
Molecular Mechanisms Regulating Vascular Endothelial Permeability

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Endothelial Barrier Structure
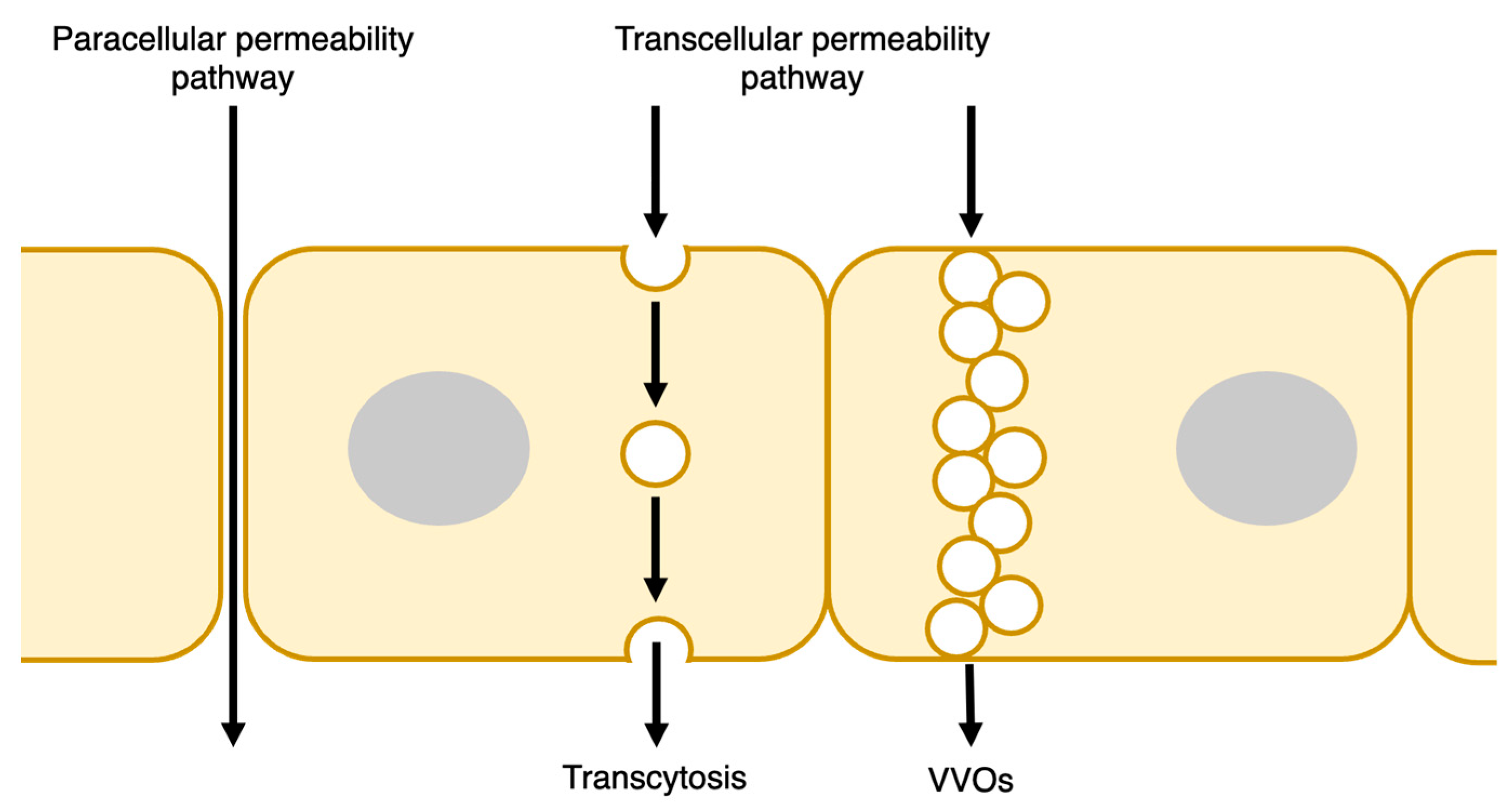
2.1. Transcellular Pathway
2.2. Paracellular Pathway
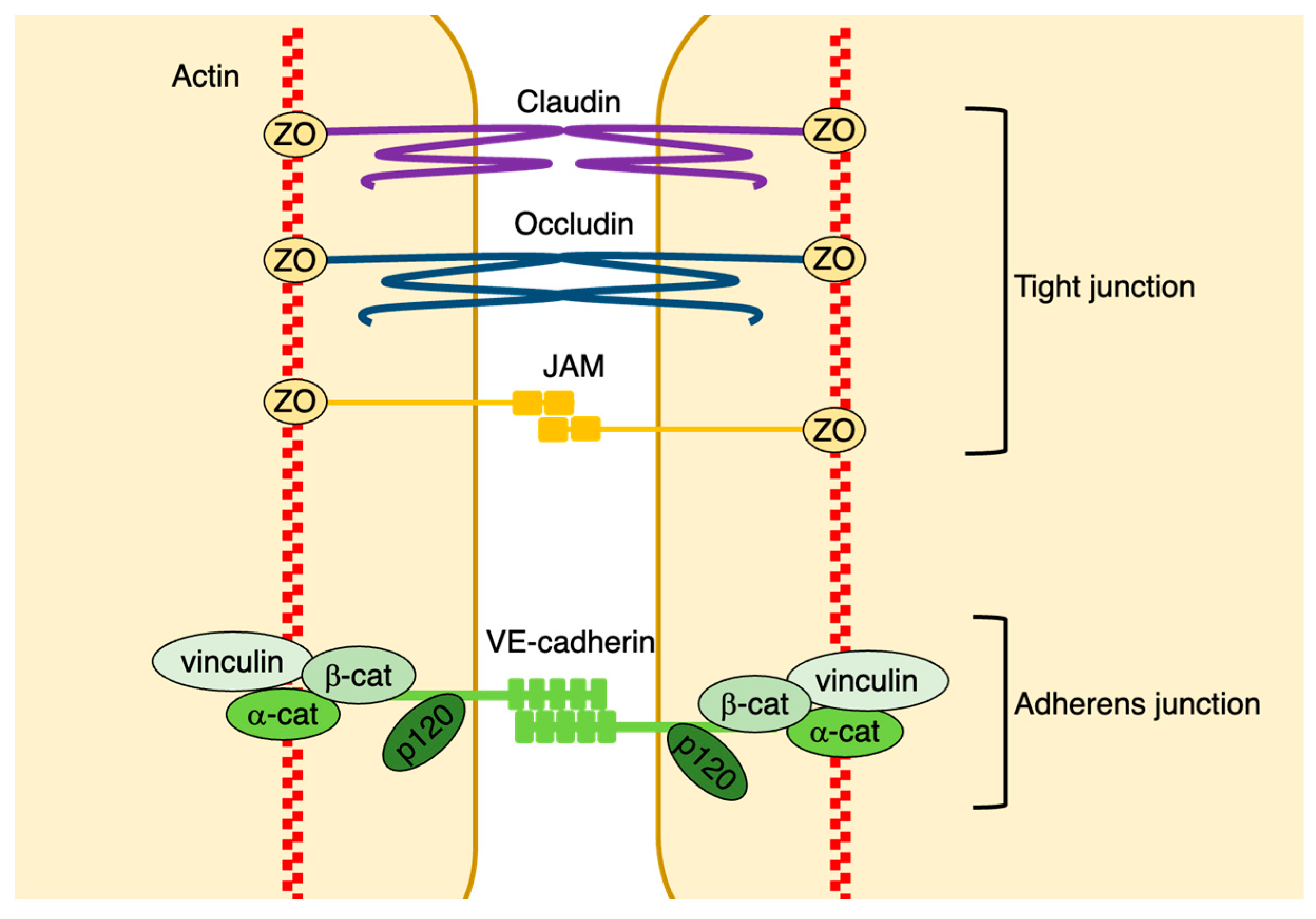
2.2.1. Adherens Junctions
2.2.2. Tight Junctions
2.2.3. Gap Junctions
3. Regulators of Vascular Endothelial Permeability
3.1. Small GTPases
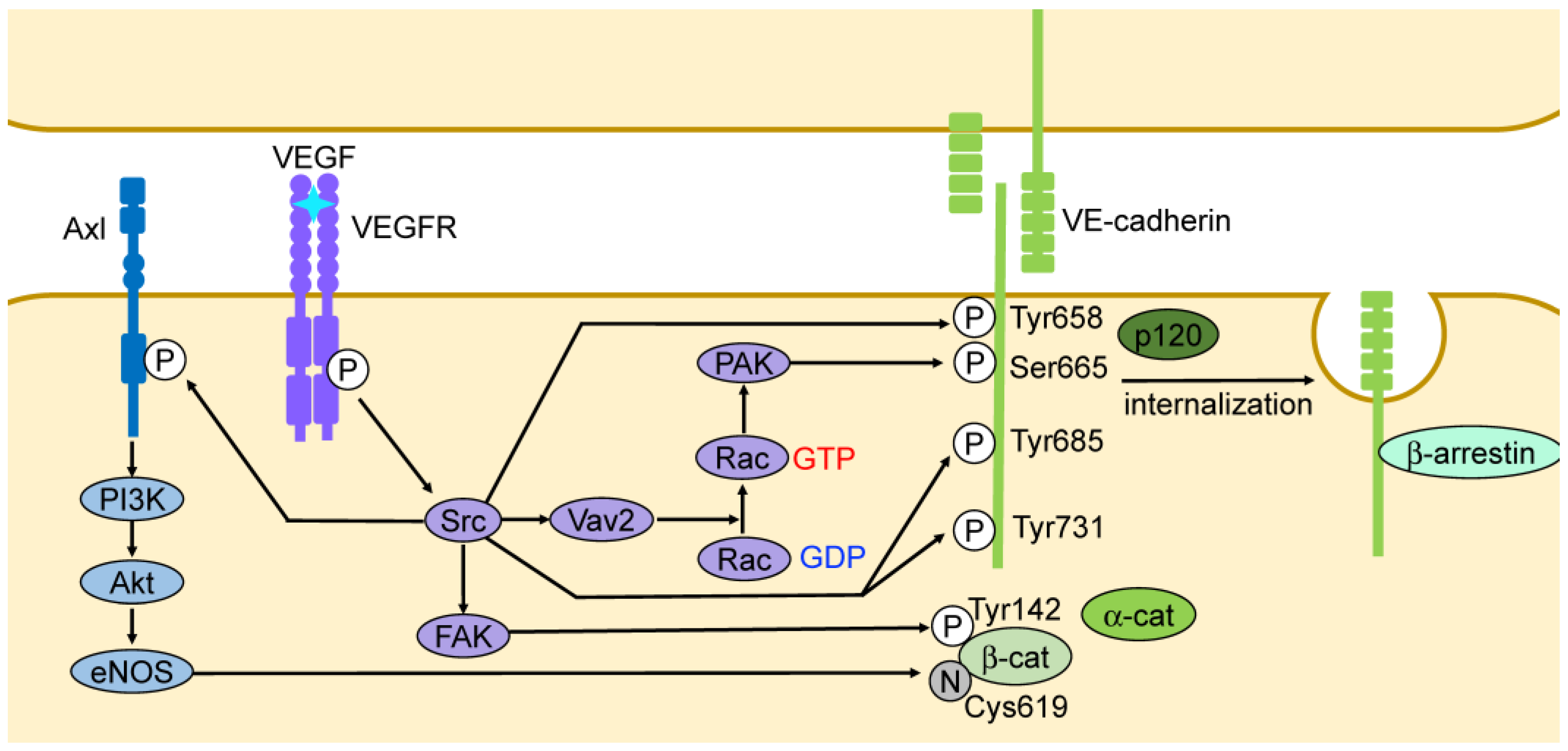
3.2. Vascular Endothelial Growth Factor (VEGF)
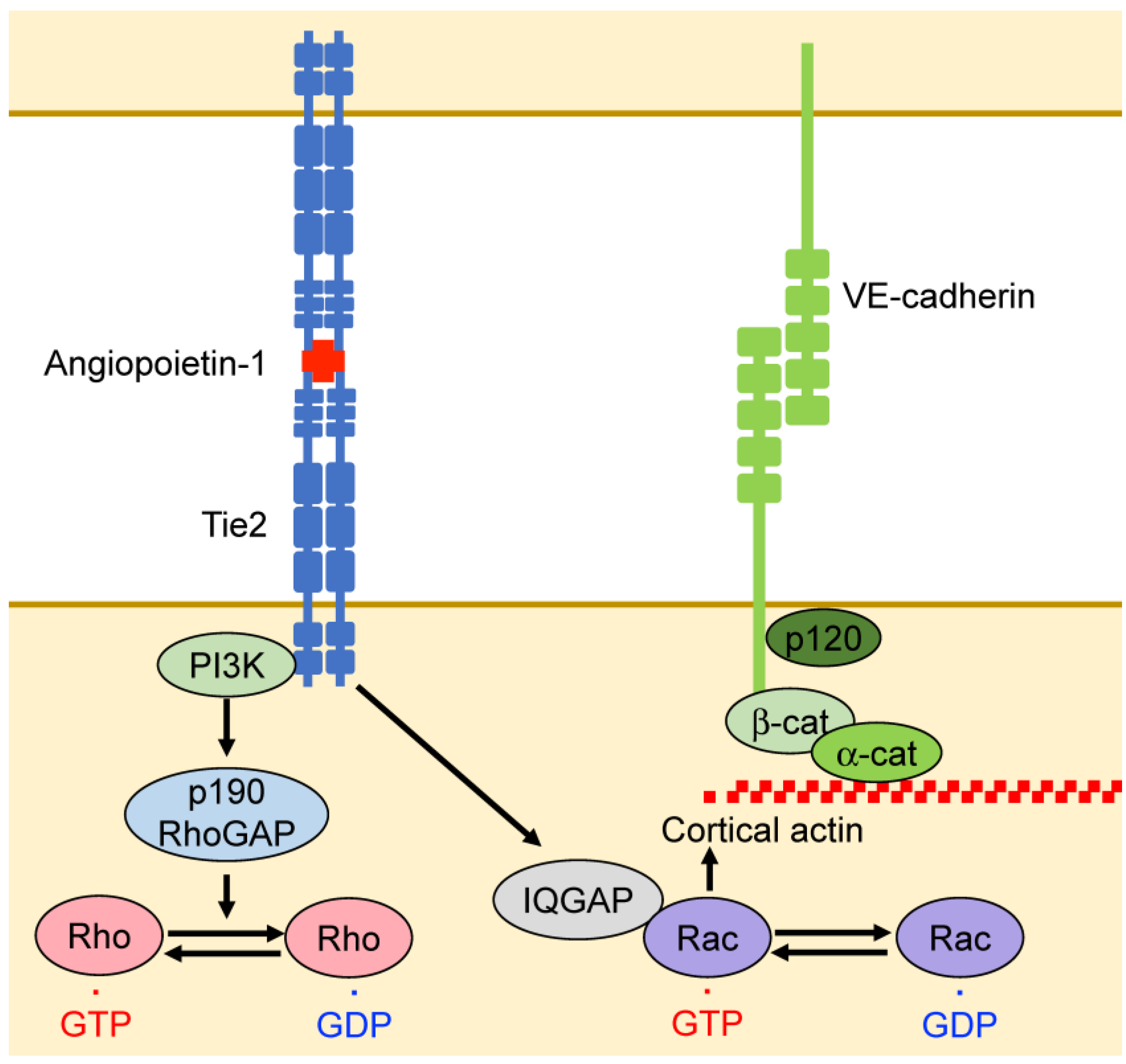
3.3. Angiopoietin
3.4. Inflammatory Mediators
3.5. Fluid Shear Stress
4. Mechanisms of Endothelial Barrier Function in Disease
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Claesson-Welsh, L.; Dejana, E.; McDonald, D.M. Permeability of the Endothelial Barrier: Identifying and Reconciling Controversies. Trends Mol. Med. 2021, 27, 314–331. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Mäkinen, T. Vascular Heterogeneity and Specialization in Development and Disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 477–494. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, Physiological, and Pathological Perspectives, Problems, and Promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.C.; Curry, F.E. Microvascular Permeability. Physiol. Rev. 1999, 79, 703–761. [Google Scholar] [CrossRef] [PubMed]
- Wettschureck, N.; Strilic, B.; Offermanns, S. Passing the Vascular Barrier: Endothelial Signaling Processes Controlling Extravasation. Physiol. Rev. 2019, 99, 1467–1525. [Google Scholar] [CrossRef] [PubMed]
- Predescu, S.A.; Predescu, D.N.; Malik, A.B. Molecular Determinants of Endothelial Transcytosis and Their Role in Endothelial Permeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L823–L842. [Google Scholar] [CrossRef] [PubMed]
- Minshall, R.D.; Sessa, W.C.; Stan, R.V.; Anderson, R.G.W.; Malik, A.B. Caveolin Regulation of Endothelial Function. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2003, 285, L1179–L1183. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, S.M.; Sugiyama, M.G.; Fung, K.Y.; Gao, Y.; Wang, C.; Levy, A.S.; Azizi, P.; Roufaiel, M.; Zhu, S.N.; Neculai, D.; et al. A Novel Assay Uncovers an Unexpected Role for SR-BI in LDL Transcytosis. Cardiovasc. Res. 2015, 108, 268–277. [Google Scholar] [CrossRef]
- Oh, P.; McIntosh, D.P.; Schnitzer, J.E. Dynamin at the Neck of Caveolae Mediates Their Budding to Form Transport Vesicles by GTP-Driven Fission from the Plasma Membrane of Endothelium. J. Cell Biol. 1998, 141, 101–114. [Google Scholar] [CrossRef]
- Minshall, R.D.; Tiruppathi, C.; Vogel, S.M.; Niles, W.D.; Gilchrist, A.; Hamm, H.E.; Malik, A.B. Endothelial Cell-Surface Gp60 Activates Vesicle Formation and Trafficking via Gi-Coupled Src Kinase Signaling Pathway. J. Cell Biol. 2000, 150, 1057–1070. [Google Scholar] [CrossRef] [PubMed]
- Predescu, S.A.; Predescu, D.N.; Shimizu, K.; Klein, I.K.; Malik, A.B. Cholesterol-Dependent Syntaxin-4 and SNAP-23 Clustering Regulates Caveolar Fusion with the Endothelial Plasma Membrane. J. Biol. Chem. 2005, 280, 37130–37138. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, A.M.; Feng, D. The Vesiculo–Vacuolar Organelle (VVO): A New Endothelial Cell Permeability Organelle. J. Histochem. Cytochem. 2001, 49, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Breviario, F.; Caveda, L.; Corada, M.; Martin-Padura, I.; Navarro, P.; Golay, J.; Introna, M.; Gulino, D.; Lampugnani, M.G.; Dejana, E. Functional Properties of Human Vascular Endothelial Cadherin (7B4/Cadherin-5), an Endothelium-Specific Cadherin. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Nanes, B.A.; Chiasson-MacKenzie, C.; Lowery, A.M.; Ishiyama, N.; Faundez, V.; Ikura, M.; Vincent, P.A.; Kowalczyk, A.P. p120-Catenin Binding Masks an Endocytic Signal Conserved in Classical Cadherins. J. Cell Biol. 2012, 199, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Garner, J.; Buckley, K.M.; Vincent, P.A.; Chiasson, C.M.; Dejana, E.; Faundez, V.; Kowalczyk, A.P. p120-Catenin Regulates Clathrin-Dependent Endocytosis of VE-Cadherin. Mol. Biol. Cell 2005, 16, 5141–5151. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, J.; de Rooij, J. Mechanical Control of the Endothelial Barrier. Cell Tissue Res. 2014, 355, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Hoelzle, M.K.; Svitkina, T. The Cytoskeletal Mechanisms of Cell–Cell Junction Formation in Endothelial Cells. Mol. Biol. Cell 2012, 23, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Orsenigo, F.; Giampietro, C.; Ferrari, A.; Corada, M.; Galaup, A.; Sigismund, S.; Ristagno, G.; Maddaluno, L.; Koh, G.Y.; Franco, D.; et al. Phosphorylation of VE-Cadherin Is Modulated by Haemodynamic Forces and Contributes to the Regulation of Vascular Permeability in Vivo. Nat. Commun. 2012, 3, 1208. [Google Scholar] [CrossRef]
- Potter, M.D.; Barbero, S.; Cheresh, D.A. Tyrosine Phosphorylation of VE-Cadherin Prevents Binding of p120- and β-Catenin and Maintains the Cellular Mesenchymal State. J. Biol. Chem. 2005, 280, 31906–31912. [Google Scholar] [CrossRef]
- Gavard, J.; Gutkind, J.S. VEGF Controls Endothelial-Cell Permeability by Promoting the β-Arrestin-Dependent Endocytosis of VE-Cadherin. Nat. Cell Biol. 2006, 8, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.L.; Nam, J.O.; Jean, C.; Lawson, C.; Walsh, C.T.; Goka, E.; Lim, S.-T.; Tomar, A.; Tancioni, I.; Uryu, S.; et al. VEGF-Induced Vascular Permeability Is Mediated by FAK. Dev. Cell 2012, 22, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Zihni, C.; Mills, C.; Matter, K.; Balda, M.S. Tight Junctions: From Simple Barriers to Multifunctional Molecular Gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef] [PubMed]
- Simionescu, M.; Simionescu, N.; Palade, G.E. Segmental Differentiations of Cell Junctions in the Vascular Endothelium. The Microvasculature. J. Cell Biol. 1975, 67, 863–885. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [PubMed]
- Dejana, E.; Orsenigo, F.; Molendini, C.; Baluk, P.; McDonald, D.M. Organization and Signaling of Endothelial Cell-to-Cell Junctions in Various Regions of the Blood and Lymphatic Vascular Trees. Cell Tissue Res. 2009, 335, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Taddei, A.; Giampietro, C.; Conti, A.; Orsenigo, F.; Breviario, F.; Pirazzoli, V.; Potente, M.; Daly, C.; Dimmeler, S.; Dejana, E. Endothelial Adherens Junctions Control Tight Junctions by VE-Cadherin-Mediated Upregulation of Claudin-5. Nat. Cell Biol. 2008, 10, 923–934. [Google Scholar] [CrossRef]
- Tornavaca, O.; Chia, M.; Dufton, N.; Almagro, L.O.; Conway, D.E.; Randi, A.M.; Schwartz, M.A.; Matter, K.; Balda, M.S. ZO-1 Controls Endothelial Adherens Junctions, Cell–Cell Tension, Angiogenesis, and Barrier Formation. J. Cell Biol. 2015, 208, 821–838. [Google Scholar] [CrossRef] [PubMed]
- Pohl, U. Connexins: Key Players in the Control of Vascular Plasticity and Function. Physiol. Rev. 2020, 100, 525–572. [Google Scholar] [CrossRef]
- Kandasamy, K.; Escue, R.; Manna, J.; Adebiyi, A.; Parthasarathi, K. Changes in Endothelial Connexin 43 Expression Inversely Correlate with Microvessel Permeability and VE-Cadherin Expression in Endotoxin-Challenged Lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L584–L592. [Google Scholar] [CrossRef]
- Yin, J.; Lv, L.; Zhai, P.; Long, T.; Zhou, Q.; Pan, H.; Botwe, G.; Wang, L.; Wang, Q.; Tan, L.; et al. Connexin 40 Regulates Lung Endothelial Permeability in Acute Lung Injury via the ROCK1-MYPT1- MLC20 Pathway. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L35–L44. [Google Scholar] [CrossRef] [PubMed]
- van Buul, J.D.; Timmerman, I. Small Rho GTPase-Mediated Actin Dynamics at Endothelial Adherens Junctions. Small GTPases 2016, 7, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Mikelis, C.M.; Simaan, M.; Ando, K.; Fukuhara, S.; Sakurai, A.; Amornphimoltham, P.; Masedunskas, A.; Weigert, R.; Chavakis, T.; Adams, R.H.; et al. RhoA and ROCK Mediate Histamine-Induced Vascular Leakage and Anaphylactic Shock. Nat. Commun. 2015, 6, 6725. [Google Scholar] [CrossRef]
- van Nieuw Amerongen, G.P.; van Delft, S.; Vermeer, M.A.; Collard, J.G.; van Hinsbergh, V.W.M. Activation of RhoA by Thrombin in Endothelial Hyperpermeability. Circ. Res. 2000, 87, 335–340. [Google Scholar] [CrossRef]
- Wojciak-Stothard, B.; Ridley, A.J. Rho GTPases and the Regulation of Endothelial Permeability. Vasc. Pharmacol. 2003, 39, 187–199. [Google Scholar] [CrossRef]
- David, S.; Ghosh, C.C.; Mukherjee, A.; Parikh, S.M. Angiopoietin-1 Requires IQ Domain GTPase-Activating Protein 1 to Activate Rac1 and Promote Endothelial Barrier Defense. Arter. Thromb. Vasc. Biol. 2011, 31, 2643–2652. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, I.; Heemskerk, N.; Kroon, J.; Schaefer, A.; van Rijssel, J.; Hoogenboezem, M.; van Unen, J.; Goedhart, J.; Gadella, T.W.J.; Yin, T.; et al. A Local VE-Cadherin and Trio-Based Signaling Complex Stabilizes Endothelial Junctions through Rac1. J. Cell Sci. 2015, 128, 3041–3054. [Google Scholar] [CrossRef]
- Knezevic, I.I.; Predescu, S.A.; Neamu, R.F.; Gorovoy, M.S.; Knezevic, N.M.; Easington, C.; Malik, A.B.; Predescu, D.N. Tiam1 and Rac1 Are Required for Platelet-Activating Factor-Induced Endothelial Junctional Disassembly and Increase in Vascular Permeability. J. Biol. Chem. 2009, 284, 5381–5394. [Google Scholar] [CrossRef]
- Monaghan-Benson, E.; Burridge, K. The Regulation of Vascular Endothelial Growth Factor-Induced Microvascular Permeability Requires Rac and Reactive Oxygen Species. J. Biol. Chem. 2009, 284, 25602–25611. [Google Scholar] [CrossRef]
- Naikawadi, R.P.; Cheng, N.; Vogel, S.M.; Qian, F.; Wu, D.; Malik, A.B.; Ye, R.D. A Critical Role for Phosphatidylinositol (3,4,5)-Trisphosphate-Dependent Rac Exchanger 1 in Endothelial Junction Disruption and Vascular Hyperpermeability. Circ. Res. 2012, 111, 1517–1527. [Google Scholar] [CrossRef]
- Cullere, X.; Shaw, S.K.; Andersson, L.; Hirahashi, J.; Luscinskas, F.W.; Mayadas, T.N. Regulation of Vascular Endothelial Barrier Function by Epac, a cAMP-Activated Exchange Factor for Rap GTPase. Blood 2005, 105, 1950–1955. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, S.; Sakurai, A.; Sano, H.; Yamagishi, A.; Somekawa, S.; Takakura, N.; Saito, Y.; Kangawa, K.; Mochizuki, N. Cyclic AMP Potentiates Vascular Endothelial Cadherin-Mediated Cell-Cell Contact to Enhance Endothelial Barrier Function through an Epac-Rap1 Signaling Pathway. Mol. Cell. Biol. 2005, 25, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, M.R.H.; Corada, M.; Dejana, E.; Bos, J.L. Epac1 Regulates Integrity of Endothelial Cell Junctions through VE-cadherin. FEBS Lett. 2005, 579, 4966–4972. [Google Scholar] [CrossRef] [PubMed]
- Noda, K.; Zhang, J.; Fukuhara, S.; Kunimoto, S.; Yoshimura, M.; Mochizuki, N. Vascular Endothelial-Cadherin Stabilizes at Cell–Cell Junctions by Anchoring to Circumferential Actin Bundles through α- and β-Catenins in Cyclic AMP-Epac-Rap1 Signal-Activated Endothelial Cells. Mol. Biol. Cell 2010, 21, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Birukova, A.A.; Zagranichnaya, T.; Fu, P.; Alekseeva, E.; Chen, W.; Jacobson, J.R.; Birukov, K.G. Prostaglandins PGE2 and PGI2 Promote Endothelial Barrier Enhancement via PKA- and Epac1/Rap1-Dependent Rac Activation. Exp. Cell Res. 2007, 313, 2504–2520. [Google Scholar] [CrossRef] [PubMed]
- Waschke, J.; Drenckhahn, D.; Adamson, R.H.; Barth, H.; Curry, F.E. cAMP Protects Endothelial Barrier Functions by Preventing Rac-1 Inhibition. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2427–H2433. [Google Scholar] [CrossRef] [PubMed]
- Birukova, A.A.; Zagranichnaya, T.; Alekseeva, E.; Bokoch, G.M.; Birukov, K.G. Epac/Rap and PKA Are Novel Mechanisms of ANP-induced Rac-mediated Pulmonary Endothelial Barrier Protection. J. Cell. Physiol. 2008, 215, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Fukuhara, S.; Moriya, T.; Obara, Y.; Nakahata, N.; Mochizuki, N. Rap1 Potentiates Endothelial Cell Junctions by Spatially Controlling Myosin II Activity and Actin Organization. J. Cell Biol. 2013, 202, 901–916. [Google Scholar] [CrossRef] [PubMed]
- Post, A.; Pannekoek, W.J.; Ponsioen, B.; Vliem, M.J.; Bos, J.L. Rap1 Spatially Controls ArhGAP29 to Inhibit Rho Signaling during Endothelial Barrier Regulation. Mol. Cell. Biol. 2015, 35, 2495–2502. [Google Scholar] [CrossRef]
- Post, A.; Pannekoek, W.J.; Ross, S.H.; Verlaan, I.; Brouwer, P.M.; Bos, J.L. Rasip1 Mediates Rap1 Regulation of Rho in Endothelial Barrier Function through ArhGAP29. Proc. Natl. Acad. Sci. USA 2013, 110, 11427–11432. [Google Scholar] [CrossRef]
- Weis, S.M.; Cheresh, D.A. Pathophysiological Consequences of VEGF-Induced Vascular Permeability. Nature 2005, 437, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and Regulation of Endothelial VEGF Receptor Signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Heinolainen, K.; Karaman, S.; D’Amico, G.; Tammela, T.; Sormunen, R.; Eklund, L.; Alitalo, K.; Zarkada, G. VEGFR3 Modulates Vascular Permeability by Controlling VEGF/VEGFR2 Signaling. Circ. Res. 2017, 120, 1414–1425. [Google Scholar] [CrossRef] [PubMed]
- Esser, S.; Lampugnani, M.G.; Corada, M.; Dejana, E.; Risau, W. Vascular Endothelial Growth Factor Induces VE-Cadherin Tyrosine Phosphorylation in Endothelial Cells. J. Cell Sci. 1998, 111, 1853–1865. [Google Scholar] [CrossRef] [PubMed]
- Lambeng, N.; Wallez, Y.; Rampon, C.; Cand, F.; Christé, G.; Gulino-Debrac, D.; Vilgrain, I.; Huber, P. Vascular Endothelial–Cadherin Tyrosine Phosphorylation in Angiogenic and Quiescent Adult Tissues. Circ. Res. 2005, 96, 384–391. [Google Scholar] [CrossRef]
- Eliceiri, B.P.; Paul, R.; Schwartzberg, P.L.; Hood, J.D.; Leng, J.; Cheresh, D.A. Selective Requirement for Src Kinases during VEGF-Induced Angiogenesis and Vascular Permeability. Mol. Cell 1999, 4, 915–924. [Google Scholar] [CrossRef]
- Jean, C.; Chen, X.L.; Nam, J.O.; Tancioni, I.; Uryu, S.; Lawson, C.; Ward, K.K.; Walsh, C.T.; Miller, N.L.G.; Ghassemian, M.; et al. Inhibition of Endothelial FAK Activity Prevents Tumor Metastasis by Enhancing Barrier Function. J. Cell Biol. 2014, 204, 247–263. [Google Scholar] [CrossRef]
- Fantin, A.; Lampropoulou, A.; Senatore, V.; Brash, J.T.; Prahst, C.; Lange, C.A.; Liyanage, S.E.; Raimondi, C.; Bainbridge, J.W.; Augustin, H.G.; et al. VEGF165-Induced Vascular Permeability Requires NRP1 for ABL-Mediated SRC Family Kinase Activation. J. Exp. Med. 2017, 214, 1049–1064. [Google Scholar] [CrossRef]
- Gioelli, N.; Neilson, L.J.; Wei, N.; Villari, G.; Chen, W.; Kuhle, B.; Ehling, M.; Maione, F.; Willox, S.; Brundu, S.; et al. Neuropilin 1 and Its Inhibitory Ligand Mini-Tryptophanyl-tRNA Synthetase Inversely Regulate VE-Cadherin Turnover and Vascular Permeability. Nat. Commun. 2022, 13, 4188. [Google Scholar] [CrossRef]
- Murakami, T.; Felinski, E.A.; Antonetti, D.A. Occludin Phosphorylation and Ubiquitination Regulate Tight Junction Trafficking and Vascular Endothelial Growth Factor-Induced Permeability. J. Biol. Chem. 2009, 284, 21036–21046. [Google Scholar] [CrossRef]
- Nawroth, R.; Poell, G.; Ranft, A.; Kloep, S.; Samulowitz, U.; Fachinger, G.; Golding, M.; Shima, D.T.; Deutsch, U.; Vestweber, D. VE-PTP and VE-Cadherin Ectodomains Interact to Facilitate Regulation of Phosphorylation and Cell Contacts. EMBO J. 2002, 21, 4885–4895. [Google Scholar] [CrossRef]
- Nottebaum, A.F.; Cagna, G.; Winderlich, M.; Gamp, A.C.; Linnepe, R.; Polaschegg, C.; Filippova, K.; Lyck, R.; Engelhardt, B.; Kamenyeva, O.; et al. VE-PTP Maintains the Endothelial Barrier via Plakoglobin and Becomes Dissociated from VE-Cadherin by Leukocytes and by VEGF. J. Exp. Med. 2008, 205, 2929–2945. [Google Scholar] [CrossRef] [PubMed]
- Broermann, A.; Winderlich, M.; Block, H.; Frye, M.; Rossaint, J.; Zarbock, A.; Cagna, G.; Linnepe, R.; Schulte, D.; Nottebaum, A.F.; et al. Dissociation of VE-PTP from VE-Cadherin Is Required for Leukocyte Extravasation and for VEGF-Induced Vascular Permeability in Vivo. J. Exp. Med. 2011, 208, 2393–2401. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Gohongi, T.; Kadambi, A.; Izumi, Y.; Ang, J.; Yun, C.-O.; Buerk, D.G.; Huang, P.L.; Jain, R.K. Predominant Role of Endothelial Nitric Oxide Synthase in Vascular Endothelial Growth Factor-Induced Angiogenesis and Vascular Permeability. Proc. Natl. Acad. Sci. USA 2001, 98, 2604–2609. [Google Scholar] [CrossRef] [PubMed]
- Thibeault, S.; Rautureau, Y.; Oubaha, M.; Faubert, D.; Wilkes, B.C.; Delisle, C.; Gratton, J.P. S-Nitrosylation of β-Catenin by eNOS-Derived NO Promotes VEGF-Induced Endothelial Cell Permeability. Mol. Cell 2010, 39, 468–476. [Google Scholar] [CrossRef]
- Pérez-Gutiérrez, L.; Ferrara, N. Biology and Therapeutic Targeting of Vascular Endothelial Growth Factor A. Nat. Rev. Mol. Cell Biol. 2023, 24, 816–834. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Li, X.; Massena, S.; Kutschera, S.; Padhan, N.; Gualandi, L.; Sundvold-Gjerstad, V.; Gustafsson, K.; Choy, W.W.; Zang, G.; et al. VEGFR2 Induces c-Src Signaling and Vascular Permeability in vivo via the Adaptor Protein TSAd. J. Exp. Med. 2012, 209, 1363–1377. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Padhan, N.; Sjöström, E.O.; Roche, F.P.; Testini, C.; Honkura, N.; Sáinz-Jaspeado, M.; Gordon, E.; Bentley, K.; Philippides, A.; et al. VEGFR2 pY949 Signalling Regulates Adherens Junction Integrity and Metastatic Spread. Nat. Commun. 2016, 7, 11017. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.; Cui, J.; Barnes, L.; Cheresh, D. Endothelial Barrier Disruption by VEGF-mediated Src Activity Potentiates Tumor Cell Extravasation and Metastasis. J. Cell Biol. 2004, 167, 223–229. [Google Scholar] [CrossRef]
- Sjöberg, E.; Melssen, M.; Richards, M.; Ding, Y.; Chanoca, C.; Chen, D.; Nwadozi, E.; Pal, S.; Love, D.T.; Ninchoji, T.; et al. Endothelial VEGFR2-PLCγ Signaling Regulates Vascular Permeability and Antitumor Immunity through eNOS/Src. J. Clin. Investig. 2023, 133, e161366. [Google Scholar] [CrossRef]
- Bentley, K.; Franco, C.A.; Philippides, A.; Blanco, R.; Dierkes, M.; Gebala, V.; Stanchi, F.; Jones, M.; Aspalter, I.M.; Cagna, G.; et al. The Role of Differential VE-cadherin Dynamics in Cell Rearrangement during Angiogenesis. Nat. Cell Biol. 2014, 16, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Szymborska, A.; Gerhardt, H. Hold Me, but Not Too Tight-Endothelial Cell-Cell Junctions in Angiogenesis. Cold Spring Harb. Perspect. Biol. 2018, 10, a029223. [Google Scholar] [CrossRef] [PubMed]
- Gordon, E.J.; Fukuhara, D.; Weström, S.; Padhan, N.; Sjöström, E.O.; van Meeteren, L.; He, L.; Orsenigo, F.; Dejana, E.; Bentley, K.; et al. The Endothelial Adaptor Molecule TSAd is Required for VEGF-induced Angiogenic Sprouting through Junctional c-Src Activation. Sci. Signal. 2016, 9, ra72. [Google Scholar] [CrossRef]
- Cao, J.; Ehling, M.; März, S.; Seebach, J.; Tarbashevich, K.; Sixta, T.; Pitulescu, M.E.; Werner, A.C.; Flach, B.; Montanez, E.; et al. Polarized Actin and VE-cadherin Dynamics Regulate Junctional Remodelling and Cell Migration during Sprouting Angiogenesis. Nat. Commun. 2017, 8, 2210. [Google Scholar] [CrossRef]
- Saharinen, P.; Eklund, L.; Alitalo, K. Therapeutic Targeting of the Angiopoietin–TIE Pathway. Nat. Rev. Drug Discov. 2017, 16, 635–661. [Google Scholar] [CrossRef]
- Saharinen, P.; Kerkelä, K.; Ekman, N.; Marron, M.; Brindle, N.; Lee, G.M.; Augustin, H.; Koh, G.Y.; Alitalo, K. Multiple Angiopoietin Recombinant Proteins Activate the Tie1 Receptor Tyrosine Kinase and Promote Its Interaction with Tie2. J. Cell Biol. 2005, 169, 239–243. [Google Scholar] [CrossRef]
- Gamble, J.R.; Drew, J.; Trezise, L.; Underwood, A.; Parsons, M.; Kasminkas, L.; Rudge, J.; Yancopoulos, G.; Vadas, M.A. Angiopoietin-1 Is an Antipermeability and Anti-Inflammatory Agent In Vitro and Targets Cell Junctions. Circ. Res. 2000, 87, 603–607. [Google Scholar] [CrossRef]
- Thurston, G.; Rudge, J.S.; Ioffe, E.; Zhou, H.; Ross, L.; Croll, S.D.; Glazer, N.; Holash, J.; McDonald, D.M.; Yancopoulos, G.D. Angiopoietin-1 Protects the Adult Vasculature against Plasma Leakage. Nat. Med. 2000, 6, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Mammoto, T.; Parikh, S.M.; Mammoto, A.; Gallagher, D.; Chan, B.; Mostoslavsky, G.; Ingber, D.E.; Sukhatme, V.P. Angiopoietin-1 Requires p190 RhoGAP to Protect against Vascular Leakage in Vivo. J. Biol. Chem. 2007, 282, 23910–23918. [Google Scholar] [CrossRef]
- Gavard, J.; Patel, V.; Gutkind, J.S. Angiopoietin-1 Prevents VEGF-induced Endothelial Permeability by Sequestering Src through mDia. Dev. Cell. 2008, 14, 25–36. [Google Scholar] [CrossRef]
- Fiedler, U.; Scharpfenecker, M.; Koidl, S.; Hegen, A.; Grunow, V.; Schmidt, J.M.; Kriz, W.; Thurston, G.; Augustin, H.G. The Tie-2 Ligand Angiopoietin-2 Is Stored in and Rapidly Released upon Stimulation from Endothelial Cell Weibel-Palade Bodies. Blood 2004, 103, 4150–4156. [Google Scholar] [CrossRef] [PubMed]
- Benest, A.V.; Kruse, K.; Savant, S.; Thomas, M.; Laib, A.M.; Loos, E.K.; Fiedler, U.; Augustin, H.G. Angiopoietin-2 Is Critical for Cytokine-Induced Vascular Leakage. PLoS ONE 2013, 8, e70459. [Google Scholar] [CrossRef] [PubMed]
- Daly, C.; Pasnikowski, E.; Burova, E.; Wong, V.; Aldrich, T.H.; Griffiths, J.; Ioffe, E.; Daly, T.J.; Fandl, J.P.; Papadopoulos, N.; et al. Angiopoietin-2 Functions as an Autocrine Protective Factor in Stressed Endothelial Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 15491–15496. [Google Scholar] [CrossRef] [PubMed]
- Wettschureck, N. Mammalian G Proteins and Their Cell Type Specific Functions. Physiol. Rev. 2005, 85, 1159–1204. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Regulation of Phosphoinositide-Specific Phospholipase C. Annu. Rev. Biochem. 2001, 70, 281–312. [Google Scholar] [CrossRef] [PubMed]
- Komarova, Y.; Malik, A.B. Regulation of Endothelial Permeability via Paracellular and Transcellular Transport Pathways. Annu. Rev. Physiol. 2010, 72, 463–493. [Google Scholar] [CrossRef] [PubMed]
- Stolwijk, J.A.; Zhang, X.; Gueguinou, M.; Zhang, W.; Matrougui, K.; Renken, C.; Trebak, M. Calcium Signaling Is Dispensable for Receptor Regulation of Endothelial Barrier Function. J. Biol. Chem. 2016, 291, 22894–22912. [Google Scholar] [CrossRef] [PubMed]
- Birukova, A.A.; Smurova, K.; Birukov, K.G.; Kaibuchi, K.; Garcia, J.G.N.; Verin, A.D. Role of Rho GTPases in Thrombin-Induced Lung Vascular Endothelial Cells Barrier Dysfunction. Microvasc. Res. 2004, 67, 64–77. [Google Scholar] [CrossRef]
- Essler, M.; Amano, M.; Kruse, H.-J.; Kaibuchi, K.; Weber, P.C.; Aepfelbacher, M. Thrombin Inactivates Myosin Light Chain Phosphatase via Rho and Its Target Rho Kinase in Human Endothelial Cells. J. Biol. Chem. 1998, 273, 21867–21874. [Google Scholar] [CrossRef]
- Szulcek, R.; Beckers, C.M.L.; Hodzic, J.; de Wit, J.; Chen, Z.; Grob, T.; Musters, R.J.P.; Minshall, R.D.; van Hinsbergh, V.W.M.; van Amerongen, G.P.N. Localized RhoA GTPase Activity Regulates Dynamics of Endothelial Monolayer Integrity. Cardiovasc. Res. 2013, 99, 471–482. [Google Scholar] [CrossRef]
- Soni, D.; Regmi, S.C.; Wang, D.-M.; DebRoy, A.; Zhao, Y.Y.; Vogel, S.M.; Malik, A.B.; Tiruppathi, C. Pyk2 Phosphorylation of VE-PTP Downstream of STIM1-Induced Ca2+ Entry Regulates Disassembly of Adherens Junctions. Am. J. Physiol.-Lung C 2017, 312, L1003–L1017. [Google Scholar] [CrossRef] [PubMed]
- Andriopoulou, P.; Navarro, P.; Zanetti, A.; Lampugnani, M.G.; Dejana, E. Histamine Induces Tyrosine Phosphorylation of Endothelial Cell-to-Cell Adherens Junctions. Arter. Thromb. Vasc. Biol. 1999, 19, 2286–2297. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.-J.; Chien, S. Effects of Disturbed Flow on Vascular Endothelium: Pathophysiological Basis and Clinical Perspectives. Physiol. Rev. 2011, 91, 327–387. [Google Scholar] [CrossRef] [PubMed]
- Noria, S.; Cowan, D.B.; Gotlieb, A.I.; Langille, B.L. Transient and Steady-State Effects of Shear Stress on Endothelial Cell Adherens Junctions. Circ. Res. 1999, 85, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Seebach, J.; Dieterich, P.; Luo, F.; Schillers, H.; Vestweber, D.; Oberleithner, H.; Galla, H.-J.; Schnittler, H.-J. Endothelial Barrier Function under Laminar Fluid Shear Stress. Lab. Investig. 2000, 80, 1819–1831. [Google Scholar] [CrossRef] [PubMed]
- Tarbell, J.M. Shear Stress and the Endothelial Transport Barrier. Cardiovasc. Res. 2010, 87, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Conway, D.E.; Coon, B.G.; Budatha, M.; Arsenovic, P.T.; Orsenigo, F.; Wessel, F.; Zhang, J.; Zhuang, Z.; Dejana, E.; Vestweber, D.; et al. VE-Cadherin Phosphorylation Regulates Endothelial Fluid Shear Stress Responses through the Polarity Protein LGN. Curr. Biol. 2017, 27, 2219–2225.e5. [Google Scholar] [CrossRef] [PubMed]
- Jahnsen, E.D.; Trindade, A.; Zaun, H.C.; Lehoux, S.; Duarte, A.; Jones, E.A.V. Notch1 Is Pan-Endothelial at the Onset of Flow and Regulated by Flow. PLoS ONE 2015, 10, e0122622. [Google Scholar] [CrossRef] [PubMed]
- Mack, J.J.; Mosqueiro, T.S.; Archer, B.J.; Jones, W.M.; Sunshine, H.; Faas, G.C.; Briot, A.; Aragón, R.L.; Su, T.; Romay, M.C.; et al. NOTCH1 Is a Mechanosensor in Adult Arteries. Nat. Commun. 2017, 8, 1620. [Google Scholar] [CrossRef]
- Polacheck, W.J.; Kutys, M.L.; Yang, J.; Eyckmans, J.; Wu, Y.; Vasavada, H.; Hirschi, K.K.; Chen, C.S. A Non-Canonical Notch Complex Regulates Adherens Junctions and Vascular Barrier Function. Nature 2017, 552, 258–262. [Google Scholar] [CrossRef]
- Caolo, V.; Debant, M.; Endesh, N.; Futers, T.S.; Lichtenstein, L.; Bartoli, F.; Parsonage, G.; Jones, E.A.; Beech, D.J. Shear Stress Activates ADAM10 Sheddase to Regulate Notch1 via the Piezo1 Force Sensor in Endothelial Cells. eLife 2020, 9, e50684. [Google Scholar] [CrossRef] [PubMed]
- Shirakura, K.; Baluk, P.; Nottebaum, A.F.; Ipe, U.; Peters, K.G.; McDonald, D.M.; Vestweber, D. Shear Stress Control of Vascular Leaks and Atheromas through Tie2 Activation by VE-PTP Sequestration. EMBO Mol. Med. 2023, 15, e16128. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.; Mahmoud, M.; Liu, R.; Andueza, A.; Kumar, S.; Kang, D.-W.; Zhang, J.; Tamargo, I.; Villa-Roel, N.; Baek, K.-I.; et al. Stable Flow-Induced Expression of KLK10 Inhibits Endothelial Inflammation and Atherosclerosis. eLife 2022, 11, e72579. [Google Scholar] [CrossRef]
- Colgan, O.C.; Ferguson, G.; Collins, N.T.; Murphy, R.P.; Meade, G.; Cahill, P.A.; Cummins, P.M. Regulation of Bovine Brain Microvascular Endothelial Tight Junction Assembly and Barrier Function by Laminar Shear Stress. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H3190–H3197. [Google Scholar] [CrossRef]
- Orr, A.W.; Stockton, R.; Simmers, M.B.; Sanders, J.M.; Sarembock, I.J.; Blackman, B.R.; Schwartz, M.A. Matrix-Specific P21-Activated Kinase Activation Regulates Vascular Permeability in Atherogenesis. J. Cell Biol. 2007, 176, 719–727. [Google Scholar] [CrossRef]
- Alfaidi, M.; Bhattarai, U.; Orr, A.W. Nck1, But Not Nck2, Mediates Disturbed Flow-Induced p21-Activated Kinase Activation and Endothelial Permeability. J. Am. Heart Assoc. 2020, 9, e016099. [Google Scholar] [CrossRef]
- Shih, Y.T.; Wei, S.Y.; Chen, J.H.; Wang, W.L.; Wu, H.Y.; Wang, M.C.; Lin, C.Y.; Lee, P.L.; Lin, C.Y.; Chiang, H.C.; et al. Vinculin Phosphorylation Impairs Vascular Endothelial Junctions Promoting Atherosclerosis. Eur. Heart J. 2022, 44, 304–318. [Google Scholar] [CrossRef]
- Weinberg, P.D. Haemodynamic Wall Shear Stress, Endothelial Permeability and Atherosclerosis—A Triad of Controversy. Front. Bioeng. Biotechnol. 2022, 10, 836680. [Google Scholar] [CrossRef]
- Ghim, M.; Alpresa, P.; Yang, S.W.; Braakman, S.T.; Gray, S.G.; Sherwin, S.J.; van Reeuwijk, M.; Weinberg, P.D. Visualization of Three Pathways for Macromolecule Transport across Cultured Endothelium and Their Modification by Flow. Am. J. Physiol.-Heart Circ. Physiol. 2017, 313, H959–H973. [Google Scholar] [CrossRef] [PubMed]
- Andueza, A.; Kumar, S.; Kim, J.; Kang, D.W.; Mumme, H.L.; Perez, J.I.; Villa-Roel, N.; Jo, H. Endothelial Reprogramming by Disturbed Flow Revealed by Single-Cell RNA and Chromatin Accessibility Study. Cell Rep. 2020, 33, 108491. [Google Scholar] [CrossRef]
- Souilhol, C.; Harmsen, M.C.; Evans, P.C.; Krenning, G. Endothelial-mesenchymal Transition in Atherosclerosis. Cardiovasc. Res. 2018, 114, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Park-Windhol, C.; D’Amore, P.A. Disorders of Vascular Permeability. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 251–281. [Google Scholar] [CrossRef] [PubMed]
- West, J.B.; Mathieu-Costello, O. Structure, Strength, Failure, and Remodeling of the Pulmonary Blood-Gas Barrier. Annu. Rev. Physiol. 1999, 61, 543–572. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, E.E.; Hong, Z.; Xiong, S.; Zhong, M.; Di, A.; Rehman, J.; Komarova, Y.A.; Malik, A.B. Endothelial Cell Piezo1 Mediates Pressure-Induced Lung Vascular Hyperpermeability via Disruption of Adherens Junctions. Proc. Natl. Acad. Sci. USA 2019, 116, 12980–12985. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.M. Angiogenesis and Remodeling of Airway Vasculature in Chronic Inflammation. Am. J. Respir. Crit. Care Med. 2001, 164, S39–S45. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.M.; Choyke, P.L. Imaging of Angiogenesis: From Microscope to Clinic. Nat. Med. 2003, 9, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Albarrán-Juárez, J.; Iring, A.; Wang, S.; Joseph, S.; Grimm, M.; Strilic, B.; Wettschureck, N.; Althoff, T.F.; Offermanns, S. Piezo1 and Gq/G11 Promote Endothelial Inflammation Depending on Flow Pattern and Integrin Activation. J. Exp. Med. 2018, 215, 2655–2672. [Google Scholar] [CrossRef] [PubMed]
- Tamargo, I.A.; Baek, K.I.; Kim, Y.; Park, C.; Jo, H. Flow-Induced Reprogramming of Endothelial Cells in Atherosclerosis. Nat. Rev. Cardiol. 2023, 20, 738–753. [Google Scholar] [CrossRef]
- Langen, U.H.; Ayloo, S.; Gu, C. Development and Cell Biology of the Blood-Brain Barrier. Annu. Rev. Cell Dev. Biol. 2019, 35, 591–613. [Google Scholar] [CrossRef]
- Di Francescomarino, S.; Sciartilli, A.; Di Valerio, V.; Di Baldassarre, A.; Gallina, S. The Effect of Physical Exercise on Endothelial Function. Sports Med. 2009, 39, 97–812. [Google Scholar] [CrossRef]
- Souza, P.S.; Gonçalves, E.D.; Pedroso, G.S.; Farias, H.R.; Junqueira, S.C.; Marcon, R.; Tuon, T.; Cola, M.; Silveira, P.C.L.; Santos, A.R.; et al. Physical Exercise Attenuates Experimental Autoimmune Encephalomyelitis by Inhibiting Peripheral Immune Response and Blood-Brain Barrier Disruption. Mol. Neurobiol. 2017, 54, 4723–4737. [Google Scholar] [CrossRef] [PubMed]
- Mokhtarzade, M.; Motl, R.; Negaresh, R.; Zimmer, P.; Khodadoost, M.; Baker, J.S.; Patel, D.; Majdinasab, N.; Ranjbar, R. Exercise-Induced Changes in Neurotrophic Factors and Markers of Blood-Brain Barrier Permeability are Moderated by Weight Status in Multiple Sclerosis. Neuropeptides 2018, 70, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Propson, N.E.; Roy, E.R.; Litvinchuk, A.; Köhl, J.; Zheng, H. Endothelial C3a Receptor Mediates Vascular Inflammation and Blood-Brain Barrier Permeability during Aging. J. Clin. Investig. 2021, 131, e140966. [Google Scholar] [CrossRef] [PubMed]
- Ya, J.; Kadir, R.R.A.; Bayraktutan, U. Delay of Endothelial Cell Senescence Protects Cerebral Barrier against Age-Related Dysfunction: Role of Senolytics and Senomorphics. Tissue Barriers 2023, 11, 2103353. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wakasugi, R.; Suzuki, K.; Kaneko-Kawano, T. Molecular Mechanisms Regulating Vascular Endothelial Permeability. Int. J. Mol. Sci. 2024, 25, 6415. https://doi.org/10.3390/ijms25126415
Wakasugi R, Suzuki K, Kaneko-Kawano T. Molecular Mechanisms Regulating Vascular Endothelial Permeability. International Journal of Molecular Sciences. 2024; 25(12):6415. https://doi.org/10.3390/ijms25126415
Chicago/Turabian StyleWakasugi, Rio, Kenji Suzuki, and Takako Kaneko-Kawano. 2024. "Molecular Mechanisms Regulating Vascular Endothelial Permeability" International Journal of Molecular Sciences 25, no. 12: 6415. https://doi.org/10.3390/ijms25126415
APA StyleWakasugi, R., Suzuki, K., & Kaneko-Kawano, T. (2024). Molecular Mechanisms Regulating Vascular Endothelial Permeability. International Journal of Molecular Sciences, 25(12), 6415. https://doi.org/10.3390/ijms25126415