
Small Molecule Therapeutics in the Pipeline Targeting for Triple-Negative Breast Cancer: Origin, Challenges, Opportunities, and Mechanisms of Action


Abstract
:1. Introduction
2. Histological and Molecular Characterization of Triple-Negative Breast Cancer
2.1. Histological Classification
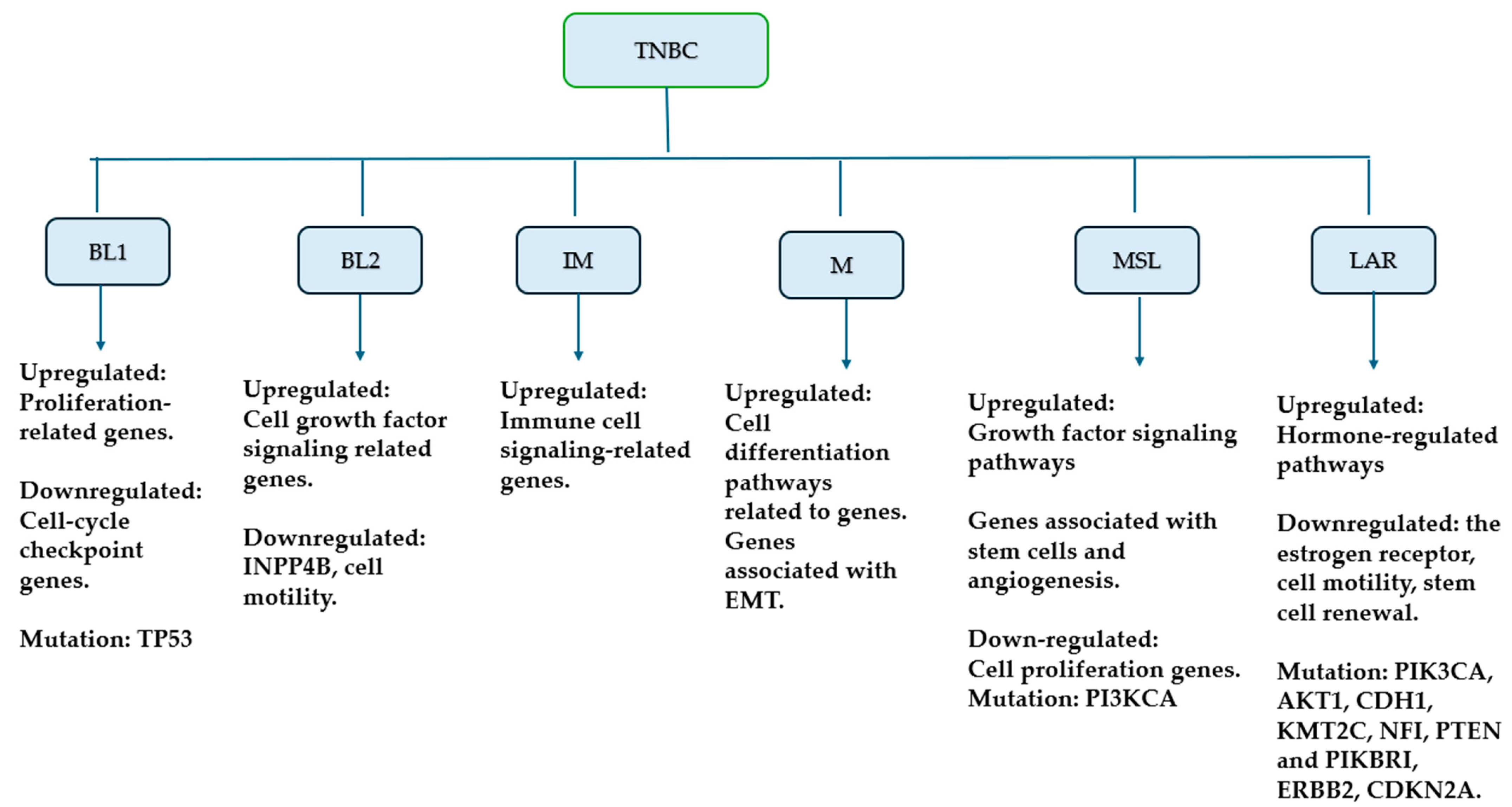
2.2. Molecular Classification
3. Signaling Pathways in TNBC
3.1. Include Targeting PI3K/AKT/mTOR Signaling Pathway
3.1.1. Buparlisib (BKM120, Norvatis)
3.1.2. Ipatasertib
3.1.3. Capivasertib (AZD5363)
3.1.4. Everolimus (RAD001)
3.2. Targeting Vascular Endothelial Growth Factor (VEGF) Pathway
3.2.1. Vandetanib
3.2.2. Apatinib (N-[4-(1-Cyano-cyclopentyl) Phenyl]-2-(4-pyridlmethyl)amino-3-pyridine Carboxamide)
3.3. Targeting Poly (ADP-Ribose) Polymerase (PARP) Pathway
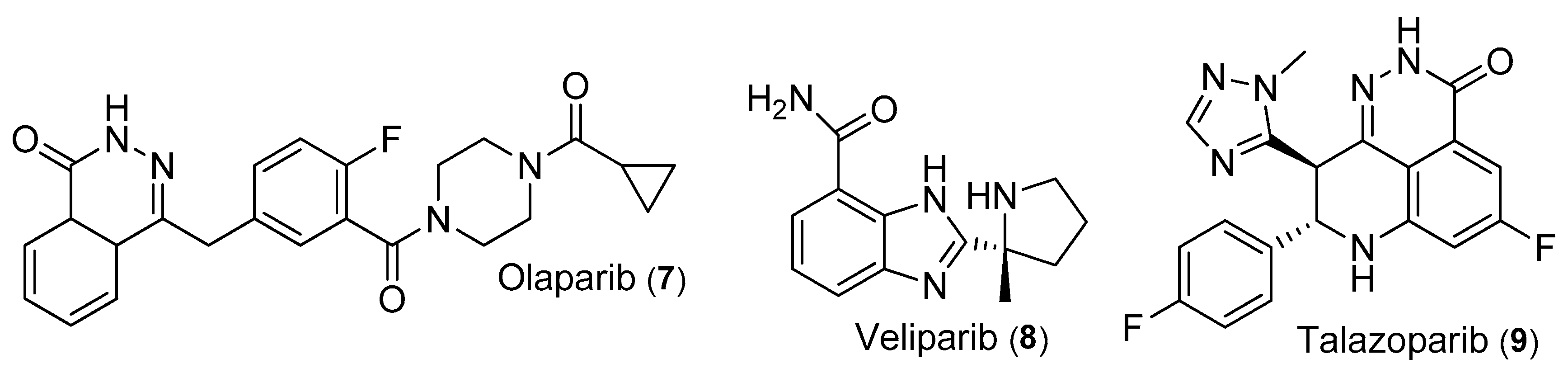
3.3.1. Olaparib
3.3.2. Veliparib (ABT-888)
3.3.3. Talazoparib
3.4. Targeting Janus Kinases (JAKs)/Signal Transducer and Activator of Transcription 3 (STAT3) Pathway
3.4.1. Ruxolitinib
3.4.2. LLL12B
3.4.3. FLU
3.4.4. Salinomycin
3.5. Targeting Mitogen-Activated Protein Kinases (MAPK) Pathway
3.5.1. E6201
3.5.2. Cobimetinib
3.5.3. Nifetepimine
3.5.4. BL-EI001
3.6. Targeting Epidermal Growth Factor Receptor (EGFR) Pathway
3.6.1. Cannabidiol (CBD)
3.6.2. Varlitinib (ASLAN001)
3.6.3. Salidroside (p-Hydroxyphenethyl-β-d-glucoside)
3.6.4. Vandetanib
3.7. Targeting Src Pathway
3.7.1. Dasatinib (BMS-354825)
3.7.2. BJ-2302
3.7.3. Compound 1j (24)
3.8. Targeting the E-Cadherin Expression
3.9. Combination Therapy with Small Molecules in TNBC
4. Challenges and Prospects
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Thilagavathi, R.; Priyankha, S.; Kannan, M.; Prakash, M.; Selvam, C. Compounds from Diverse Natural Origin against Triple-Negative Breast Cancer: A Comprehensive Review. Chem. Biol. Drug Des. 2023, 101, 218–243. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, D. Farmer to Pharmacist: Curcumin as an Anti-Invasive and Antimetastatic Agent for the Treatment of Cancer1. Front. Chem. 2014, 2, 113. [Google Scholar] [CrossRef]
- Taborda Ribas, H.; Sogayar, M.C.; Dolga, A.M.; Winnischofer, S.M.B.; Trombetta-Lima, M. Lipid Profile in Breast Cancer: From Signaling Pathways to Treatment Strategies. Biochimie 2024, 219, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Tarighati, E.; Keivan, H.; Mahani, H. A Review of Prognostic and Predictive Biomarkers in Breast Cancer. Clin. Exp. Med. 2023, 23, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zubair, M.; Wang, S.; Ali, N. Advanced Approaches to Breast Cancer Classification and Diagnosis. Front. Pharmacol. 2021, 11, 632079. [Google Scholar] [CrossRef]
- Misra, G.; Qaisar, S.; Singh, P. CRISPR-Based Therapeutic Targeting of Signaling Pathways in Breast Cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 166872. [Google Scholar] [CrossRef]
- Nguyen, H.-M.; Paulishak, W.; Oladejo, M.; Wood, L. Dynamic Tumor Microenvironment, Molecular Heterogeneity, and Distinct Immunologic Portrait of Triple-Negative Breast Cancer: An Impact on Classification and Treatment Approaches. Breast Cancer Tokyo Jpn. 2023, 30, 167–186. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-Q.; Liu, J.; Zhao, S.-Q.; Zhu, K.; Gong, Z.-Q.; Xu, R.; Lu, H.-M.; Zhou, R.-B.; Zhao, G.; Yin, D.-C.; et al. Recent Treatment Progress of Triple Negative Breast Cancer. Prog. Biophys. Mol. Biol. 2020, 151, 40–53. [Google Scholar] [CrossRef]
- Chai, C.; Wu, H.H.; Abuetabh, Y.; Sergi, C.; Leng, R. Regulation of the Tumor Suppressor PTEN in Triple-Negative Breast Cancer. Cancer Lett. 2022, 527, 41–48. [Google Scholar] [CrossRef]
- Kong, X.; Qi, Y.; Wang, X.; Jiang, R.; Wang, J.; Fang, Y.; Gao, J.; Chu Hwang, K. Nanoparticle Drug Delivery Systems and Their Applications as Targeted Therapies for Triple Negative Breast Cancer. Prog. Mater. Sci. 2023, 134, 101070. [Google Scholar] [CrossRef]
- Liao, M.; Zhang, J.; Wang, G.; Wang, L.; Liu, J.; Ouyang, L.; Liu, B. Small-Molecule Drug Discovery in Triple Negative Breast Cancer: Current Situation and Future Directions. J. Med. Chem. 2021, 64, 2382–2418. [Google Scholar] [CrossRef] [PubMed]
- Nagayama, A.; Vidula, N.; Ellisen, L.; Bardia, A. Novel Antibody-Drug Conjugates for Triple Negative Breast Cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920915980. [Google Scholar] [CrossRef] [PubMed]
- Fraguas-Sánchez, A.I.; Fernández-Carballido, A.; Simancas-Herbada, R.; Martin-Sabroso, C.; Torres-Suárez, A.I. CBD Loaded Microparticles as a Potential Formulation to Improve Paclitaxel and Doxorubicin-Based Chemotherapy in Breast Cancer. Int. J. Pharm. 2020, 574, 118916. [Google Scholar] [CrossRef] [PubMed]
- Keenan, T.E.; Tolaney, S.M. Role of Immunotherapy in Triple-Negative Breast Cancer–PubMed. J. Natl. Compr. Cancer Netw. 2020, 18, 479–489. [Google Scholar] [CrossRef]
- Mediratta, K.; El-Sahli, S.; D’Costa, V.; Wang, L. Current Progresses and Challenges of Immunotherapy in Triple-Negative Breast Cancer. Cancers 2020, 12, 3529. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, X. Classification of Triple-Negative Breast Cancer Based on Pathway Enrichment Levels. Med. Oncol. Northwood Lond. Engl. 2023, 40, 157. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small Molecules in Targeted Cancer Therapy: Advances, Challenges, and Future Perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Mahgoub, R.E.; Atatreh, N.; Ghattas, M.A. Chapter Three–Using Filters in Virtual Screening: A Comprehensive Guide to Minimize Errors and Maximize Efficiency. In Annual Reports in Medicinal Chemistry; Caballero, J., Ed.; Virtual Screening and Drug Docking; Academic Press: New York, NY, USA, 2022; Volume 59, pp. 99–136. [Google Scholar]
- Zubair, T.; Bandyopadhyay, D. Small Molecule EGFR Inhibitors as Anti-Cancer Agents: Discovery, Mechanisms of Action, and Opportunities. Int. J. Mol. Sci. 2023, 24, 2651. [Google Scholar] [CrossRef] [PubMed]
- Shetu, S.A.; James, N.; Rivera, G.; Bandyopadhyay, D. Molecular Research in Pancreatic Cancer: Small Molecule Inhibitors, Their Mechanistic Pathways and Beyond. Curr. Issues Mol. Biol. 2023, 45, 1914–1949. [Google Scholar] [CrossRef] [PubMed]
- Shetu, S.A.; Bandyopadhyay, D. Small-Molecule RAS Inhibitors as Anticancer Agents: Discovery, Development, and Mechanistic Studies. Int. J. Mol. Sci. 2022, 23, 3706. [Google Scholar] [CrossRef] [PubMed]
- Rock, J.; Garcia, D.; Espino, O.; Shetu, S.A.; Chan-Bacab, M.J.; Moo-Puc, R.; Patel, N.B.; Rivera, G.; Bandyopadhyay, D. Benzopyrazine-Based Small Molecule Inhibitors As Trypanocidal and Leishmanicidal Agents: Green Synthesis, In Vitro, and In Silico Evaluations. Front. Chem. 2021, 9, 725892. [Google Scholar] [CrossRef] [PubMed]
- Popovic, L.S.; Matovina-Brko, G.; Popovic, M.; Punie, K.; Cvetanovic, A.; Lambertini, M. Targeting Triple-Negative Breast Cancer: A Clinical Perspective. Oncol. Res. 2023, 31, 221–238. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.M.H.; Mueller, K.; Gartner, E.; Boerner, J. Dasatinib Is Synergistic with Cetuximab and Cisplatin in Triple-Negative Breast Cancer Cells. J. Surg. Res. 2013, 185, 231–239. [Google Scholar] [CrossRef]
- Stecklein, S.R.; Barlow, W.; Pusztai, L.; Timms, K.; Kennedy, R.; Logan, G.E.; Seitz, R.; Badve, S.; Gökmen-Polar, Y.; Porter, P.; et al. Dual Prognostic Classification of Triple-Negative Breast Cancer by DNA Damage Immune Response and Homologous Recombination Deficiency. JCO Precis. Oncol. 2023, 7, e2300197. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Collet, L.; Rediti, M.; Debien, V.; De Caluwé, A.; Venet, D.; Romano, E.; Rothé, F.; Sotiriou, C.; Buisseret, L. Predictive Biomarkers for Response to Immunotherapy in Triple Negative Breast Cancer: Promises and Challenges. J. Clin. Med. 2023, 12, 953. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, S.; Inubushi, S.; Miki, M.; Nakamura, H.; Baba, M.; Yamashita, Y.; Yamamoto, M.; Inoue, S.; Tanino, H.; Kunihisa, T. Target-Oriented Classification of Triple-Negative Breast Cancer. Anticancer Res. 2023, 43, 5067–5072. [Google Scholar] [CrossRef]
- Zhao, S.; Zuo, W.-J.; Shao, Z.-M.; Jiang, Y.-Z. Molecular Subtypes and Precision Treatment of Triple-Negative Breast Cancer. Ann. Transl. Med. 2020, 8, 499. [Google Scholar] [CrossRef]
- Kashyap, D.; Bal, A.; Irinike, S.; Khare, S.; Bhattacharya, S.; Das, A.; Singh, G. Heterogeneity of the Tumor Microenvironment Across Molecular Subtypes of Breast Cancer. Appl. Immunohistochem. Mol. Morphol. 2023, 31, 533–543. [Google Scholar] [CrossRef]
- Zagami, P.; Carey, L.A. Triple Negative Breast Cancer: Pitfalls and Progress. NPJ Breast Cancer 2022, 8, 95. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, H.; Merkher, Y.; Chen, L.; Liu, N.; Leonov, S.; Chen, Y. Recent Advances in Therapeutic Strategies for Triple-Negative Breast Cancer. J. Hematol. Oncol. 2022, 15, 121. [Google Scholar] [CrossRef]
- Brewster, A.M.; Chavez-MacGregor, M.; Brown, P. Epidemiology, Biology, and Treatment of Triple-Negative Breast Cancer in Women of African Ancestry. Lancet Oncol. 2014, 15, e625–e634. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, Y.; Xue, J.; Li, J.; Yi, J.; Bu, J.; Zhang, Z.; Qiu, P.; Gu, X. Advances in Immunotherapy for Triple-Negative Breast Cancer. Mol. Cancer 2023, 22, 145. [Google Scholar] [CrossRef]
- Chen, L.; Zhimin, S.; Wang, Z.; Yang, W.; Jiang, Y.; Wu, S.; Wu, J.; Di, G.; Liu, G.; Yu, K.; et al. The Overall Survival Analysis of FUTURE-C-PLUS: Combination of Famitinib with Camrelizumab plus Nab-Paclitaxel as First-Line Treatment for Advanced, Immunomodulatory Triple-Negative Breast Cancer—An Open-Label, Single-Arm, Phase 2 Trial. J. Clin. Oncol. 2023, 41, 1086. [Google Scholar] [CrossRef]
- Thompson, K.J.; Leon-Ferre, R.A.; Sinnwell, J.P.; Zahrieh, D.M.; Suman, V.J.; Metzger, F.O.; Asad, S.; Stover, D.G.; Carey, L.; Sikov, W.M.; et al. Luminal Androgen Receptor Breast Cancer Subtype and Investigation of the Microenvironment and Neoadjuvant Chemotherapy Response. NAR Cancer 2022, 4, zcac018. [Google Scholar] [CrossRef]
- Asemota, S.; Effah, W.; Young, K.L.; Holt, J.; Cripe, L.; Ponnusamy, S.; Thiyagarajan, T.; Hwang, D.-J.; He, Y.; Mcnamara, K.; et al. Identification of a Targetable JAK-STAT Enriched Androgen Receptor and Androgen Receptor Splice Variant Positive Triple-Negative Breast Cancer Subtype. Cell Rep. 2023, 42, 113461. [Google Scholar] [CrossRef]
- Ellis, H.; Ma, C.X. PI3K Inhibitors in Breast Cancer Therapy. Curr. Oncol. Rep. 2019, 21, 110. [Google Scholar] [CrossRef]
- Cerma, K.; Piacentini, F.; Moscetti, L.; Barbolini, M.; Canino, F.; Tornincasa, A.; Caggia, F.; Cerri, S.; Molinaro, A.; Dominici, M.; et al. Targeting PI3K/AKT/mTOR Pathway in Breast Cancer: From Biology to Clinical Challenges. Biomedicines 2023, 11, 109. [Google Scholar] [CrossRef]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR Signaling Transduction Pathway and Targeted Therapies in Cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Murphy, C.J.; Karreth, F.A.; Emdal, K.B.; White, F.M.; Elemento, O.; Toker, A.; Wulf, G.M.; Cantley, L.C. Identifying and Targeting Sporadic Oncogenic Genetic Aberrations in Mouse Models of Triple-Negative Breast Cancer. Cancer Discov. 2018, 8, 354–369. [Google Scholar] [CrossRef]
- Yuan, Y.; Yost, S.E.; Cui, Y.; Ruel, C.; Murga, M.; Tang, A.; Martinez, N.; Schmolze, D.; Waisman, J.; Patel, N.; et al. Phase I Trial of Ipatasertib Plus Carboplatin, Carboplatin/Paclitaxel, or Capecitabine and Atezolizumab in Metastatic Triple-Negative Breast Cancer. Oncologist 2023, 28, e498–e507. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Abraham, J.; Chan, S.; Wheatley, D.; Brunt, A.M.; Nemsadze, G.; Baird, R.D.; Park, Y.H.; Hall, P.S.; Perren, T.; et al. Capivasertib Plus Paclitaxel Versus Placebo Plus Paclitaxel As First-Line Therapy for Metastatic Triple-Negative Breast Cancer: The PAKT Trial. J. Clin. Oncol. 2020, 38, 423–433. [Google Scholar] [CrossRef]
- Garrido-Castro, A.C.; Saura, C.; Barroso-Sousa, R.; Guo, H.; Ciruelos, E.; Bermejo, B.; Gavilá, J.; Serra, V.; Prat, A.; Paré, L.; et al. Phase 2 Study of Buparlisib (BKM120), a Pan-Class I PI3K Inhibitor, in Patients with Metastatic Triple-Negative Breast Cancer. Breast Cancer Res. 2020, 22, 120. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.L.B.; Han, H.S.; Gradishar, W.J. Targeting the PI3K/AKT/mTOR Pathway in Triple-Negative Breast Cancer: A Review. Breast Cancer Res. Treat. 2018, 169, 397–406. [Google Scholar] [CrossRef]
- Martín, M.; Chan, A.; Dirix, L.; O’Shaughnessy, J.; Hegg, R.; Manikhas, A.; Shtivelband, M.; Krivorotko, P.; Batista López, N.; Campone, M.; et al. A Randomized Adaptive Phase II/III Study of Buparlisib, a Pan-Class I PI3K Inhibitor, Combined with Paclitaxel for the Treatment of HER2– Advanced Breast Cancer (BELLE-4). Ann. Oncol. 2017, 28, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Criscitiello, C.; Viale, G.; Curigliano, G.; Goldhirsch, A. Profile of Buparlisib and Its Potential in the Treatment of Breast Cancer: Evidence to Date. Breast Cancer Targets Ther. 2018, 10, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Wongchenko, M.J.; Kim, S.-B.; Saura, C.; Oliveira, M.; Lipson, D.; Kennedy, M.; Greene, M.; Breese, V.; Mani, A.; Xu, N.; et al. Circulating Tumor DNA and Biomarker Analyses From the LOTUS Randomized Trial of First-Line Ipatasertib and Paclitaxel for Metastatic Triple-Negative Breast Cancer. JCO Precis. Oncol. 2020, 4, 1012–1024. [Google Scholar] [CrossRef]
- Oliveira, M.; Saura, C.; Nuciforo, P.; Calvo, I.; Andersen, J.; Passos-Coelho, J.L.; Gil Gil, M.; Bermejo, B.; Patt, D.A.; Ciruelos, E.; et al. FAIRLANE, a Double-Blind Placebo-Controlled Randomized Phase II Trial of Neoadjuvant Ipatasertib plus Paclitaxel for Early Triple-Negative Breast Cancer. Ann. Oncol. 2019, 30, 1289–1297. [Google Scholar] [CrossRef]
- Andrikopoulou, A.; Chatzinikolaou, S.; Panourgias, E.; Kaparelou, M.; Liontos, M.; Dimopoulos, M.-A.; Zagouri, F. The Emerging Role of Capivasertib in Breast Cancer. Breast 2022, 63, 157–167. [Google Scholar] [CrossRef]
- Royce, M.E.; Osman, D. Everolimus in the Treatment of Metastatic Breast Cancer. Breast Cancer Basic Clin. Res. 2015, 9, BCBCR.S29268. [Google Scholar] [CrossRef] [PubMed]
- Euceda, L.R.; Hill, D.K.; Stokke, E.; Hatem, R.; El Botty, R.; Bièche, I.; Marangoni, E.; Bathen, T.F.; Moestue, S.A. Metabolic Response to Everolimus in Patient-Derived Triple-Negative Breast Cancer Xenografts. J. Proteome Res. 2017, 16, 1868–1879. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Kong, S.-Y.; Kwon, Y.; Kim, M.K.; Sim, S.H.; Joo, J.; Lee, K.S. Phase I/II Clinical Trial of Everolimus Combined with Gemcitabine/Cisplatin for Metastatic Triple-Negative Breast Cancer. J. Cancer 2018, 9, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.C.; Novik, Y.; Stein, S.; Volm, M.; Meyers, M.; Smith, J.; Omene, C.; Speyer, J.; Schneider, R.; Jhaveri, K.; et al. Phase 2 Trial of Everolimus and Carboplatin Combination in Patients with Triple Negative Metastatic Breast Cancer. Breast Cancer Res. 2014, 16, R32. [Google Scholar] [CrossRef]
- Zhu, X.; Zhou, W. The Emerging Regulation of VEGFR-2 in Triple-Negative Breast Cancer. Front. Endocrinol. 2015, 6, 164766. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.L.; Isakoff, S.J.; Klement, G.; Downing, S.R.; Chen, W.Y.; Hannagan, K.; Gelman, R.; Winer, E.P.; Burstein, H.J. Combination Antiangiogenic Therapy in Advanced Breast Cancer: A Phase 1 Trial of Vandetanib, a VEGFR Inhibitor, and Metronomic Chemotherapy, with Correlative Platelet Proteomics. Breast Cancer Res. Treat. 2012, 136, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yu, J.; Jiao, S.; Wang, W.; Zhang, F.; Sun, S. Vandetanib (ZD6474) Induces Antiangiogenesis through mTOR–HIF-1 Alpha–VEGF Signaling Axis in Breast Cancer Cells. OncoTargets Ther. 2018, 11, 8543–8553. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Shi, M.; Wang, Y.; Chen, J.; Ou, Y. Apatinib Enhanced Anti-Tumor Activity of Cisplatin on Triple-Negative Breast Cancer through Inhibition of VEGFR-2. Pathol. Res. Pract. 2019, 215, 152422. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, J.; Xu, B.; Jiang, Z.; Ragaz, J.; Tong, Z.; Zhang, Q.; Wang, X.; Feng, J.; Pang, D.; et al. Multicenter Phase II Study of Apatinib, a Novel VEGFR Inhibitor in Heavily Pretreated Patients with Metastatic Triple-Negative Breast Cancer. Int. J. Cancer 2014, 135, 1961–1969. [Google Scholar] [CrossRef]
- Layman, R.M.; Arun, B. PARP Inhibitors in Triple-Negative Breast Cancer Including Those With BRCA Mutations. Cancer J. 2021, 27, 67–75. [Google Scholar] [CrossRef]
- Thorsell, A.G.; Ekblad, T.; Karlberg, T.; Löw, M.; Pinto, A.F.; Trésaugues, L.; Moche, M.; Cohen, M.S.; Schüler, H. Structural Basis for Potency and Promiscuity in Poly(ADP-ribose) Polymerase (PARP) and Tankyrase Inhibitors. J. Med. Chem. 2017, 60, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Somlo, G.; Frankel, P.H.; Arun, B.K.; Ma, C.X.; Garcia, A.A.; Cigler, T.; Cream, L.V.; Harvey, H.A.; Sparano, J.A.; Nanda, R.; et al. Efficacy of the PARP Inhibitor Veliparib with Carboplatin or as a Single Agent in Patients with Germline BRCA1- or BRCA2-Associated Metastatic Breast Cancer: California Cancer Consortium Trial NCT01149083. Clin. Cancer Res. 2017, 23, 4066–4076. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.; Rugo, H.; Ettl, J.; Hurvitz, S.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.; Martin, M.; et al. Abstract GS6-07: EMBRACA: A Phase 3 Trial Comparing Talazoparib, an Oral PARP Inhibitor, to Physician’s Choice of Therapy in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. Cancer Res. 2018, 78, GS6-07. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Loap, P.; Loirat, D.; Berger, F.; Cao, K.; Ricci, F.; Jochem, A.; Raizonville, L.; Mosseri, V.; Fourquet, A.; Kirova, Y. Combination of Olaparib with Radiotherapy for Triple-Negative Breast Cancers: One-Year Toxicity Report of the RADIOPARP Phase I Trial. Int. J. Cancer 2021, 149, 1828–1832. [Google Scholar] [CrossRef] [PubMed]
- Marijon, H.; Gery, S.; Chang, H.; Landesman, Y.; Shacham, S.; Lee, D.H.; de Gramont, A.; Koeffler, H.P. Selinexor, a Selective Inhibitor of Nuclear Export, Enhances the Anti-Tumor Activity of Olaparib in Triple Negative Breast Cancer Regardless of BRCA1 Mutation Status. Oncotarget 2021, 12, 1749–1762. [Google Scholar] [CrossRef] [PubMed]
- Diéras, V.; Han, H.S.; Kaufman, B.; Wildiers, H.; Friedlander, M.; Ayoub, J.-P.; Puhalla, S.L.; Bondarenko, I.; Campone, M.; Jakobsen, E.H.; et al. Veliparib with Carboplatin and Paclitaxel in BRCA-Mutated Advanced Breast Cancer (BROCADE3): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2020, 21, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
- Kummar, S.; Wade, J.L.; Oza, A.M.; Sullivan, D.; Chen, A.P.; Gandara, D.R.; Ji, J.; Kinders, R.J.; Wang, L.; Allen, D.; et al. Randomized Phase II Trial of Cyclophosphamide and the Oral Poly (ADP-Ribose) Polymerase Inhibitor Veliparib in Patients with Recurrent, Advanced Triple-Negative Breast Cancer. Invest. New Drugs 2016, 34, 355–363. [Google Scholar] [CrossRef]
- Loibl, S.; Sikov, W.; Huober, J.; Rugo, H.S.; Wolmark, N.; O’Shaughnessy, J.; Maag, D.; Untch, M.; Golshan, M.; Lorenzo, J.P.; et al. 119O Event-Free Survival (EFS), Overall Survival (OS), and Safety of Adding Veliparib (V) plus Carboplatin (Cb) or Carboplatin Alone to Neoadjuvant Chemotherapy in Triple-Negative Breast Cancer (TNBC) after ≥4 Years of Follow-up: BrighTNess, a Randomized Phase III Trial. Ann. Oncol. 2021, 32, S408. [Google Scholar] [CrossRef]
- Loibl, S.; O’Shaughnessy, J.; Untch, M.; Sikov, W.M.; Rugo, H.S.; McKee, M.D.; Huober, J.; Golshan, M.; von Minckwitz, G.; Maag, D.; et al. Addition of the PARP Inhibitor Veliparib plus Carboplatin or Carboplatin Alone to Standard Neoadjuvant Chemotherapy in Triple-Negative Breast Cancer (BrighTNess): A Randomised, Phase 3 Trial. Lancet Oncol. 2018, 19, 497–509. [Google Scholar] [CrossRef]
- Sharma, P.; Rodler, E.; Barlow, W.E.; Gralow, J.; Huggins-Puhalla, S.L.; Anders, C.K.; Goldstein, L.J.; Brown-Glaberman, U.A.; Huynh, T.-T.; Szyarto, C.S.; et al. Results of a Phase II Randomized Trial of Cisplatin +/- Veliparib in Metastatic Triple-Negative Breast Cancer (TNBC) and/or Germline BRCA-Associated Breast Cancer (SWOG S1416). J. Clin. Oncol. 2020, 38, 1001. [Google Scholar] [CrossRef]
- de Bono, J.; Ramanathan, R.K.; Mina, L.; Chugh, R.; Glaspy, J.; Rafii, S.; Kaye, S.; Sachdev, J.; Heymach, J.; Smith, D.C.; et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov. 2017, 7, 620–629. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Telli, M.L.; Rugo, H.S.; Mailliez, A.; Ettl, J.; Grischke, E.-M.; Mina, L.A.; Balmaña, J.; Fasching, P.A.; Hurvitz, S.A.; et al. A Phase II Study of Talazoparib after Platinum or Cytotoxic Nonplatinum Regimens in Patients with Advanced Breast Cancer and Germline BRCA1/2 Mutations (ABRAZO). Clin. Cancer Res. 2019, 25, 2717–2724. [Google Scholar] [CrossRef]
- Qin, J.-J.; Yan, L.; Zhang, J.; Zhang, W.-D. STAT3 as a Potential Therapeutic Target in Triple Negative Breast Cancer: A Systematic Review. J. Exp. Clin. Cancer Res. 2019, 38, 195. [Google Scholar] [CrossRef]
- Pan, L.; Chen, X.; Rassool, F.V.; Li, C.; Lin, J. LLL12B, a Novel Small-Molecule STAT3 Inhibitor, Induces Apoptosis and Suppresses Cell Migration and Tumor Growth in Triple-Negative Breast Cancer Cells. Biomedicines 2022, 10, 2003. [Google Scholar] [CrossRef]
- Stover, D.G.; Gil Del Alcazar, C.R.; Brock, J.; Guo, H.; Overmoyer, B.; Balko, J.; Xu, Q.; Bardia, A.; Tolaney, S.M.; Gelman, R.; et al. Phase II Study of Ruxolitinib, a Selective JAK1/2 Inhibitor, in Patients with Metastatic Triple-Negative Breast Cancer. NPJ Breast Cancer 2018, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Kim, J.Y.; Oh, E.; Lee, N.; Cho, Y.; Seo, J.H. Salinomycin Promotes Anoikis and Decreases the CD44+/CD24- Stem-Like Population via Inhibition of STAT3 Activation in MDA-MB-231 Cells. PLoS ONE 2015, 10, e0141919. [Google Scholar] [CrossRef] [PubMed]
- Guvenir Celik, E.; Eroglu, O. Combined Treatment with Ruxolitinib and MK-2206 Inhibits the JAK2/STAT5 and PI3K/AKT Pathways via Apoptosis in MDA-MB-231 Breast Cancer Cell Line. Mol. Biol. Rep. 2023, 50, 319–329. [Google Scholar] [CrossRef]
- Oh, E.; Kim, Y.-J.; An, H.; Sung, D.; Cho, T.-M.; Farrand, L.; Jang, S.; Seo, J.H.; Kim, J.Y. Flubendazole Elicits Anti-Metastatic Effects in Triple-Negative Breast Cancer via STAT3 Inhibition. Int. J. Cancer 2018, 143, 1978–1993. [Google Scholar] [CrossRef]
- Lee, J.; Lim, B.; Pearson, T.; Choi, K.; Fuson, J.A.; Bartholomeusz, C.; Paradiso, L.J.; Myers, T.; Tripathy, D.; Ueno, N.T. Anti-Tumor and Anti-Metastasis Efficacy of E6201, a MEK1 Inhibitor, in Preclinical Models of Triple-Negative Breast Cancer. Breast Cancer Res. Treat. 2019, 175, 339–351. [Google Scholar] [CrossRef]
- Miles, D.; Kim, S.-B.; McNally, V.A.; Simmons, B.P.; Wongchenko, M.; Hsu, J.J.; Brufsky, A.M. COLET (NCT02322814): A Multistage, Phase 2 Study Evaluating the Safety and Efficacy of Cobimetinib (C) in Combination with Paclitaxel (P) as First-Line Treatment for Patients (Pts) with Metastatic Triple-Negative Breast Cancer (TNBC). J. Clin. Oncol. 2016, 34, TPS1100. [Google Scholar] [CrossRef]
- Liu, B.; Fu, L.; Zhang, C.; Zhang, L.; Zhang, Y.; Ouyang, L.; He, G.; Huang, J. Computational Design, Chemical Synthesis, and Biological Evaluation of a Novel ERK Inhibitor (BL-EI001) with Apoptosis-Inducing Mechanisms in Breast Cancer. Oncotarget 2015, 6, 6762–6775. [Google Scholar] [CrossRef]
- Brufsky, A.; Kim, S.-B.; Zvirbule, Z.; Dirix, L.Y.; Eniu, A.E.; Carabantes, F.; Izarzugaza, Y.; Mebis, J.; Sohn, J.; Wongchenko, M.; et al. Phase II COLET Study: Atezolizumab (A) + Cobimetinib (C) + Paclitaxel (P)/Nab-Paclitaxel (nP) as First-Line (1L) Treatment (Tx) for Patients (Pts) with Locally Advanced or Metastatic Triple-Negative Breast Cancer (mTNBC). J. Clin. Oncol. 2019, 37, 1013. [Google Scholar] [CrossRef]
- Brufsky, A.; Kim, S.B.; Zvirbule, Ž.; Eniu, A.; Mebis, J.; Sohn, J.H.; Wongchenko, M.; Chohan, S.; Amin, R.; Yan, Y.; et al. A Phase II Randomized Trial of Cobimetinib plus Chemotherapy, with or without Atezolizumab, as First-Line Treatment for Patients with Locally Advanced or Metastatic Triple-Negative Breast Cancer (COLET): Primary Analysis. Ann. Oncol. 2021, 32, 652–660. [Google Scholar] [CrossRef]
- Ghosh, A.; Bhowmik, A.; Bhandary, S.; Putatunda, S.; Laskar, A.; Biswas, A.; Dolui, S.; Banerjee, B.; Khan, R.; Das, N.; et al. Formulation and Antitumorigenic Activities of Nanoencapsulated Nifetepimine: A Promising Approach in Treating Triple Negative Breast Carcinoma. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 1973–1985. [Google Scholar] [CrossRef]
- Ghosh, S.; Adhikary, A.; Chakraborty, S.; Bhattacharjee, P.; Mazumder, M.; Putatunda, S.; Gorain, M.; Chakraborty, A.; Kundu, G.C.; Das, T.; et al. Cross-Talk between Endoplasmic Reticulum (ER) Stress and the MEK/ERK Pathway Potentiates Apoptosis in Human Triple Negative Breast Carcinoma Cells: ROLE OF A DIHYDROPYRIMIDONE, NIFETEPIMINE. J. Biol. Chem. 2015, 290, 3936–3949. [Google Scholar] [CrossRef]
- Li, Y.; Zhan, Z.; Yin, X.; Fu, S.; Deng, X. Targeted Therapeutic Strategies for Triple-Negative Breast Cancer. Front. Oncol. 2021, 11, 731535. [Google Scholar] [CrossRef]
- Elbaz, M.; Nasser, M.W.; Ravi, J.; Wani, N.A.; Ahirwar, D.K.; Zhao, H.; Oghumu, S.; Satoskar, A.R.; Shilo, K.; Carson, W.E.; et al. Modulation of the Tumor Microenvironment and Inhibition of EGF/EGFR Pathway: Novel Anti-Tumor Mechanisms of Cannabidiol in Breast Cancer. Mol. Oncol. 2015, 9, 906–919. [Google Scholar] [CrossRef]
- Liu, C.-Y.; Chu, P.-Y.; Huang, C.-T.; Chen, J.-L.; Yang, H.-P.; Wang, W.-L.; Lau, K.-Y.; Lee, C.-H.; Lan, T.-Y.; Huang, T.-T.; et al. Varlitinib Downregulates HER/ERK Signaling and Induces Apoptosis in Triple Negative Breast Cancer Cells. Cancers 2019, 11, 105. [Google Scholar] [CrossRef]
- Zhao, G.; Shi, A.; Fan, Z.; Du, Y. Salidroside Inhibits the Growth of Human Breast Cancer in Vitro and in Vivo. Oncol. Rep. 2015, 33, 2553–2560. [Google Scholar] [CrossRef]
- Sun, A.-Q.; Ju, X.-L. Inhibitory Effects of Salidroside on MCF-7 Breast Cancer Cells in Vivo. J. Int. Med. Res. 2020, 48, 0300060520968353. [Google Scholar] [CrossRef]
- Almeida, C.F.; Teixeira, N.; Correia-da-Silva, G.; Amaral, C. Cannabinoids in Breast Cancer: Differential Susceptibility According to Subtype. Molecules 2021, 27, 156. [Google Scholar] [CrossRef]
- Shrivastava, A.; Kuzontkoski, P.M.; Groopman, J.E.; Prasad, A. Cannabidiol Induces Programmed Cell Death in Breast Cancer Cells by Coordinating the Cross-Talk between Apoptosis and Autophagy. Mol. Cancer Ther. 2011, 10, 1161–1172. [Google Scholar] [CrossRef]
- Patel, N.; Kommineni, N.; Surapaneni, S.K.; Kalvala, A.; Yaun, X.; Gebeyehu, A.; Arthur, P.; Duke, L.C.; York, S.B.; Bagde, A.; et al. Cannabidiol Loaded Extracellular Vesicles Sensitize Triple-Negative Breast Cancer to Doxorubicin in Both in-Vitro and in Vivo Models. Int. J. Pharm. 2021, 607, 120943. [Google Scholar] [CrossRef]
- Kang, D.Y.; Sp, N.; Kim, D.H.; Joung, Y.H.; Lee, H.G.; Park, Y.M.; Yang, Y.M. Salidroside Inhibits Migration, Invasion and Angiogenesis of MDA-MB 231 TNBC Cells by Regulating EGFR/Jak2/STAT3 Signaling via MMP2. Int. J. Oncol. 2018, 53, 877–885. [Google Scholar] [CrossRef]
- Hatem, R.; Labiod, D.; Château-Joubert, S.; de Plater, L.; El Botty, R.; Vacher, S.; Bonin, F.; Servely, J.-L.; Dieras, V.; Bièche, I.; et al. Vandetanib as a Potential New Treatment for Estrogen Receptor-Negative Breast Cancers. Int. J. Cancer 2016, 138, 2510–2521. [Google Scholar] [CrossRef]
- Tam, S.; Al-Zubaidi, Y.; Rahman, M.K.; Bourget, K.; Zhou, F.; Murray, M. The Ixabepilone and Vandetanib Combination Shows Synergistic Activity in Docetaxel-Resistant MDA-MB-231 Breast Cancer Cells. Pharmacol. Rep. 2022, 74, 998–1010. [Google Scholar] [CrossRef]
- Wu, Z.-H.; Lin, C.; Liu, M.-M.; Zhang, J.; Tao, Z.-H.; Hu, X.-C. Src Inhibition Can Synergize with Gemcitabine and Reverse Resistance in Triple Negative Breast Cancer Cells via the AKT/c-Jun Pathway. PLoS ONE 2016, 11, e0169230. [Google Scholar] [CrossRef]
- Qian, X.-L.; Zhang, J.; Li, P.-Z.; Lang, R.-G.; Li, W.-D.; Sun, H.; Liu, F.-F.; Guo, X.-J.; Gu, F.; Fu, L. Dasatinib Inhibits C-Src Phosphorylation and Prevents the Proliferation of Triple-Negative Breast Cancer (TNBC) Cells Which Overexpress Syndecan-Binding Protein (SDCBP). PLoS ONE 2017, 12, e0171169. [Google Scholar] [CrossRef]
- Gasch, C.; Ffrench, B.; O’OLeary, J.J.; Gallegher, M.F. Catching Moving Targets: Cancer Stem Cell Hierarchies, Therapy-Resistance & Considerations for Clinical Intervention|Molecular Cancer. Mol. Cancer 2017, 16, 1–15. Available online: https://link.springer.com/article/10.1186/s12943-017-0601-3 (accessed on 19 March 2024).
- Turke, A.B.; Zejnullahu, K.; Wu, Y.-L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and Clonal Selection of MET Amplification in EGFR Mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef]
- Finn, R.S.; Dering, J.; Ginther, C.; Wilson, C.A.; Glaspy, P.; Tchekmedyian, N.; Slamon, D.J. Dasatinib, an Orally Active Small Molecule Inhibitor of Both the Src and Abl Kinases, Selectively Inhibits Growth of Basal-Type/“Triple-Negative” Breast Cancer Cell Lines Growing in Vitro. Breast Cancer Res. Treat. 2007, 105, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Bengala, C.; Ibrahim, N.; Roché, H.; Sparano, J.; Strauss, L.C.; Fairchild, J.; Sy, O.; Goldstein, L.J. Dasatinib as a Single Agent in Triple-Negative Breast Cancer: Results of an Open-Label Phase 2 Study. Clin. Cancer Res. 2011, 17, 6905–6913. [Google Scholar] [CrossRef]
- Nautiyal, J.; Majumder, P.; Patel, B.B.; Lee, F.Y.; Majumdar, A.P.N. Src Inhibitor Dasatinib Inhibits Growth of Breast Cancer Cells by Modulating EGFR Signaling. Cancer Lett. 2009, 283, 143–151. [Google Scholar] [CrossRef]
- Tian, J.; Raffa, F.A.; Dai, M.; Moamer, A.; Khadang, B.; Hachim, I.Y.; Bakdounes, K.; Ali, S.; Jean-Claude, B.; Lebrun, J.-J. Dasatinib Sensitises Triple Negative Breast Cancer Cells to Chemotherapy by Targeting Breast Cancer Stem Cells. Br. J. Cancer 2018, 119, 1495–1507. [Google Scholar] [CrossRef]
- Gautam, J.; Banskota, S.; Lee, H.; Lee, Y.-J.; Jeon, Y.H.; Kim, J.-A.; Jeong, B.-S. Down-Regulation of Cathepsin S and Matrix Metalloproteinase-9 via Src, a Non-Receptor Tyrosine Kinase, Suppresses Triple-Negative Breast Cancer Growth and Metastasis. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Zhang, C.-H.; Zheng, M.-W.; Li, Y.-P.; Lin, X.-D.; Huang, M.; Zhong, L.; Li, G.-B.; Zhang, R.-J.; Lin, W.-T.; Jiao, Y.; et al. Design, Synthesis, and Structure–Activity Relationship Studies of 3-(Phenylethynyl)-1H-Pyrazolo[3,4-d]Pyrimidin-4-Amine Derivatives as a New Class of Src Inhibitors with Potent Activities in Models of Triple Negative Breast Cancer. J. Med. Chem. 2015, 58, 3957–3974. [Google Scholar] [CrossRef]
- Bajrami, I.; Marlow, R.; van de Ven, M.; Brough, R.; Pemberton, H.N.; Frankum, J.; Song, F.; Rafiq, R.; Konde, A.; Krastev, D.B.; et al. E-Cadherin/ROS1 Inhibitor Synthetic Lethality in Breast Cancer. Cancer Discov. 2018, 8, 498–515. [Google Scholar] [CrossRef]
- Kashiwagi, S.; Yashiro, M.; Takashima, T.; Nomura, S.; Noda, S.; Kawajiri, H.; Ishikawa, T.; Wakasa, K.; Hiwakara, K. Significant of E-cadherin in Triple Negative Breast Cancer. Br. J. Canc. 2010, 103, 249–255. [Google Scholar] [CrossRef]
- Lu, L.; Niu, Z.; Chao, Z.; Fu, C.; Chen, K.; Shi, Y. Exploring the therapeutic potential of ADC combination for triple-negative breast cancer. Cell. Mol. Life Sci. 2023, 80, 350. [Google Scholar] [CrossRef]
- Mele, L.; Del Vecchio, V.; Liccardo, D.; Prisco, C.; Schwerdtfeger, M.; Robinson, N.; Desiderio, V.; Tirino, V.; Papaccio, G.; La Noce, M. The Role of Autophagy in Resistance to Targeted Therapies. Cancer Treat. Rev. 2020, 88, 102043. [Google Scholar] [CrossRef]
- Hussain, S.; Singh, A.; Nazir, S.U.; Tulsyan, S.; Khan, A.; Kumar, R.; Bashir, N.; Tanwar, P.; Mehrotra, R. Cancer Drug Resistance: A Fleet to Conquer. J. Cell. Biochem. 2019, 120, 14213–14225. [Google Scholar] [CrossRef]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-Label Clinical Trial of Niraparib Combined With Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132–1140. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S/N | Small Molecule | Preclinical and Clinical Results | Clinical Stage | Reference |
|---|---|---|---|---|
| 1 | Buparlisib | p110α (IC50; 52 nM), p110β (IC50; 166 nM), p110δ (IC50; 116 nM), p110γ (IC50; 262 nM) | NCT01576666 (phase 1) | [43] |
| 2 | Ipatasertib, | ORR = 33% | Phase I | [44] |
| 3 | Capivasertib | Median PFS = 5.9 months OS = 19.1 months | PAKT trial | [45] |
| 4 | Everolimus | (IC50 = 5−6 nM) | NCT01931163 (phase 2), NCT02616848 (phase 1), NCT02456857 (phase 2), NCT02120469 (phase 1), NCT02890069 (phase 1) | [11] |
| S/N | Small Molecule | Preclinical and Clinical Results | Clinical Stage | Reference |
|---|---|---|---|---|
| 7 | Olaparib | IC50 = 5 nM | Phase I, II | [11] |
| 8 | Veliparib | PFS = 5.7 months OS = 13.7 months | Phase II | [64] |
| 9 | Talazoparib | PFS = 8.6 months ORR = 62.6% | EMBRACA Phase III | [65] |
| S/N | Small Molecule | Pre-Clinical and Clinical Results | Clinical Stage | Reference |
|---|---|---|---|---|
| 10 | Ruxolitinib | IC50 = 3 μM | NCT01562873 (phase 1) | [11] |
| 11 | LLL12B | Decreased CD44+/CD24− Stem-Like Population | [78] | |
| 12 | Flubendazole | MDA-MB-231 (IC50 = 0.25 μM), Hs578T (IC50 = 0.125 μM), BT-549 (IC50 = 0.125 μM) | [11] | |
| 13 | Salinomycin | MDA-MB-231 (IC50 = 0.5−10 μM) | [11] |
| S/N | Small Molecules | Preclinical and Clinical Results | Clinical Stage | Reference |
|---|---|---|---|---|
| 14 | E6201 | MDA-MB-231 (IC50 = 0.25 μM), SUM149 (IC50 = 0.21 μM), SUM159 (IC50 = 2.36 μM) | None | [83] |
| 15 | Cobimetinib | IC50 = 4.2 nM | None | [84] |
| 16 | Nifetepimine | IC50 = 50 μM | None | [80] |
| 17 | BL-EI001 | IC50 = 5 μM | None | [85] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
James, N.; Owusu, E.; Rivera, G.; Bandyopadhyay, D. Small Molecule Therapeutics in the Pipeline Targeting for Triple-Negative Breast Cancer: Origin, Challenges, Opportunities, and Mechanisms of Action. Int. J. Mol. Sci. 2024, 25, 6285. https://doi.org/10.3390/ijms25116285
James N, Owusu E, Rivera G, Bandyopadhyay D. Small Molecule Therapeutics in the Pipeline Targeting for Triple-Negative Breast Cancer: Origin, Challenges, Opportunities, and Mechanisms of Action. International Journal of Molecular Sciences. 2024; 25(11):6285. https://doi.org/10.3390/ijms25116285
Chicago/Turabian StyleJames, Nneoma, Esther Owusu, Gildardo Rivera, and Debasish Bandyopadhyay. 2024. "Small Molecule Therapeutics in the Pipeline Targeting for Triple-Negative Breast Cancer: Origin, Challenges, Opportunities, and Mechanisms of Action" International Journal of Molecular Sciences 25, no. 11: 6285. https://doi.org/10.3390/ijms25116285