, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Huppertz, B.; Weyand, I.; Bauer, P.J. Ca2+ binding capacity of cytoplasmic proteins from rod photoreceptors is mainly due to arrestin. J. Biol. Chem. 1990, 265, 9470–9475. [Google Scholar] [CrossRef] [PubMed]
- Palczewski, K.; Hargrave, P.A. Studies of ligand binding to arrestin. J. Biol. Chem. 1991, 266, 4201–4206. [Google Scholar] [CrossRef] [PubMed]
- Yamaki, K.; Takahashi, Y.; Sakuragi, S.; Matsubara, K. Molecular cloning of the S-antigen cDNA from bovine retina. Biochem. Biophys. Res. Commun. 1987, 142, 904–910. [Google Scholar] [CrossRef]
- Shinohara, T.; Dietzschold, B.; Craft, C.M.; Wistow, G.; Early, J.J.; Donoso, L.A.; Horwitz, J.; Tao, R. Primary and sec-ondary structure of bovine retinal S antigen (48 kDa protein). Proc. Natl. Acad. Sci. USA 1987, 84, 6975–6979. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.; Syed, M.; Bugra, K.; Whelan, J.; McGinnis, J.; Shinohara, T. Structural analysis of mouse S-antigen. Gene 1988, 73, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Noel, J.P.; Hamm, H.E.; Sigler, P.B. The 2.2 A crystal structure of transducin-alpha complexed with GTP gamma S. Nature 1993, 366, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Sondek, J.; Lambright, D.G.; Noel, J.P.; Hamm, H.E.; Sigler, P.B. GTPase mechanism of Gproteins from the 1.7-A crystal structure of transducin alpha-GDP-AIF-4. Nature 1994, 372, 276–279. [Google Scholar] [CrossRef]
- Hirsch, J.A.; Schubert, C.; Gurevich, V.V.; Sigler, P.B. The 2.8 A crystal structure of visual arrestin: A model for arrestin’s regulation. Cell 1999, 97, 257–269. [Google Scholar] [CrossRef]
- Granzin, J.; Wilden, U.; Choe, H.-W.; Labahn, J.; Krafft, B.; Büldt, G. X-ray crystal structure of arrestin from bovine rod outer segments. Nature 1998, 391, 918–921. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, K.P.; Lamb, T.D. Rhodopsin, light-sensor of vision. Prog. Retin. Eye Res. 2023, 93, 101116. [Google Scholar] [CrossRef] [PubMed]
- Broekhuyse, R.M.; Tolhuizen, E.F.J.; Janssen, A.P.M.; Winkens, H.J. Light induced shift and binding of S-antigen in retinal rods. Curr. Eye Res. 1985, 4, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Philp, N.J.; Chang, W.; Long, K. Light-stimulated protein movement in rod photoreceptor cells of the rat retina. FEBS Lett. 1987, 225, 127–132. [Google Scholar] [CrossRef]
- Whelan, J.P.; McGinnis, J.F. Light-dependent subcellular movement of photoreceptor proteins. J. Neurosci. Res. 1988, 20, 263–270. [Google Scholar] [CrossRef]
- Nair, K.S.; Hanson, S.M.; Mendez, A.; Gurevich, E.V.; Kennedy, M.J.; Shestopalov, V.I.; Vishnivetskiy, S.A.; Chen, J.; Hurley, J.B.; Gurevich, V.V.; et al. Light-dependent redistribution of arrestin in vertebrate rods is an ener-gy-independent process governed by protein-protein interactions. Neuron 2005, 46, 555–567. [Google Scholar] [CrossRef]
- Rosenzweig, D.H.; Nair, K.S.; Wei, J.; Wang, Q.; Garwin, G.; Saari, J.C.; Chen, C.-K.; Smrcka, A.V.; Swaroop, A.; Lem, J.; et al. Subunit Dissociation and Diffusion Determine the Subcellular Localization of Rod and Cone Transducins. J. Neurosci. 2007, 27, 5484–5494. [Google Scholar] [CrossRef] [PubMed]
- Slepak, V.Z.; Hurley, J.B. Mechanism of light-induced translocation of arrestin and transducin in photoreceptors: Inter-action-restricted diffusion. IUBMB Life 2008, 60, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Hanson, S.M.; Song, X.; Vishnivetskiy, S.A.; Gurevich, E.V. The functional cycle of visual arrestins in photoreceptor cells. Prog. Retin. Eye Res. 2011, 30, 405–430. [Google Scholar] [CrossRef]
- Dixon, R.A.; Kobilka, B.K.; Strader, D.J.; Benovic, J.L.; Dohlman, H.G.; Frielle, T.; Bolanowski, M.A.; Bennett, C.D.; Rands, E.; Diehl, R.E.; et al. Cloning of the gene and cDNA for mammalian beta-adrenergic receptor and homology with rhodopsin. Nature 1986, 321, 75–79. [Google Scholar] [CrossRef]
- Ovchinnikov, Y. Rhodopsin and bacteriorhodopsin: Structure—Function relationships. FEBS Lett. 1982, 148, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.G.; Schiöth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef]
- Shichi, H.; Somers, R. Light-dependent phosphorylation of rhodopsin. Purification and properties of rhodopsin kinase. J. Biol. Chem. 1978, 253, 7040–7046. [Google Scholar] [CrossRef] [PubMed]
- Benovic, J.L.; DeBlasi, A.; Stone, W.C.; Caron, M.G.; Lefkowitz, R.J. Beta-adrenergic receptor kinase: Primary structure delineates a multigene family. Science 1989, 246, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, W.; Inglese, J.; Palczewski, K.; Onorato, J.J.; Caron, M.G.; Lefkowitz, R.J. The receptor kinase family: Primary structure of rhodopsin kinase reveals similarities to the beta-adrenergic receptor kinase. Proc. Natl. Acad. Sci. USA 1991, 88, 8715–8719. [Google Scholar] [CrossRef] [PubMed]
- Benovic, J.L.; Kühn, H.; Weyand, I.; Codina, J.; Caron, M.G.; Lefkowitz, R.J. Functional desensitization of the isolated β-adrenergic receptor by the β-adrenergic receptor kinase: Potential role of an analog of the retinal protein arrestin (48 kDa protein). Proc. Natl. Acad. Sci. USA 1987, 84, 8879–8882. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.J.; Benovic, J.L.; Codina, J.; Caron, M.G.; Lefkowitz, R.J. beta-Arrestin: A protein that regulates beta-adrenergic receptor function. Science 1990, 248, 1547–1550. [Google Scholar] [CrossRef]
- Lohse, M.; Andexinger, S.; Pitcher, J.; Trukawinski, S.; Codina, J.; Faure, J.; Caron, M.; Lefkowitz, R. Receptor-specific desensitization with purified proteins. Kinase dependence and receptor specificity of beta-arrestin and arrestin in the beta 2-adrenergic receptor and rhodopsin systems. J. Biol. Chem. 1992, 267, 8558–8564. [Google Scholar] [CrossRef]
- Craft, C.; Whitmore, D.; Wiechmann, A. Cone arrestin identified by targeting expression of a functional family. J. Biol. Chem. 1994, 269, 4613–4619. [Google Scholar] [CrossRef]
- Murakami, A.; Yajima, T.; Sakuma, H.; McLaren, M.J.; Inana, G. X-Arrestin: A new retinal arrestin mapping to the X chromosome. FEBS Lett. 1993, 334, 203–209. [Google Scholar] [CrossRef]
- Chan, S.; Rubin, W.W.; Mendez, A.; Liu, X.; Song, X.; Hanson, S.M.; Craft, C.M.; Gurevich, V.V.; Burns, M.E.; Chen, J. Functional Comparisons of Visual Arrestins in Rod Photoreceptors of Transgenic Mice. Investig. Opthalmology Vis. Sci. 2007, 48, 1968–1975. [Google Scholar] [CrossRef] [PubMed]
- Nikonov, S.S.; Brown, B.M.; Davis, J.A.; Zuniga, F.I.; Bragin, A.; Pugh, E.N.; Craft, C.M. Mouse Cones Require an Arrestin for Normal Inactivation of Phototransduction. Neuron 2008, 59, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Attramadal, H.; Arriza, J.; Aoki, C.; Dawson, T.; Codina, J.; Kwatra, M.; Snyder, S.; Caron, M.; Lefkowitz, R. Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family. J. Biol. Chem. 1992, 267, 17882–17890. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, B.; Kaufman, K.; Chazenbalk, G. Cloning of a member of the arrestin family from a human thyroid cDNA library. Mol. Cell. Endocrinol. 1992, 84, R39–R43. [Google Scholar] [CrossRef] [PubMed]
- Barak, L.S.; Ferguson, S.S.; Zhang, J.; Caron, M.G. A beta-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J. Biol. Chem. 1997, 272, 27497–27500. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Dion, S.B.; Onorato, J.J.; Ptasienski, J.; Kim, C.M.; Sterne-Marr, R.; Hosey, M.M.; Benovic, J.L. Arrestin interaction with G protein-coupled receptors. Direct binding studies of wild type and mutant arrestins with rhodopsin, b2-adrenergic, and m2 muscarinic cholinergic receptors. J. Biol. Chem. 1995, 270, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.; Richardson, R.; Kim, C.; Hosey, M.; Benovic, J. Binding of wild type and chimeric arrestins to the m2 muscarinic cholinergic receptor. J. Biol. Chem. 1993, 268, 16879–16882. [Google Scholar] [CrossRef] [PubMed]
- Sterne-Marr, R.; Gurevich, V.V.; Goldsmith, P.; Bodine, R.C.; Sanders, C.; Donoso, L.A.; Benovic, J.L. Polypeptide var-iants of beta-arrestin and arrestin3. J. Biol. Chem. 1993, 268, 15640–15648. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Gurevich, V.V.; Vishnivetskiy, S.A.; Sigler, P.B.; Schubert, C. Crystal structure of beta-arrestin at 1.9 A: Possible mechanism of receptor binding and membrane translocation. Structure 2001, 9, 869–880. [Google Scholar] [CrossRef]
- Milano, S.K.; Pace, H.C.; Kim, Y.-M.; Brenner, C.; Benovic, J.L. Scaffolding Functions of Arrestin-2 Revealed by Crystal Structure and Mutagenesis. Biochemistry 2002, 41, 3321–3328. [Google Scholar] [CrossRef]
- Sutton, R.B.; Vishnivetskiy, S.A.; Robert, J.; Hanson, S.M.; Raman, D.; Knox, B.E.; Kono, M.; Navarro, J.; Gurevich, V.V. Crystal Structure of Cone Arrestin at 2.3Å: Evolution of Receptor Specificity. J. Mol. Biol. 2005, 354, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Gimenez, L.E.; Gurevich, V.V.; Spiller, B.W. Crystal Structure of Arrestin-3 Reveals the Basis of the Difference in Receptor Binding Between Two Non-visual Subtypes. J. Mol. Biol. 2011, 406, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.; Van Eps, N.; Eger, B.T.; Rauscher, S.; Yedidi, R.S.; Moroni, T.; West, G.M.; Robinson, K.A.; Griffin, P.R.; Mitchell, J.; et al. A Novel Polar Core and Weakly Fixed C-Tail in Squid Arrestin Provide New Insight into In-teraction with Rhodopsin. J. Mol. Biol. 2018, 430, 4102–4118. [Google Scholar] [CrossRef] [PubMed]
- Sander, C.L.; Luu, J.; Kim, K.; Furkert, D.; Jang, K.; Reichenwallner, J.; Kang, M.; Lee, H.-J.; Eger, B.T.; Choe, H.-W.; et al. Structural evidence for visual arrestin priming via complexation of phosphoinositols. Structure 2021, 30, 263–277.e5. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Rojas, R.; Bonifacino, J.S.; Hurley, J.H. The retromer subunit Vps26 has an arrestin fold and binds Vps35 through its C-terminal domain. Nat. Struct. Mol. Biol. 2006, 13, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.; Chen, C.; Kim, C.; Benovic, J. Visual arrestin binding to rhodopsin. Intramolecular interaction between the basic N. terminus and acidic C terminus of arrestin may regulate binding selectivity. J. Biol. Chem. 1994, 269, 8721–8727. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Gurevich, E.V. Solo vs. Chorus: Monomers and Oligomers of Arrestin Proteins. Int. J. Mol. Sci. 2022, 23, 7253. [Google Scholar] [CrossRef]
- Hanson, S.M.; Vishnivetskiy, S.A.; Hubbell, W.L.; Gurevich, V.V. Opposing Effects of Inositol Hexakisphosphate on Rod Arrestin and Arrestin2 Self-Association. Biochemistry 2007, 47, 1070–1075. [Google Scholar] [CrossRef]
- Kim, M.; Hanson, S.M.; Vishnivetskiy, S.A.; Song, X.; Cleghorn, W.M.; Hubbell, W.L.; Gurevich, V.V. Robust Self-Association Is a Common Feature of Mammalian Visual Arrestin-1. Biochemistry 2011, 50, 2235–2242. [Google Scholar] [CrossRef] [PubMed]
- Hanson, S.M.; Van Eps, N.; Francis, D.J.; Altenbach, C.; Vishnivetskiy, S.A.; Arshavsky, V.Y.; Klug, C.S.; Hubbell, W.L.; Gurevich, V.V. Structure and function of the visual arrestin oligomer. EMBO J. 2007, 26, 1726–1736. [Google Scholar] [CrossRef]
- Hanson, S.M.; Dawson, E.S.; Francis, D.J.; Van Eps, N.; Klug, C.S.; Hubbell, W.L.; Meiler, J.; Gurevich, V.V. A Model for the Solution Structure of the Rod Arrestin Tetramer. Structure 2008, 16, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhuo, Y.; Sharma, P.; Perez, I.; Francis, D.J.; Chakravarthy, S.; Vishnivetskiy, S.A.; Berndt, S.; Hanson, S.M.; Zhan, X.; et al. An Eight Amino Acid Segment Controls Oligomerization and Preferred Conformation of the two Non-visual Arrestins. J. Mol. Biol. 2020, 433, 166790. [Google Scholar] [CrossRef] [PubMed]
- Milano, S.K.; Kim, Y.M.; Stefano, F.P.; Benovic, J.L.; Brenner, C. Nonvisual arrestin oligomerization and cellular local-ization are regulated by inositol hexakisphosphate binding. J. Biol. Chem. 2006, 281, 9812–9823. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Perry, N.A.; Vishnivetskiy, S.A.; Berndt, S.; Gilbert, N.C.; Zhuo, Y.; Singh, P.K.; Tholen, J.; Ohi, M.D.; Gurevich, E.V.; et al. Structural basis of arrestin-3 activation and signaling. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hanson, S.M.; Cleghorn, W.M.; Francis, D.J.; Vishnivetskiy, S.A.; Raman, D.; Song, X.; Nair, K.S.; Slepak, V.Z.; Klug, C.S.; Gurevich, V.V. Arrestin Mobilizes Signaling Proteins to the Cytoskeleton and Redirects their Activity. J. Mol. Biol. 2007, 368, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Vishnivetskiy, S.; Seo, J.; Chen, J.; Gurevich, E.; Gurevich, V. Arrestin-1 expression level in rods: Balancing functional performance and photoreceptor health. Neuroscience 2011, 174, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Schubert, C.; Hirsch, J.A.; Gurevich, V.V.; Engelman, D.M.; Sigler, P.B.; Fleming, K.G. Visual Arrestin Activity May Be Regulated by Self-association. J. Biol. Chem. 1999, 274, 21186–21190. [Google Scholar] [CrossRef] [PubMed]
- Samaranayake, S.; Vishnivetskiy, S.A.; Shores, C.R.; Thibeault, K.C.; Kook, S.; Chen, J.; Burns, M.E.; Gurevich, E.V.; Gurevich, V.V. Biological Role of Arrestin-1 Oligomerization. J. Neurosci. 2020, 40, 8055–8069. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Raman, D.; Gurevich, E.V.; Vishnivetskiy, S.A.; Gurevich, V.V. Visual and Both Non-visual Arrestins in Their “Inactive” Conformation Bind JNK3 and Mdm2 and Relocalize Them from the Nucleus to the Cytoplasm. J. Biol. Chem. 2006, 281, 21491–21499. [Google Scholar] [CrossRef]
- Storez, H.; Scott, M.G.; Issafras, H.; Burtey, A.; Benmerah, A.; Muntaner, O.; Piolot, T.; Tramier, M.; Coppey-Moisan, M.; Bouvier, M.; et al. Homo- and hetero-oligomerization of beta-arrestins in living cells. J. Biol. Chem. 2005, 280, 40210–40215. [Google Scholar] [CrossRef]
- Boularan, C.; Scott, M.G.; Bourougaa, K.; Bellal, M.; Esteve, E.; Thuret, A.; Benmerah, A.; Tramier, M.; Coppey-Moisan, M.; Labbé-Jullié, C.; et al. beta-arrestin 2 oligomerization controls the Mdm2-dependent inhibition of p53. Proc. Natl. Acad. Sci. USA 2007, 104, 18061–18066. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.R.; Baillie, G.S.; Bhari, N.; Houslay, T.M.; Pitt, A.M.; Adams, D.R.; Kolch, W.; Houslay, M.D.; Milligan, G. Mu-tations of beta-arrestin 2 that limit self-association also interfere with interactions with the beta2-adrenoceptor and the ERK1/2 MAPKs: Implications for beta2-adrenoceptor signalling via the ERK1/2 MAPKs. Biochem. J. 2008, 413, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.A.; Liu, T.; Fang, C.C.; Castaño, M.A.; Kee, T.; Yrigoin, K.; Yan, Y.; Cazzaro, S.; Matlack, J.; Wang, X.; et al. β-Arrestin2 oligomers impair the clearance of pathological tau and increase tau aggregates. Proc. Natl. Acad. Sci. USA 2020, 117, 5006–5015. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Gurevich, E.V. Plethora of functions packed into 45 kDa arrestins: Biological implications and possible therapeutic strategies. Cell. Mol. Life Sci. 2019, 76, 4413–4421. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; McClatchy, D.B.; Shukla, A.K.; Zhao, Y.; Chen, M.; Shenoy, S.K.; Yates, J.R.; Lefkowitz, R.J. Functional spe-cialization of beta-arrestin interactions revealed by proteomic analysis. Proc. Natl. Acad. Sci. USA 2007, 104, 12011–12016. [Google Scholar] [CrossRef] [PubMed]
- Wilden, U. Duration and Amplitude of the Light-Induced cGMP Hydrolysis in Vertebrate Photoreceptors Are Regulated by Multiple Phosphorylation of Rhodopsin and by Arrestin Binding. Biochemistry 1995, 34, 1446–1454. [Google Scholar] [CrossRef] [PubMed]
- Krupnick, J.G.; Gurevich, V.V.; Benovic, J.L. Mechanism of quenching of phototransduction. Binding competition be-tween arrestin and transducin for phosphorhodopsin. J. Biol. Chem. 1997, 272, 18125–18131. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.; Choi, H.J.; Rosenbaum, D.M.; Kobilka, T.S.; Thian, F.S.; Edwards, P.C.; Burghammer, M.; Ratnala, V.R.; Sanishvili, R.; Fischetti, R.F.; et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature 2007, 450, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Zhou, X.E.; Gao, X.; He, Y.; Liu, W.; Ishchenko, A.; Barty, A.; White, T.A.; Yefanov, O.; Han, G.W.; et al. Crystal structure of rhodopsin bound to arrestin determined by femtosecond X-ray laser. Nature 2015, 523, 561–567. [Google Scholar] [CrossRef]
- Farrens, D.L.; Altenbach, C.; Yang, K.; Hubbell, W.L.; Khorana, H.G. Requirement of rigid-body motion of transmem-brane helices for light activation of rhodopsin. Science 1996, 274, 768–770. [Google Scholar] [CrossRef]
- Ohguro, H.; Palczewski, K.; Walsh, K.A.; Johnson, R.S. Topographic study of arrestin using differential chemical modi-fications and hydrogen/deuterium exchange. Protein Sci. 1994, 3, 2428–2434. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Benovic, J.L. Visual arrestin binding to rhodopsin: Diverse functional roles of positively charged residues within the phosphorylation-recignition region of arrestin. J. Biol. Chem. 1995, 270, 6010–6016. [Google Scholar] [CrossRef] [PubMed]
- Hanson, S.M.; Gurevich, V.V. The Differential Engagement of Arrestin Surface Charges by the Various Functional Forms of the Receptor. J. Biol. Chem. 2006, 281, 3458–3462. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Benovic, J.L. Visual arrestin interaction with rhodopsin: Sequential multisite binding ensures strict se-lectivity towards light-activated phosphorylated rhodopsin. J. Biol. Chem. 1993, 268, 11628–11638. [Google Scholar] [CrossRef] [PubMed]
- Aydin, Y.; Böttke, T.; Lam, J.H.; Ernicke, S.; Fortmann, A.; Tretbar, M.; Zarzycka, B.; Gurevich, V.V.; Katritch, V.; Coin, I. Structural details of a class B GPCR-arrestin complex revealed by genetically encoded crosslinkers in living cells. Nat. Commun. 2023, 14, 1151. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.; Benovic, J. Cell-free expression of visual arrestin. Truncation mutagenesis identifies multiple domains involved in rhodopsin interaction. J. Biol. Chem. 1992, 267, 21919–21923. [Google Scholar] [CrossRef]
- Zhou, X.E.; He, Y.; de Waal, P.W.; Gao, X.; Kang, Y.; Van Eps, N.; Yin, Y.; Pal, K.; Goswami, D.; White, T.A.; et al. Identification of Phosphorylation Codes for Arrestin Recruitment by G Protein-Coupled Receptors. Cell 2017, 170, 457–469.e13. [Google Scholar] [CrossRef]
- Yin, W.; Li, Z.; Jin, M.; Yin, Y.L.; de Waal, P.W.; Pal, K.; Yin, Y.; Gao, X.; He, Y.; Gao, J.; et al. A complex structure of arrestin-2 bound to a G pro-tein-coupled receptor. Cell Res. 2019, 29, 971–983. [Google Scholar] [CrossRef]
- Staus, D.P.; Hu, H.; Robertson, M.J.; Kleinhenz, A.L.W.; Wingler, L.M.; Capel, W.D.; Latorraca, N.R.; Lefkowitz, R.J.; Skiniotis, G. Structure of the M2 muscarinic receptor–β-arrestin complex in a lipid nanodisc. Nature 2020, 579, 297–302. [Google Scholar] [CrossRef]
- Huang, W.; Masureel, M.; Qu, Q.; Janetzko, J.; Inoue, A.; Kato, H.E.; Robertson, M.J.; Nguyen, K.C.; Glenn, J.S.; Skiniotis, G.; et al. Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature 2020, 579, 303–308. [Google Scholar] [CrossRef]
- Lee, Y.; Warne, T.; Nehmé, R.; Pandey, S.; Dwivedi-Agnihotri, H.; Chaturvedi, M.; Edwards, P.C.; García-Nafría, J.; Leslie, A.G.W.; Shukla, A.K.; et al. Molecular basis of β-arrestin coupling to formoterol-bound β(1)-adrenoceptor. Nature 2020, 583, 862–866. [Google Scholar] [CrossRef] [PubMed]
- Bous, J.; Fouillen, A.; Orcel, H.; Trapani, S.; Cong, X.; Fontanel, S.; Saint-Paul, J.; Lai-Kee-Him, J.; Urbach, S.; Sibille, N.; et al. Structure of the vasopressin hormone-V2 receptor-β-arrestin1 ternary complex. Sci. Adv. 2022, 8, eabo7761. [Google Scholar] [CrossRef]
- Cao, C.; Barros-Álvarez, X.; Zhang, S.; Kim, K.; Dämgen, M.A.; Panova, O.; Suomivuori, C.-M.; Fay, J.F.; Zhong, X.; Krumm, B.E.; et al. Signaling snapshots of a serotonin receptor activated by the prototypical psychedelic LSD. Neuron 2022, 110, 3154–3167.e7. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Schafer, C.T.; Mukherjee, S.; Gustavsson, M.; Agrawal, P.; Yao, X.Q.; Kossiakoff, A.A.; Handel, T.M.; Tesmer, J.J.G. ACKR3-arrestin2/3 complexes reveal molecular consequences of GRK-dependent barcoding. bioRxiv 2023, 2023, 549504. [Google Scholar]
- Liao, Y.-Y.; Zhang, H.; Shen, Q.; Cai, C.; Ding, Y.; Shen, D.-D.; Guo, J.; Qin, J.; Dong, Y.; Zhang, Y.; et al. Snapshot of the cannabinoid receptor 1-arrestin complex unravels the biased signaling mechanism. Cell 2023, 186, 5784–5797.e17. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Gurevich, E.V. The molecular acrobatics of arrestin activation. Trends Pharmacol. Sci. 2004, 25, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Karnam, P.C.; Vishnivetskiy, S.A.; Gurevich, V.V. Structural Basis of Arrestin Selectivity for Active Phosphorylated G Protein-Coupled Receptors. Int. J. Mol. Sci. 2021, 22, 12481. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Benovic, J.L. Mechanism of Phosphorylation-Recognition by Visual Arrestin and the Transition of Arrestin into a High Affinity Binding State. Mol. Pharmacol. 1997, 51, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Celver, J.; Vishnivetskiy, S.A.; Chavkin, C.; Gurevich, V.V. Conservation of the Phosphate-sensitive Elements in the Arrestin Family of Proteins. J. Biol. Chem. 2002, 277, 9043–9048. [Google Scholar] [CrossRef]
- Kovoor, A.; Celver, J.; Abdryashitov, R.I.; Chavkin, C.; Gurevich, V.V. Targeted construction of phosphoryla-tion-independent b-arrestin mutants with constitutive activity in cells. J. Biol. Chem. 1999, 274, 6831–6834. [Google Scholar] [CrossRef]
- Palczewski, K.; Buczyłko, J.; Imami, N.; McDowell, J.; Hargrave, P. Role of the carboxyl-terminal region of arrestin in binding to phosphorylated rhodopsin. J. Biol. Chem. 1991, 266, 15334–15339. [Google Scholar] [CrossRef] [PubMed]
- Ostermaier, M.K.; Peterhans, C.; Jaussi, R.; Deupi, X.; Standfuss, J. Functional map of arrestin-1 at single amino acid resolution. Proc. Natl. Acad. Sci. USA 2014, 111, 1825–1830. [Google Scholar] [CrossRef]
- Schleicher, A.; Kuhn, H.; Hofmann, K.P. Kinetics, binding constant, and activation energy of the 48-kDa pro-tein-rhodopsin complex by extra-metarhodopsin II. Biochemistry 1989, 28, 1770–1775. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.H.; Thomsen, A.R.B.; Cahill, T.J., 3rd; Huang, R.; Huang, L.Y.; Marcink, T.; Clarke, O.B.; Heissel, S.; Masoudi, A.; Ben-Hail, D.; et al. Structure of an endosomal signaling GPCR-G pro-tein-β-arrestin megacomplex. Nat. Struct. Mol. Biol. 2019, 26, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Vishnivetskiy, S.A.; Hosey, M.M.; Benovic, J.L.; Gurevich, V.V. Mapping the arrestin-receptor interface: Structural elements responsible for receptor specificity of arrestin proteins. J. Biol. Chem. 2004, 279, 1262–1268. [Google Scholar] [CrossRef]
- Vishnivetskiy, S.A.; Schubert, C.; Climaco, G.C.; Gurevich, Y.V.; Velez, M.-G.; Gurevich, V.V. An additional phos-phate-binding element in arrestin molecule: Implications for the mechanism of arrestin activation. J. Biol. Chem. 2000, 275, 41049–41057. [Google Scholar] [CrossRef] [PubMed]
- Gray-Keller, M.P.; Detwiler, P.B.; Benovic, J.L.; Gurevich, V.V. Arrestin with a single amino acid sustitution quenches light-activated rhodopsin in a phosphorylation0independent fasion. Biochemistry 1997, 36, 7058–7063. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V. The selectivity of visual arrestin for light-activated phosphorhodopsin is controlled by multiple nonre-dundant mechanisms. J. Biol. Chem. 1998, 273, 15501–15506. [Google Scholar] [CrossRef]
- Vishnivetskiy, S.A.; Huh, E.K.; Gurevich, E.V.; Gurevich, V.V. The finger loop as an activation sensor in arrestin. J. Neurochem. 2020, 157, 1138–1152. [Google Scholar] [CrossRef]
- Vishnivetskiy, S.A.; Huh, E.K.; Karnam, P.C.; Oviedo, S.; Gurevich, E.V.; Gurevich, V.V. The Role of Arrestin-1 Middle Loop in Rhodopsin Binding. Int. J. Mol. Sci. 2022, 23, 13887. [Google Scholar] [CrossRef]
- Vishnivetskiy, S.A.; Paz, C.L.; Schubert, C.; Hirsch, J.A.; Sigler, P.B.; Gurevich, V.V. How does arrestin respond to the phosphorylated state of rhodopsin? J. Biol. Chem. 1999, 274, 11451–11454. [Google Scholar] [CrossRef] [PubMed]
- Vishnivetskiy, S.A.; Weinstein, L.D.; Zheng, C.; Gurevich, E.V.; Gurevich, V.V. Functional Role of Arrestin-1 Residues Interacting with Unphosphorylated Rhodopsin Elements. Int. J. Mol. Sci. 2023, 24, 8903. [Google Scholar] [CrossRef] [PubMed]
- Vishnivetskiy, S.A.; Zheng, C.; May, M.B.; Karnam, P.C.; Gurevich, E.; Gurevich, V.V. Lysine in the lariat loop of arrestins does not serve as phosphate sensor. J. Neurochem. 2020, 156, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Gurevich, E.V.; Gurevich, V.V. The nature of the arrestin x receptor complex determines the ultimate fate of the internalized receptor. J. Biol. Chem. 2003, 278, 11623–11632. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M.D.; Balius, A.M.; Yamaguchi, K.; Freilich, E.R.; Barak, L.S.; Kwatra, M.M. Human substance P receptor lacking the C-terminal domain remains competent to desensitize and internalize. J. Neurochem. 2003, 84, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Jala, V.R.; Shao, W.H.; Haribabu, B. Phosphorylation-independent beta-arrestin translocation and internalization of leukotriene B4 receptors. J. Biol. Chem. 2005, 280, 4880–4887. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.-R.; Kushmerick, C.; Seo, J.B.; Koh, D.-S.; Hille, B. Muscarinic receptor regulates extracellular signal regulated kinase by two modes of arrestin binding. Proc. Natl. Acad. Sci. USA 2017, 114, E5579–E5588. [Google Scholar] [CrossRef] [PubMed]
- Gimenez, L.E.; Kook, S.; Vishnivetskiy, S.A.; Ahmed, M.R.; Gurevich, E.V.; Gurevich, V.V. Role of Receptor-attached Phosphates in Binding of Visual and Non-visual Arrestins to G Protein-coupled Receptors. J. Biol. Chem. 2012, 287, 9028–9040. [Google Scholar] [CrossRef] [PubMed]
- Bentrop, J.; Paulsen, R. Light-modulated ADP-ribosylation, protein phosphorylation and protein binding in isolated fly photoreceptor membranes. Eur. J. Biochem. 1986, 161, 61–67. [Google Scholar] [CrossRef]
- Alloway, P.G.; Dolph, P.J. A role for the light-dependent phosphorylation of visual arrestin. Proc. Natl. Acad. Sci. USA 1999, 96, 6072–6077. [Google Scholar] [CrossRef]
- Satoh, A.K.; Xia, H.; Yan, L.; Liu, C.-H.; Hardie, R.C.; Ready, D.F. Arrestin Translocation Is Stoichiometric to Rhodopsin Isomerization and Accelerated by Phototransduction in Drosophila Photoreceptors. Neuron 2010, 67, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Weis, W.I.; Kobilka, B.K. The Molecular Basis of G Protein–Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef]
- Chen, Q.; Plasencia, M.; Li, Z.; Mukherjee, S.; Patra, D.; Chen, C.-L.; Klose, T.; Yao, X.-Q.; Kossiakoff, A.A.; Chang, L.; et al. Structures of rhodopsin in complex with G-protein-coupled receptor kinase 1. Nature 2021, 595, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Gurevich, E.V. The structural basis of arrestin-mediated regulation of G-protein-coupled receptors. Pharmacol. Ther. 2006, 110, 465–502. [Google Scholar] [CrossRef] [PubMed]
- Peterson, Y.K.; Luttrell, L.M. The Diverse Roles of Arrestin Scaffolds in G Protein–Coupled Receptor Signaling. Pharmacol. Rev. 2017, 69, 256–297. [Google Scholar] [CrossRef] [PubMed]
- Min, K.; Yoon, H.-J.; Park, J.Y.; Baidya, M.; Dwivedi-Agnihotri, H.; Maharana, J.; Chaturvedi, M.; Chung, K.Y.; Shukla, A.K.; Lee, H.H. Crystal Structure of β-Arrestin 2 in Complex with CXCR7 Phosphopeptide. Structure 2020, 28, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Pals-Rylaarsdam, R.; Benovic, J.L.; Hosey, M.M.; Onorato, J.J. Agonist-Receptor-Arrestin, an Alternative Ternary Complex with High Agonist Affinity. J. Biol. Chem. 1997, 272, 28849–28852. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Weinstein, L.D.; Nguyen, K.K.; Grewal, A.; Gurevich, E.V.; Gurevich, V.V. GPCR Binding and JNK3 Activation by Arrestin-3 Have Different Structural Requirements. Cells 2023, 12, 1563. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.K.; Manglik, A.; Kruse, A.C.; Xiao, K.; Reis, R.I.; Tseng, W.C.; Staus, D.P.; Hilger, D.; Uysal, S.; Huang, L.Y.; et al. Structure of active beta-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature 2013, 497, 137–141. [Google Scholar] [CrossRef]
- Gurevich, E.V.; Gurevich, V.V. Arrestins are ubiquitous regulators of cellular signaling pathways. Genome Biol. 2006, 7, 236. [Google Scholar] [CrossRef]
- Sente, A.; Peer, R.; Srivastava, A.; Baidya, M.; Lesk, A.M.; Balaji, S.; Shukla, A.K.; Babu, M.M.; Flock, T. Molecular mechanism of modulating arrestin conformation by GPCR phosphorylation. Nat. Struct. Mol. Biol. 2018, 25, 541. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Gurevich, V.V.; Preninger, A.; Hamm, H.E.; Bader, M.-F.; Fazleabas, A.T.; Birnbaumer, L.; Hunzicker-Dunn, M. Aspartic Acid 564 in the Third Cytoplasmic Loop of the Luteinizing Hormone/Choriogonadotropin Receptor Is Crucial for Phosphorylation-independent Interaction with Arrestin2. J. Biol. Chem. 2002, 277, 17916–17927. [Google Scholar] [CrossRef]
- Dolph, P.J.; Ranganathan, R.; Colley, N.J.; Hardy, R.W.; Socolich, M.; Zuker, C.S. Arrestin Function in Inactivation of G Protein-Coupled Receptor Rhodopsin in Vivo. Science 1993, 260, 1910–1916. [Google Scholar] [CrossRef]
- Palczewski, K.; Pulvermüller, A.; Buczyłko, J.; Hofmann, K. Phosphorylated rhodopsin and heparin induce similar conformational changes in arrestin. J. Biol. Chem. 1991, 266, 18649–18654. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.; Milam, A.; Dugger, D.; Arendt, A.; Hargrave, P.; Palczewski, K. A splice variant of arrestin. Molecular cloning and localization in bovine retina. J. Biol. Chem. 1994, 269, 15407–15410. [Google Scholar] [CrossRef] [PubMed]
- Pulvermüller, A.; Maretzki, D.; Rudnicka-Nawrot, M.; Smith, W.C.; Palczewski, K.; Hofmann, K.P. Functional Differences in the Interaction of Arrestin and Its Splice Variant, p44, with Rhodopsin. Biochemistry 1997, 36, 9253–9260. [Google Scholar] [CrossRef] [PubMed]
- Hanson, S.M.; Francis, D.J.; Vishnivetskiy, S.A.; Kolobova, E.A.; Hubbell, W.L.; Klug, C.S.; Gurevich, V.V. Differential interaction of spin-labeled arrestin with inactive and active phosphorhodopsin. Proc. Natl. Acad. Sci. USA 2006, 103, 4900–4905. [Google Scholar] [CrossRef] [PubMed]
- Vishnivetskiy, S.A.; Francis, D.; Van Eps, N.; Kim, M.; Hanson, S.M.; Klug, C.S.; Hubbell, W.L.; Gurevich, V.V. The Role of Arrestin α-Helix I in Receptor Binding. J. Mol. Biol. 2010, 395, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, Y.; Vishnivetskiy, S.A.; Zhan, X.; Gurevich, V.V.; Klug, C.S. Identification of receptor binding-induced confor-mational changes in non-visual arrestins. J. Biol. Chem. 2014, 289, 20991–21002. [Google Scholar] [CrossRef]
- Asher, W.B.; Terry, D.S.; Gregorio, G.G.A.; Kahsai, A.W.; Borgia, A.; Xie, B.; Modak, A.; Zhu, Y.; Jang, W.; Govindaraju, A.; et al. GPCR-mediated β-arrestin activation deconvoluted with single-molecule precision. Cell 2022, 185, 1661–1675.e16. [Google Scholar] [CrossRef]
- Kim, Y.J.; Hofmann, K.P.; Ernst, O.P.; Scheerer, P.; Choe, H.-W.; Sommer, M.E. Crystal structure of pre-activated arrestin p44. Nature 2013, 497, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Vishnivetskiy, S.A.; Van Eps, N.; Alexander, N.S.; Cleghorn, W.M.; Zhan, X.; Hanson, S.M.; Morizumi, T.; Ernst, O.P.; Meiler, J.; et al. Conformation of receptor-bound visual arrestin. Proc. Natl. Acad. Sci. USA 2012, 109, 18407–18412. [Google Scholar] [CrossRef] [PubMed]
- Vishnivetskiy, S.A.; Baameur, F.; Findley, K.R.; Gurevich, V.V. Critical Role of the Central 139-Loop in Stability and Binding Selectivity of Arrestin-1. J. Biol. Chem. 2013, 288, 11741–11750. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Coffa, S.; Fu, H.; Gurevich, V.V. How does arrestin assemble MAP kinases into a signaling complex? J. Biol. Chem. 2009, 284, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Perez, A.; Gimenez, L.E.; Vishnivetskiy, S.A.; Gurevich, V.V. Arrestin-3 binds the MAP kinase JNK3α2 via multiple sites on both domains. Cell. Signal. 2014, 26, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.R.; Zhan, X.; Song, X.; Kook, S.; Gurevich, V.V.; Gurevich, E.V. Ubiquitin Ligase Parkin Promotes Mdm2–Arrestin Interaction but Inhibits Arrestin Ubiquitination. Biochemistry 2011, 50, 3749–3763. [Google Scholar] [CrossRef] [PubMed]
- Lokits, A.D.; Indrischek, H.; Meiler, J.; Hamm, H.E.; Stadler, P.F. Tracing the evolution of the heterotrimeric G protein α subunit in Metazoa. BMC Evol. Biol. 2018, 18, 51. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, E.V.; Tesmer, J.J.; Mushegian, A.; Gurevich, V.V. G protein-coupled receptor kinases: More than just kinases and not only for GPCRs. Pharmacol. Ther. 2012, 133, 40–69. [Google Scholar] [CrossRef]
- Vishnivetskiy, S.A.; Gimenez, L.E.; Francis, D.J.; Hanson, S.M.; Hubbell, W.L.; Klug, C.S.; Gurevich, V.V. Few Residues within an Extensive Binding Interface Drive Receptor Interaction and Determine the Specificity of Arrestin Proteins. J. Biol. Chem. 2011, 286, 24288–24299. [Google Scholar] [CrossRef]
- Gimenez, L.E.; Vishnivetskiy, S.A.; Baameur, F.; Gurevich, V.V. Manipulation of Very Few Receptor Discriminator Residues Greatly Enhances Receptor Specificity of Non-visual Arrestins. J. Biol. Chem. 2012, 287, 29495–29505. [Google Scholar] [CrossRef]
- Gimenez, L.E.; Babilon, S.; Wanka, L.; Beck-Sickinger, A.G.; Gurevich, V.V. Mutations in arrestin-3 differentially affect binding to neuropeptide Y receptor subtypes. Cell. Signal. 2014, 26, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Gimenez, L.E.; Vishnivetskiy, S.A.; Gurevich, V.V. Targeting individual GPCRs with redesigned nonvisual arrestins. Handb. Exp. Pharmacol. 2014, 219, 153–170. [Google Scholar] [PubMed]
- Böttke, T.; Ernicke, S.; Serfling, R.; Ihling, C.; Burda, E.; Gurevich, V.V.; Sinz, A.; Coin, I. Exploring GPCR-arrestin inter-faces with genetically encoded crosslinkers. EMBO Rep. 2020, 21, e50437. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- Nobles, K.N.; Xiao, K.; Ahn, S.; Shukla, A.K.; Lam, C.M.; Rajagopal, S.; Strachan, R.T.; Huang, T.Y.; Bressler, E.A.; Hara, M.R.; et al. Distinct phosphorylation sites on the β(2)-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signal 2011, 4, ra51. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.R.; Reiter, E.; Ahn, S.; Kim, J.; Chen, W.; Lefkowitz, R.J. Different G protein-coupled receptor kinases govern G protein and beta-arrestin mediated signaling of V2 vasopressin receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 1448–1453. [Google Scholar] [CrossRef] [PubMed]
- Eiger, D.S.; Smith, J.S.; Shi, T.; Stepniewski, T.M.; Tsai, C.-F.; Honeycutt, C.; Boldizsar, N.; Gardner, J.; Nicora, C.D.; Moghieb, A.M.; et al. Phosphorylation barcodes direct biased chemokine signaling at CXCR3. Cell Chem. Biol. 2023, 30, 362. [Google Scholar] [CrossRef] [PubMed]
- Latorraca, N.R.; Masureel, M.; Hollingsworth, S.A.; Heydenreich, F.M.; Suomivuori, C.-M.; Brinton, C.; Townshend, R.J.L.; Bouvier, M.; Kobilka, B.K.; Dror, R.O. How GPCR Phosphorylation Patterns Orchestrate Arrestin-Mediated Signaling. Cell 2020, 183, 1813–1825.e18. [Google Scholar] [CrossRef] [PubMed]
- Kaya, A.I.; Perry, N.A.; Gurevich, V.V.; Iverson, T.M. Phosphorylation barcode-dependent signal bias of the dopamine D1 receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 14139–14149. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.-Y.; Lu, S.-S.; Chang, S.-L.; Liao, F. The Phosphorylation of CCR6 on Distinct Ser/Thr Residues in the Carboxyl Terminus Differentially Regulates Biological Function. Front. Immunol. 2018, 9, 415. [Google Scholar] [CrossRef]
- Prihandoko, R.; Bradley, S.J.; Tobin, A.B.; Butcher, A.J. Determination of GPCR Phosphorylation Status: Establishing a Phosphorylation Barcode. Curr. Protoc. Pharmacol. 2015, 69, 2.13.1–2.13.26. [Google Scholar] [CrossRef]
- Vishnivetskiy, S.A.; Raman, D.; Wei, J.; Kennedy, M.J.; Hurley, J.B.; Gurevich, V.V. Regulation of Arrestin Binding by Rhodopsin Phosphorylation Level. J. Biol. Chem. 2007, 282, 32075–32083. [Google Scholar] [CrossRef]
- Mendez, A.; Burns, M.E.; Roca, A.; Lem, J.; Wu, L.W.; Simon, M.I.; Baylor, D.A.; Chen, J. Rapid and reproducible de-activation of rhodopsin requires multiple phosphorylation sites. Neuron 2000, 28, 153–164. [Google Scholar] [CrossRef]
- Gurevich, E.V.; Gurevich, V.V. GRKs as Modulators of Neurotransmitter Receptors. Cells 2020, 10, 52. [Google Scholar] [CrossRef]
- Kim, J.; Ahn, S.; Ren, X.R.; Whalen, E.J.; Reiter, E.; Wei, H.; Lefkowitz, R.J. Functional antagonism of different G pro-tein-coupled receptor kinases for beta-arrestin-mediated angiotensin II receptor signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 1442–1447. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Staus, D.P.; Wingler, L.M.; Ahn, S.; Pani, B.; Capel, W.D.; Lefkowitz, R.J. G protein-coupled receptor kinases (GRKs) orchestrate biased agonism at the β2-adrenergic receptor. Sci. Signal 2018, 11, 544. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.S.; Downey, W.E., 3rd; Colapietro, A.M.; Barak, L.S.; Ménard, L.; Caron, M.G. Role of beta-arrestin in medi-ating agonist-promoted G protein-coupled receptor internalization. Science 1996, 271, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Goodman, O.B., Jr.; Krupnick, J.G.; Santini, F.; Gurevich, V.V.; Penn, R.B.; Gagnon, A.W.; Keen, J.H.; Benovic, J.L. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature 1996, 383, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Laporte, S.A.; Oakley, R.H.; Zhang, J.; Holt, J.A.; Ferguson, S.S.G.; Caron, M.G.; Barak, L.S. The β2-adrenergic recep-tor/arrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc. Natl. Acad. Sci. USA 1999, 96, 3712–3717. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Benovic, J.L. Differential Roles of Arrestin-2 Interaction with Clathrin and Adaptor Protein 2 in G Protein-coupled Receptor Trafficking. J. Biol. Chem. 2002, 277, 30760–30768. [Google Scholar] [CrossRef]
- Ferguson, S.S.; Barak, L.S.; Zhang, J.; Caron, M.G. G-protein-coupled receptor regulation: Role of G-protein-coupled receptor kinases and arrestins. Can. J. Physiol. Pharmacol. 1996, 74, 1095–1110. [Google Scholar] [CrossRef] [PubMed]
- Orsini, M.J.; Benovic, J.L. Characterization of dominant negative arrestins that inhibit beta2-adrenergic receptor inter-nalization by distinct mechanisms. J. Biol. Chem. 1998, 273, 34616–34622. [Google Scholar] [CrossRef] [PubMed]
- Cleghorn, W.M.; Branch, K.M.; Kook, S.; Arnette, C.; Bulus, N.; Zent, R.; Kaverina, I.; Gurevich, E.V.; Weaver, A.M.; Gurevich, V.V. Arrestins regulate cell spreading and motility via focal adhesion dynamics. Mol. Biol. Cell 2015, 26, 622–635. [Google Scholar] [CrossRef]
- Lee, K.B.; Ptasienski, J.A.; Pals-Rylaarsdam, R.; Gurevich, V.V.; Hosey, M.M. Arrestin Binding to the M2 Muscarinic Acetylcholine Receptor Is Precluded by an Inhibitory Element in the Third Intracellular Loop of the Receptor. J. Biol. Chem. 2000, 275, 9284–9289. [Google Scholar] [CrossRef] [PubMed]
- Roseberry, A.G.; Hosey, M.M. Internalization of the M2 muscarinic acetylcholine receptor proceeds through an atypical pathway in HEK293 cells that is independent of clathrin and caveolae. J. Cell Sci. 2001, 114, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Pals-Rylaarsdam, R.; Gurevich, V.V.; Lee, K.B.; Ptasienski, J.; Benovic, J.L.; Hosey, M.M. Internalization of the m2 muscarinic acetylcholine receptor: Arrestin-independent and -dependent pathways. J. Biol. Chem. 1997, 272, 23682–23689. [Google Scholar] [CrossRef] [PubMed]
- Key, T.A.; Foutz, T.D.; Gurevich, V.V.; Sklar, L.A.; Prossnitz, E.R. N-Formyl Peptide Receptor Phosphorylation Domains Differentially Regulate Arrestin and Agonist Affinity. J. Biol. Chem. 2003, 278, 4041–4047. [Google Scholar] [CrossRef]
- Bennett, T.A.; Maestas, D.C.; Prossnitz, E.R. Arrestin Binding to the G Protein-coupled N-Formyl Peptide Receptor Is Regulated by the Conserved “DRY” Sequence. J. Biol. Chem. 2000, 275, 24590–24594. [Google Scholar] [CrossRef] [PubMed]
- Vines, C.M.; Revankar, C.M.; Maestas, D.C.; LaRusch, L.L.; Cimino, D.F.; Kohout, T.A.; Lefkowitz, R.J.; Prossnitz, E.R. N-Formyl Peptide Receptors Internalize but Do Not Recycle in the Absence of Arrestins. J. Biol. Chem. 2003, 278, 41581–41584. [Google Scholar] [CrossRef]
- Berthouze, M.; Venkataramanan, V.; Li, Y.; Shenoy, S.K. The deubiquitinases USP33 and USP20 coordinate beta2 adrenergic receptor recycling and resensitization. EMBO J. 2009, 28, 1684–1696. [Google Scholar] [CrossRef]
- Shenoy, S.K.; McDonald, P.H.; Kohout, T.A.; Lefkowitz, R.J. Regulation of receptor fate by ubiquitination of activated beta 2-adrenergic receptor and beta-arrestin. Science 2001, 294, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Gurevich, E.V. The New Face of Active Receptor Bound Arrestin Attracts New Partners. Structure 2003, 11, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Ferguson, S.S.; Daaka, Y.; Miller, W.E.; Maudsley, S.; Della Rocca, G.J.; Lin, F.; Kawakatsu, H.; Owada, K.; Luttrell, D.K.; et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science 1999, 283, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Roudabush, F.L.; Choy, E.W.; Miller, W.E.; Field, M.E.; Pierce, K.L.; Lefkowitz, R.J. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc. Natl. Acad. Sci. USA 2001, 98, 2449–2454. [Google Scholar] [CrossRef] [PubMed]
- McDonald, P.H.; Chow, C.W.; Miller, W.E.; Laporte, S.A.; Field, M.E.; Lin, F.T.; Davis, R.J.; Lefkowitz, R.J. Beta-arrestin 2: A receptor-regulated MAPK scaffold for the activation of JNK3. Science 2000, 290, 1574–1577. [Google Scholar] [CrossRef]
- DeFea, K.A.; Vaughn, Z.D.; O’Bryan, E.M.; Nishijima, D.; Déry, O.; Bunnett, N.W. The proliferative and antiapoptotic effects of substance P are facilitated by formation of a beta -arrestin-dependent scaffolding complex. Proc. Natl. Acad. Sci. USA 2000, 97, 11086–11091. [Google Scholar] [CrossRef] [PubMed]
- DeFea, K.A.; Zalevsky, J.; Thoma, M.S.; Déry, O.; Mullins, R.D.; Bunnett, N.W. beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell. Biol. 2000, 148, 1267–1281. [Google Scholar] [CrossRef]
- DeWire, S.M.; Ahn, S.; Lefkowitz, R.J.; Shenoy, S.K. Beta-arrestins and cell signaling. Annu. Rev. Physiol. 2007, 69, 483–510. [Google Scholar] [CrossRef]
- Wu, N.; Hanson, S.M.; Francis, D.J.; Vishnivetskiy, S.A.; Thibonnier, M.; Klug, C.S.; Shoham, M.; Gurevich, V.V. Arrestin Binding to Calmodulin: A Direct Interaction Between Two Ubiquitous Signaling Proteins. J. Mol. Biol. 2006, 364, 955–963. [Google Scholar] [CrossRef]
- Breitman, M.; Kook, S.; Gimenez, L.E.; Lizama, B.N.; Palazzo, M.C.; Gurevich, E.V.; Gurevich, V.V. Silent scaffolds: Inhibition of c-Jun N-terminal kinase 3 activity in the cell by a dominant-negative arrestin-3 mutant. J. Biol. Chem. 2012, 287, 19653–19664. [Google Scholar] [CrossRef]
- Song, X.; Gurevich, E.V.; Gurevich, V.V. Cone arrestin binding to JNK3 and Mdm2: Conformational preference and localization of interaction sites. J. Neurochem. 2007, 103, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-P.; Brown, B.M.; Craft, C.M. Visual Arrestin 1 Acts as a Modulator for N-Ethylmaleimide-Sensitive Factor in the Photoreceptor Synapse. J. Neurosci. 2010, 30, 9381–9391. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.S.; Simpson, C.; Dyka, F.M.; Dinculescu, A.; Smith, W.C. A Modified Arrestin1 Increases Lactate Production in the Retina and Slows Retinal Degeneration. Hum. Gene Ther. 2022, 33, 695–707. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.C.; Bolch, S.; Dugger, D.R.; Li, J.; Esquenazi, I.; Arendt, A.; Benzenhafer, D.; McDowell, J.H. Interaction of Arrestin with Enolase1 in Photoreceptors. Investig. Opthalmology Vis. Sci. 2011, 52, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- Moaven, H.; Koike, Y.; Jao, C.C.; Gurevich, V.V.; Langen, R.; Chen, J. Visual arrestin interaction with clathrin adaptor AP-2 regulates photoreceptor survival in the vertebrate retina. Proc. Natl. Acad. Sci. USA 2013, 110, 9463–9468. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, E.; Benovic, J.; Gurevich, V. Arrestin2 and arrestin3 are differentially expressed in the rat brain during postnatal development. Neuroscience 2002, 109, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, E.V.; Benovic, J.L.; Gurevich, V.V. Arrestin2 expression selectively increases during neural differentiation. J. Neurochem. 2004, 91, 1404–1416. [Google Scholar] [CrossRef] [PubMed]
- Hanson, S.M.; Francis, D.J.; Vishnivetskiy, S.A.; Klug, C.S.; Gurevich, V.V. Visual Arrestin Binding to Microtubules Involves a Distinct Conformational Change. J. Biol. Chem. 2006, 281, 9765–9772. [Google Scholar] [CrossRef] [PubMed]
- Vishnivetskiy, S.A.; Hirsch, J.A.; Velez, M.-G.; Gurevich, Y.V.; Gurevich, V.V. Transition of arrestin in the active re-ceptor-binding state requires an extended interdomain hinge. J. Biol. Chem. 2002, 277, 43961–43968. [Google Scholar] [CrossRef]
- Bayburt, T.H.; Vishnivetskiy, S.A.; McLean, M.; Morizumi, T.; Huang, C.-c.; Tesmer, J.J.; Ernst, O.P.; Sligar, S.G.; Gurevich, V.V. Rhodopsin monomer is sufficient for normal rhodopsin kinase (GRK1) phosphorylation and arrestin-1 binding. J. Biol. Chem. 2011, 286, 1420–1428. [Google Scholar] [CrossRef]
- Widmann, C.; Gibson, S.; Jarpe, M.B.; Johnson, G.L. Mitogen-Activated Protein Kinase: Conservation of a Three-Kinase Module from Yeast to Human. Physiol. Rev. 1999, 79, 143–180. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L. Defining MAPK Interactomes. ACS Chem. Biol. 2011, 6, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Coffey, E.T. Nuclear and cytosolic JNK signalling in neurons. Nat. Rev. Neurosci. 2014, 15, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Levchenko, A.; Bruck, J.; Sternberg, P.W. Scaffold proteins may biphasically affect the levels of mitogen-activated protein kinase signaling and reduce its threshold properties. Proc. Natl. Acad. Sci. USA 2000, 97, 5818–5823. [Google Scholar] [CrossRef] [PubMed]
- Dickens, M.; Rogers, J.S.; Cavanagh, J.; Raitano, A.; Xia, Z.; Halpern, J.R.; Greenberg, M.E.; Sawyers, C.L.; Davis, R.J. A Cytoplasmic Inhibitor of the JNK Signal Transduction Pathway. Science 1997, 277, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.E.; McDonald, P.H.; Cai, S.F.; Field, M.E.; Davis, R.J.; Lefkowitz, R.J. Identification of a motif in the carboxyl terminus of beta -arrestin2 responsible for activation of JNK3. J. Biol. Chem. 2001, 276, 27770–27777. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Kaoud, T.S.; Dalby, K.N.; Gurevich, V.V. Nonvisual arrestins function as simple scaffolds assembling the MKK4-JNK3alpha2 signaling complex. Biochemistry 2011, 50, 10520–10529. [Google Scholar] [CrossRef]
- Zhan, X.; Kaoud, T.S.; Kook, S.; Dalby, K.N.; Gurevich, V.V. JNK3 Enzyme Binding to Arrestin-3 Differentially Affects the Recruitment of Upstream Mitogen-activated Protein (MAP) Kinase Kinases. J. Biol. Chem. 2013, 288, 28535–28547. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Tsakem, E.L.; Breitman, M.; Gurevich, V.V. Identification of arrestin-3-specific residues necessary for JNK3 acti-vation. J. Biol. Chem. 2011, 286, 27894–27901. [Google Scholar] [CrossRef]
- Zhan, X.; Stoy, H.; Kaoud, T.S.; Perry, N.A.; Chen, Q.; Perez, A.; Els-Heindl, S.; Slagis, J.V.; Iverson, T.M.; Beck-Sickinger, A.G.; et al. Peptide mini-scaffold facilitates JNK3 activation in cells. Sci. Rep. 2016, 6, 21025. [Google Scholar] [CrossRef]
- Perry-Hauser, N.A.; Kaoud, T.S.; Stoy, H.; Zhan, X.; Chen, Q.; Dalby, K.N.; Iverson, T.M.; Gurevich, V.V.; Gurevich, E.V. Short Arrestin-3-Derived Peptides Activate JNK3 in Cells. Int. J. Mol. Sci. 2022, 23, 8679. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Lynch, M.J.; Huston, E.; Beyermann, M.; Eichhorst, J.; Adams, D.R.; Klusmann, E.; Houslay, M.D.; Baillie, G.S. MEK1 binds directly to betaarrestin1, influencing both its phosphorylation by ERK and the timing of its iso-prenaline-stimulated internalization. J. Biol. Chem. 2009, 284, 11425–11435. [Google Scholar] [CrossRef] [PubMed]
- Coffa, S.; Breitman, M.; Hanson, S.M.; Callaway, K.; Kook, S.; Dalby, K.N.; Gurevich, V.V. The Effect of Arrestin Con-formation on the Recruitment of c-Raf1, MEK1, and ERK1/2 Activation. PLoS ONE 2011, 6, e28723. [Google Scholar] [CrossRef]
- Naor, Z.; Benard, O.; Seger, R. Activation of MAPK cascades by G-protein-coupled receptors: The case of gonadotro-pin-releasing hormone receptor. Trends Endocrinol. Metab. 2000, 11, 91–99. [Google Scholar] [CrossRef]
- Zheng, K.; Smith, J.S.; Eiger, D.S.; Warman, A.; Choi, I.; Honeycutt, C.C.; Boldizsar, N.; Gundry, J.N.; Pack, T.F.; Inoue, A.; et al. Biased agonists of the chemokine receptor CXCR3 differentially signal through Gαi:β-arrestin complexes. Sci. Signal 2022, 15, eabg5203. [Google Scholar] [CrossRef] [PubMed]
- O’Hayre, M.; Eichel, K.; Avino, S.; Zhao, X.; Steffen, D.J.; Feng, X.; Kawakami, K.; Aoki, J.; Messer, K.; Sunahara, R.; et al. Genetic evidence that β-arrestins are dispensable for the initiation of β2-adrenergic receptor signaling to ERK. Sci. Signal 2017, 10, 484. [Google Scholar] [CrossRef]
- Luttrell, L. ‘Location, location, location’: Activation and targeting of MAP kinases by G protein-coupled receptors. J. Mol. Endocrinol. 2003, 30, 117–126. [Google Scholar] [CrossRef]
- Grundmann, M.; Merten, N.; Malfacini, D.; Inoue, A.; Preis, P.; Simon, K.; Rüttiger, N.; Ziegler, N.; Benkel, T.; Schmitt, N.K.; et al. Lack of beta-arrestin signaling in the absence of active G proteins. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Curto, E.; Inoue, A.; Jenkins, L.; Raihan, S.Z.; Prihandoko, R.; Tobin, A.B.; Milligan, G. Targeted Elimination of G Proteins and Arrestins Defines Their Specific Contributions to Both Intensity and Duration of G Protein-coupled Receptor Signaling. J. Biol. Chem. 2016, 291, 27147–27159. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Wang, J.; Plouffe, B.; Smith, J.S.; Yamani, L.; Kaur, S.; Jean-Charles, P.-Y.; Gauthier, C.; Lee, M.-H.; Pani, B.; et al. Manifold roles of beta-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci. Signal 2018, 11, eaat7650. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E.V. Arrestins and G proteins in cellular signaling: The coin has two sides. Sci. Signal. 2018, 11, eaav1646. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Gurevich, E.V. Arrestin-mediated signaling: Is there a controversy? World J. Biol. Chem. 2018, 9, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.; Kahsai, A.W.; Pakharukova, N.; Huang, L.-Y.; Lefkowitz, R.J. The GPCR–β-arrestin complex allosterically activates C-Raf by binding its amino terminus. J. Biol. Chem. 2021, 297, 101369. [Google Scholar] [CrossRef] [PubMed]
- Kahsai, A.W.; Shah, K.S.; Shim, P.J.; Lee, M.A.; Shreiber, B.N.; Schwalb, A.M.; Zhang, X.; Kwon, H.Y.; Huang, L.Y.; Soderblom, E.J.; et al. Signal transduction at GPCRs: Allosteric activation of the ERK MAPK by β-arrestin. Proc. Natl. Acad. Sci. USA 2023, 120, e2303794120. [Google Scholar] [CrossRef] [PubMed]
- Bruchas, M.R.; Macey, T.A.; Lowe, J.D.; Chavkin, C. Kappa Opioid Receptor Activation of p38 MAPK Is GRK3- and Arrestin-dependent in Neurons and Astrocytes. J. Biol. Chem. 2006, 281, 18081–18089. [Google Scholar] [CrossRef] [PubMed]
- Berndt, S.; Liebscher, I. New Structural Perspectives in G Protein-Coupled Receptor-Mediated Src Family Kinase Activa-tion. Int. J. Mol. Sci. 2021, 22, 6489. [Google Scholar] [CrossRef] [PubMed]
- Barlic, J.; Andrews, J.D.; Kelvin, A.A.; Bosinger, S.E.; DeVries, M.E.; Xu, L.; Dobransky, T.; Feldman, R.D.; Ferguson, S.S.; Kelvin, D.J. Regulation of tyrosine kinase activation and granule release through beta-arrestin by CXCRI. Nat. Immunol. 2000, 1, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Huang, J.; Dalle, S.; Ugi, S.; Usui, I.; Luttrell, L.M.; Miller, W.E.; Lefkowitz, R.J.; Olefsky, J.M. beta-Arrestin-mediated recruitment of the Src family kinase Yes mediates endothelin-1-stimulated glucose transport. J. Biol. Chem. 2001, 276, 43663–43667. [Google Scholar] [CrossRef] [PubMed]
- Galet, C.; Ascoli, M. Arrestin-3 is essential for the activation of Fyn by the luteinizing hormone receptor (LHR) in MA-10 cells. Cell. Signal. 2008, 20, 1822–1829. [Google Scholar] [CrossRef]
- Cheung, R.; Malik, M.; Ravyn, V.; Tomkowicz, B.; Ptasznik, A.; Collman, R.G. An arrestin-dependent multi-kinase sig-naling complex mediates MIP-1beta/CCL4 signaling and chemotaxis of primary human macrophages. J. Leukoc. Biol. 2009, 86, 833–845. [Google Scholar] [CrossRef]
- Miller, W.E.; Maudsley, S.; Ahn, S.; Khan, K.D.; Luttrell, L.M.; Lefkowitz, R.J. beta-arrestin1 interacts with the catalytic domain of the tyrosine kinase c-SRC. Role of beta-arrestin1-dependent targeting of c-SRC in receptor endocytosis. J. Biol. Chem. 2000, 275, 11312–11319. [Google Scholar] [CrossRef] [PubMed]
- Pakharukova, N.; Masoudi, A.; Pani, B.; Staus, D.P.; Lefkowitz, R.J. Allosteric activation of proto-oncogene kinase Src by GPCR–beta-arrestin complexes. J. Biol. Chem. 2020, 295, 16773–16784. [Google Scholar] [CrossRef] [PubMed]
- Perez, I.; Berndt, S.; Agarwal, R.; Castro, M.A.; Vishnivetskiy, S.A.; Smith, J.C.; Sanders, C.R.; Gurevich, V.V.; Iverson, T. A Model for the Signal Initiation Complex Between Arrestin-3 and the Src Family Kinase Fgr. J. Mol. Biol. 2021, 434, 167400. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Doshi, A.; Lei, M.; Eck, M.J.; Harrison, S.C. Crystal Structures of c-Src Reveal Features of Its Autoinhibitory Mechanism. Mol. Cell 1999, 3, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K.; Xiao, K.; Venkataramanan, V.; Snyder, P.M.; Freedman, N.J.; Weissman, A.M. Nedd4 mediates ago-nist-dependent ubiquitination, lysosomal targeting, and degradation of the beta2-adrenergic receptor. J. Biol. Chem. 2008, 283, 22166–22176. [Google Scholar] [CrossRef] [PubMed]
- Girnita, L.; Shenoy, S.K.; Sehat, B.; Vasilcanu, R.; Girnita, A.; Lefkowitz, R.J.; Larsson, O. [43]-Arrestin is crucial for ubiquitination and down-regulation of the insulin-like growth factor-1 receptor by acting as adaptor for the MDM2 E3 ligase. J. Biol. Chem. 2005, 280, 24412–24419. [Google Scholar] [CrossRef] [PubMed]
- Girnita, L.; Shenoy, S.K.; Sehat, B.; Vasilcanu, R.; Vasilcanu, D.; Girnita, A.; Lefkowitz, R.J.; Larsson, O. Beta-arrestin and Mdm2 mediate IGF-1 receptor-stimulated ERK activation and cell cycle progression. J. Biol. Chem. 2007, 282, 11329–11338. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K.; Modi, A.S.; Shukla, A.K.; Xiao, K.; Berthouze, M.; Ahn, S.; Wilkinson, K.D.; Miller, W.E.; Lefkowitz, R.J. Beta-arrestin-dependent signaling and trafficking of 7-transmembrane receptors is reciprocally regulated by the deubiquitinase USP33 and the E3 ligase Mdm2. Proc. Natl. Acad. Sci. USA 2009, 106, 6650–6655. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, D.; Trejo, J.; Benovic, J.L.; Marchese, A. Arrestin-2 interacts with the ubiquitin-protein isopeptide ligase atro-phin-interacting protein 4 and mediates endosomal sorting of the chemokine receptor CXCR4. J. Biol. Chem. 2007, 282, 36971–36979. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Nguyen, K.K.; Vishnivetskiy, S.A.; Gurevich, V.V.; Gurevich, E.V. Arrestin-3 binds parkin and enhances parkin-dependent mitophagy. J. Neurochem. 2023, in press. [Google Scholar] [CrossRef]
- Kim, S.A.; Heinze, K.G.; Waxham, M.N.; Schwille, P. Intracellular calmodulin availability accessed with two-photon cross-correlation. Proc. Natl. Acad. Sci. USA 2003, 101, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.G.; Le Rouzic, E.; Périanin, A.; Pierotti, V.; Enslen, H.; Benichou, S.; Marullo, S.; Benmerah, A. Differential nu-cleocytoplasmic shuttling of beta-arrestins. Characterization of a leucine-rich nuclear export signal in beta-arrestin2. J. Biol. Chem. 2002, 277, 37693–37701. [Google Scholar] [CrossRef] [PubMed]
- McDonald, P.H.; Cote, N.L.; Lin, F.T.; Premont, R.T.; Pitcher, J.A.; Lefkowitz, R.J. Identification of NSF as a be-ta-arrestin1-binding protein. Implications for beta2-adrenergic receptor regulation. J. Biol. Chem. 1999, 274, 10677–10680. [Google Scholar] [CrossRef] [PubMed]
- Kook, S.; Zhan, X.; Kaoud, T.S.; Dalby, K.N.; Gurevich, V.V.; Gurevich, E.V. Arrestin-3 binds JNK1α1 and JNK2α2 and facilitates the activation of these ubiquitous JNK isoforms in cells via scaffolding. J. Biol. Chem. 2014, 289, 37332–37342. [Google Scholar]
- Zhuang, T.; Chen, Q.; Cho, M.-K.; Vishnivetskiy, S.A.; Iverson, T.M.; Gurevich, V.V.; Sanders, C.R. Involvement of distinct arrestin-1 elements in binding to different functional forms of rhodopsin. Proc. Natl. Acad. Sci. USA 2012, 110, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Gilbert, N.C.; Perry, N.A.; Zhuo, Y.; Vishnivetskiy, S.A.; Berndt, S.; Klug, C.S.; Gurevich, V.V.; Iverson, T.M. Structural basis for arrestin-3 activation by inositol hexakisphosphate. Cell, 2016; in review. [Google Scholar]
- Gurevich, V.V.; Gurevich, E.V. Extensive shape shifting underlies functional versatility of arrestins. Curr. Opin. Cell Biol. 2014, 27, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Nuber, S.; Zabel, U.; Lorenz, K.; Nuber, A.; Milligan, G.; Tobin, A.B.; Lohse, M.J.; Hoffmann, C. β-Arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature 2016, 531, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-H.; Appleton, K.M.; Strungs, E.G.; Kwon, J.Y.; Morinelli, T.A.; Peterson, Y.K.; Laporte, S.A.; Luttrell, L.M. The conformational signature of β-arrestin2 predicts its trafficking and signalling functions. Nature 2016, 531, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Tobin, A.B.; Butcher, A.J.; Kong, K.C. Location, location, location...site-specific GPCR phosphorylation offers a mecha-nism for cell-type-specific signalling. Trends Pharmacol. Sci. 2008, 29, 413–420. [Google Scholar] [CrossRef]
- Lau, E.K.; Trester-Zedlitz, M.; Trinidad, J.C.; Kotowski, S.J.; Krutchinsky, A.N.; Burlingame, A.L.; von Zastrow, M. Quantitative Encoding of the Effect of a Partial Agonist on Individual Opioid Receptors by Multisite Phosphorylation and Threshold Detection. Sci. Signal. 2011, 4, ra52. [Google Scholar] [CrossRef]
- Yang, F.; Yu, X.; Liu, C.; Qu, C.-X.; Gong, Z.; Liu, H.-D.; Li, F.-H.; Wang, H.-M.; He, D.-F.; Yi, F.; et al. Phospho-selective mechanisms of arrestin conformations and functions revealed by unnatural amino acid incorporation and 19F-NMR. Nat. Commun. 2015, 6, 8202. [Google Scholar] [CrossRef] [PubMed]
- Coffa, S.; Breitman, M.; Spiller, B.W.; Gurevich, V.V. A single mutation in arrestin-2 prevents ERK1/2 activation by re-ducing c-Raf1 binding. Biochemistry 2011, 50, 6951–6958. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, Y.; Gurevich, V.V.; Vishnivetskiy, S.A.; Klug, C.S.; Marchese, A. A non–GPCR-binding partner interacts with a novel surface on β-arrestin1 to mediate GPCR signaling. J. Biol. Chem. 2020, 295, 14111–14124. [Google Scholar] [CrossRef] [PubMed]
- Kook, S.; Vishnivetskiy, S.A.; Gurevich, V.V.; Gurevich, E.V. Cleavage of arrestin-3 by caspases attenuates cell death by precluding arrestin-dependent JNK activation. Cell. Signal. 2018, 54, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Kook, S.; Zhan, X.; Cleghorn, W.M.; Benovic, J.L.; Gurevich, V.V. Caspase-cleaved arrestin-2 and BID cooperatively facilitate cytochrome C release and cell death. Cell Death Differ. 2013, 21, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Cleghorn, W.M.; Bulus, N.; Kook, S.; Gurevich, V.V.; Zent, R.; Gurevich, E.V. Non-visual arrestins regulate the focal adhesion formation via small GTPases RhoA and Rac1 independently of GPCRs. Cell. Signal. 2017, 42, 259–269. [Google Scholar] [CrossRef]
- Perry, N.A.; Fialkowski, K.P.; Kaoud, T.S.; Kaya, A.I.; Chen, A.L.; Taliaferro, J.M.; Gurevich, V.V.; Dalby, K.N.; Iverson, T. Arrestin-3 interaction with maternal embryonic leucine-zipper kinase. Cell. Signal. 2019, 63, 109366. [Google Scholar] [CrossRef] [PubMed]
- Vishnivetskiy, S.A.; Chen, Q.; Palazzo, M.C.; Brooks, E.K.; Altenbach, C.; Iverson, T.M.; Hubbell, W.L.; Gurevich, V.V. Engineering Visual Arrestin-1 with Special Functional Characteristics. J. Biol. Chem. 2013, 288, 3394–3405. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Vishnivetskiy, S.A.; Gross, O.P.; Emelianoff, K.; Mendez, A.; Chen, J.; Gurevich, E.V.; Burns, M.E.; Gurevich, V.V. Enhanced arrestin facilitates recovery and protects rods lacking rhodopsin phosphorylation. Curr. Biol. 2009, 19, 700–705. [Google Scholar] [CrossRef]
- Samaranayake, S.; Song, X.; Vishnivetskiy, S.A.; Chen, J.; Gurevich, E.V.; Gurevich, V.V. Enhanced Mutant Compensates for Defects in Rhodopsin Phosphorylation in the Presence of Endogenous Arrestin-1. Front. Mol. Neurosci. 2018, 11, 203. [Google Scholar] [CrossRef]
- Song, X.; Seo, J.; Baameur, F.; Vishnivetskiy, S.A.; Chen, Q.; Kook, S.; Kim, M.; Brooks, E.K.; Altenbach, C.; Hong, Y.; et al. Rapid degeneration of rod photoreceptors expressing self-association-deficient arrestin-1 mutant. Cell. Signal. 2013, 25, 2613–2624. [Google Scholar] [CrossRef] [PubMed]
- Krupnick, J.G.; Santini, F.; Gagnon, A.W.; Keen, J.H.; Benovic, J.L. Modulation of the arrestin-clathrin interaction in cells. Characterization of beta-arrestin dominant-negative mutants. J. Biol. Chem. 1997, 272, 32507–32512. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.M.; Gao, E.; Chuprun, J.K.; Koch, W.J. GRK2 in the Heart: A GPCR Kinase and Beyond. Antioxid. Redox Signal. 2014, 21, 2032–2043. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurevich, V.V. Arrestins: A Small Family of Multi-Functional Proteins. Int. J. Mol. Sci. 2024, 25, 6284. https://doi.org/10.3390/ijms25116284
Gurevich VV. Arrestins: A Small Family of Multi-Functional Proteins. International Journal of Molecular Sciences. 2024; 25(11):6284. https://doi.org/10.3390/ijms25116284
Chicago/Turabian StyleGurevich, Vsevolod V. 2024. "Arrestins: A Small Family of Multi-Functional Proteins" International Journal of Molecular Sciences 25, no. 11: 6284. https://doi.org/10.3390/ijms25116284
APA StyleGurevich, V. V. (2024). Arrestins: A Small Family of Multi-Functional Proteins. International Journal of Molecular Sciences, 25(11), 6284. https://doi.org/10.3390/ijms25116284