

Dynamic Changes in Ion Channels during Myocardial Infarction and Therapeutic Challenges
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
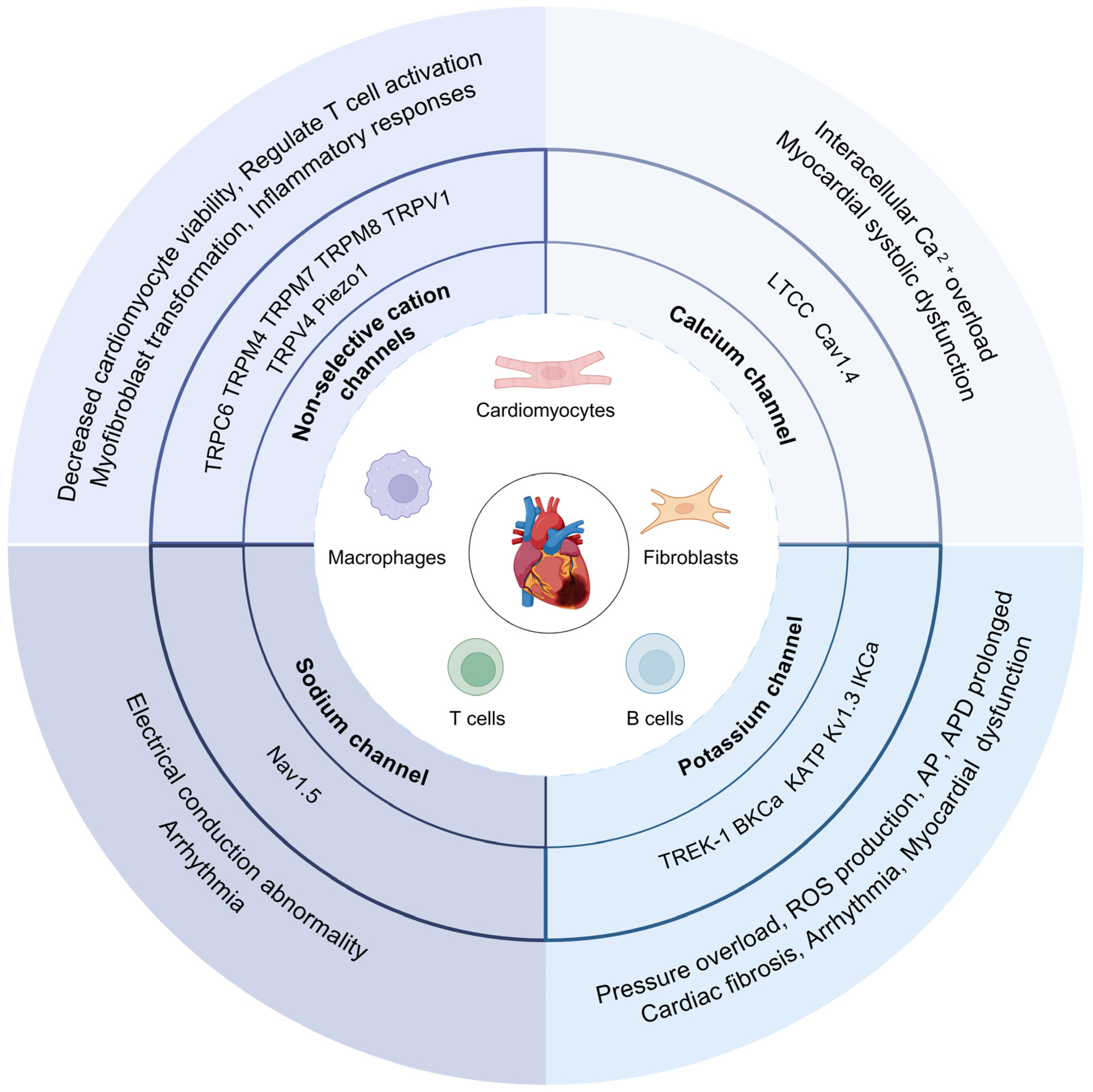
2. Ion Channel Changes after MI
2.1. Ion Current Remodeling after MI
2.2. Ca2+ Channels after MI
2.3. K+ Channels after MI
2.4. Na+ Channels after MI
3. Ion Channels in Cells of Myocardium
3.1. Cardiomyocytes
3.2. Cardiac Fibroblasts
3.3. Cardiac Immune Cells
4. Application of Ion Channel Therapy in MI
4.1. Drugs Targeting Ca2+ Channels
4.2. Drugs Targeting K+ Channels
4.3. Drugs Targeting Na+ Channels
4.4. Basic Ion Channel Physiology and Clinical Applications
5. Perspectives and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nerbonne, J.M.; Kass, R.S. Molecular Physiology of Cardiac Repolarization. Physiol. Rev. 2005, 85, 1205–1253. [Google Scholar] [CrossRef] [PubMed]
- Fozzard, H.A. Heart: Excitation-contraction coupling. Annu. Rev. Physiol. 1977, 39, 201–220. [Google Scholar] [CrossRef]
- Kanno, S.; Saffitz, J.E. The role of myocardial gap junctions in electrical conduction and arrhythmogenesis. Cardiovasc. Pathol. Pathol. 2001, 10, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Matthews, G.D.; Martin, C.A.; Grace, A.A.; Zhang, Y.; Huang, C.L. Regional variations in action potential alternans in isolated murine Scn5a+/− hearts during dynamic pacing. Acta Physiol. 2010, 200, 129–146. [Google Scholar] [CrossRef]
- Deshmukh, T.; Kumar, S.; Chong, J. Cardiac Inflammation after Myocardial Infarction and its Impact on Ventricular Arrhythmias. Heart Lung Circ. 2021, 30, 783–785. [Google Scholar] [CrossRef] [PubMed]
- Fabritz, L.; Herzig, S. Can T-type calcium channels make a change of heart after myocardial infarction? Fiction or fact, and for better or for worse? Cardiovasc. Res. 2011, 91, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Wiecha, J.; Hombach, V. Cellular electrophysiological properties in myocardial infarction. Eur. Heart J. 1993, 14, 9–19. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Q.; Tao, B.; Angelini, M.; Ramadoss, S.; Sun, B.; Wang, P.; Krokhaleva, Y.; Ma, F.; Gu, Y.; et al. Fibroblasts in heart scar tissue directly regulate cardiac excitability and arrhythmogenesis. Science 2023, 381, 1480–1487. [Google Scholar] [CrossRef]
- Ajijola, O.; Tung, R.; Shivkumar, K. Ventricular tachycardia in ischemic heart disease substrates. Indian Heart J. 2014, 66, S24–S34. [Google Scholar] [CrossRef]
- Glazer, A.; Wada, Y.; Li, B.; Muhammad, A.; Kalash, O.; O’Neill, M.; Shields, T.; Hall, L.; Short, L.; Blair, M.; et al. High-Throughput Reclassification of SCN5A Variants. Am. J. Hum. Genet. 2020, 107, 111–123. [Google Scholar] [CrossRef]
- Marshall, I.; Boyfield, I.; McNulty, S. Ratiometric Ca2+ measurements using the FlexStation® Scanning Fluorometer. Methods Mol. Biol. 2013, 937, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Wickenden, A.; Priest, B.; Erdemli, G. Ion channel drug discovery: Challenges and future directions. Future Med. Chem. 2012, 4, 661–679. [Google Scholar] [CrossRef] [PubMed]
- Darré, L.; Furini, S.; Domene, C. Permeation and dynamics of an open-activated TRPV1 channel. J. Mol. Biol. 2015, 427, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Dworakowska, B.; Dołowy, K. Ion channels-related diseases. Acta Biochim. Pol. 2000, 47, 685–703. [Google Scholar] [CrossRef] [PubMed]
- Douguet, D.; Patel, A.; Xu, A.; Vanhoutte, P.M.; Honoré, E. Piezo Ion Channels in Cardiovascular Mechanobiology. Trends Pharmacol. Sci. 2019, 40, 956–970. [Google Scholar] [CrossRef]
- Joseph, N.; Reicher, B.; Barda-Saad, M. The calcium feedback loop and T cell activation: How cytoskeleton networks control intracellular calcium flux. Biochim. Biophys. Acta 2014, 1838, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Prado, W.A. Involvement of calcium in pain and antinociception. Braz. J. Med. Biol. Res. 2001, 34, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S.; Maguy, A.; Le Bouter, S.; Yeh, Y.H. Arrhythmogenic ion-channel remodeling in the heart: Heart failure, myocardial infarction, and atrial fibrillation. Physiol. Rev. 2007, 87, 425–456. [Google Scholar] [CrossRef] [PubMed]
- Friedman, P.L.; Fenoglio, J.J.; Wit, A.L. Time course for reversal of electrophysiological and ultrastructural abnormalities in subendocardial Purkinje fibers surviving extensive myocardial infarction in dogs. Circ. Res. 1975, 36, 127–144. [Google Scholar] [CrossRef]
- Janse, M.J.; Wit, A.L.; Nassal, M.M.J.; Wan, X.; Dale, Z.; Deschênes, I.; Wilson, L.D.; Piktel, J.S.; Huang, C.L.-H.; Foster, M.N.; et al. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol. Rev. 1989, 69, 1049–1169. [Google Scholar] [CrossRef]
- Shaw, R.M.; Rudy, Y. Electrophysiologic effects of acute myocardial ischemia: A theoretical study of altered cell excitability and action potential duration. Cardiovasc. Res. 1997, 35, 256–272. [Google Scholar] [CrossRef] [PubMed]
- Beardslee, M.A.; Lerner, D.L.; Tadros, P.N.; Laing, J.G.; Beyer, E.C.; Yamada, K.A.; Kléber, A.G.; Schuessler, R.B.; Saffitz, J.E. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ. Res. 2000, 87, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.M.; Boyden, P.A. Reduced inward rectifying and increased E-4031-sensitive K+ current density in arrhythmogenic subendocardial purkinje myocytes from the infarcted heart. J. Cardiovasc. Electrophysiol. 1998, 9, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Boyden, P.A.; Pinto, J.M. Reduced calcium currents in subendocardial Purkinje myocytes that survive in the 24- and 48-hour infarcted heart. Circulation 1994, 89, 2747–2759. [Google Scholar] [CrossRef] [PubMed]
- Jeck, C.; Pinto, J.; Boyden, P. Transient outward currents in subendocardial Purkinje myocytes surviving in the infarcted heart. Circulation 1995, 92, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Lue, W.M.; Boyden, P.A. Abnormal electrical properties of myocytes from chronically infarcted canine heart. Alterations in Vmax and the transient outward current. Circulation 1992, 85, 1175–1188. [Google Scholar] [CrossRef] [PubMed]
- Peters, N.S.; Coromilas, J.; Severs, N.J.; Wit, A.L. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation 1997, 95, 988–996. [Google Scholar] [CrossRef]
- Gardner, P.I.; Ursell, P.C.; Fenoglio, J.J., Jr.; Wit, A.L. Electrophysiologic and anatomic basis for fractionated electrograms recorded from healed myocardial infarcts. Circulation 1985, 72, 596–611. [Google Scholar] [CrossRef] [PubMed]
- Ursell, P.C.; I Gardner, P.; Albala, A.; Fenoglio, J.J., Jr.; Wit, A.L. Structural and electrophysiological changes in the epicardial border zone of canine myocardial infarcts during infarct healing. Circ. Res. 1985, 56, 436–451. [Google Scholar] [CrossRef]
- Bo, T.; Zhebo, L.; Fang, W.; Suzhen, F.; Shengyu, C.; Hao, X.; Lin, X. Over-expression of Kv4.3 gene reverses cardiac remodeling and transient-outward K+ current (Ito) reduction via CaMKII inhibition in myocardial infarction. Biomed. Pharmacother. 2020, 132, 110896. [Google Scholar] [CrossRef]
- Rossow, C.F.; Minami, E.; Chase, E.G.; Murry, C.E.; Santana, L.F. NFATc3-induced reductions in voltage-gated K+ currents after myocardial infarction. Circ. Res. 2004, 94, 1340–1350. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Gaur, H.; Bhargava, A. Cardiac voltage gated calcium channels and their regulation by β-adrenergic signaling. Life Sci. 2018, 194, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Seagar, M.; Jones, J.; Reber, B.; Catterall, W. Subunit structure of dihydropyridine-sensitive calcium channels from skeletal muscle. Proc. Natl. Acad. Sci. USA 1987, 84, 5478–5482. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Perez-Reyes, E.; Snutch, T.P.; Striessnig, J. International Union of Pharmacology. XLVIII. Nomenclature and Structure-Function Relationships of Voltage-Gated Calcium Channels. Pharmacol. Rev. 2005, 57, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Dolphin, A.C. Voltage-gated calcium channels: Their discovery, function and importance as drug targets. Brain Neurosci. Adv. 2018, 2. [Google Scholar] [CrossRef] [PubMed]
- Terrar, D.A. Structure and Function of Calcium Channels and the Actions of Anaesthetics. Br. J. Anaesth. 1993, 71, 39–46. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Zhang, J.; Yu, Y.; Jizi, L.; Wang, W.; Li, M. New Insights Into Interactions of Presynaptic Calcium Channel Subtypes and SNARE Proteins in Neurotransmitter Release. Front. Mol. Neurosci. 2018, 11, 213. [Google Scholar] [CrossRef]
- Napolitano, C.; Antzelevitch, C.; Priori, S. Phenotypical Manifestations of Mutations in the Genes Encoding Subunits of the Cardiac Voltage–Dependent L-Type Calcium Channel. Circ. Res. 2011, 108, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C.; Sibley, D.R. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef]
- Catterall, W.A. Voltage-Gated Calcium Channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef]
- Gurkoff, G.; Shahlaie, K.; Lyeth, B.; Berman, R. Voltage-Gated Calcium Channel Antagonists and Traumatic Brain Injury. Pharmaceuticals 2013, 6, 788–812. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Wang, F.; Wang, W.; Makarewich, C.; Zhang, H.; Kubo, H.; Berretta, R.; Barr, L.; Molkentin, J.; Houser, S. Ca2+ influx through L-type Ca2+ channels and transient receptor potential channels activates pathological hypertrophy signaling. J. Mol. Cell. Cardiol. 2012, 53, 657–667. [Google Scholar] [CrossRef]
- Santos, P.; Barcellos, L.; Mill, J.; Masuda, M. Ventricular action potential and L-type calcium channel in infarct-induced hypertrophy in rats. J. Cardiovasc. Electrophysiol. 1995, 6, 1004–1014. [Google Scholar] [CrossRef]
- Zheng, M.; Li, X.; Tang, K.; Sharma, N.; Wyatt, T.; Patel, K.; Gao, L.; Bidasee, K.; Rozanski, G. Pyruvate restores β-adrenergic sensitivity of L-type Ca2+ channels in failing rat heart: Role of protein phosphatase. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1352–H1360. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Alonso, J.L.; Bhargava, A.; O’Hara, T.; Glukhov, A.V.; Schobesberger, S.; Bhogal, N.; Sikkel, M.B.; Mansfield, C.; Korchev, Y.E.; Lyon, A.R.; et al. Microdomain-Specific Modulation of L-Type Calcium Channels Leads to Triggered Ventricular Arrhythmia in Heart Failure. Circ. Res. 2016, 119, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Manning, J.R.; Chelvarajan, L.; Levitan, B.M.; Withers, C.N.; Nagareddy, P.R.; Haggerty, C.M.; Fornwalt, B.K.; Gao, E.; Tripathi, H.; Abdel-Latif, A.; et al. Rad GTPase deletion attenuates post-ischemic cardiac dysfunction and remodeling. JACC Basic. Transl. Sci. 2018, 3, 83–96. [Google Scholar] [CrossRef]
- Lu, L.; Sirish, P.; Zhang, Z.; Woltz, R.L.; Li, N.; Timofeyev, V.; Knowlton, A.A.; Zhang, X.-D.; Yamoah, E.N.; Chiamvimonvat, N. Regulation of Gene Transcription by Voltage-gated L-type Calcium Channel, Cav1.3. J. Biol. Chem. 2015, 290, 4663–4676. [Google Scholar] [CrossRef]
- Senatore, A.; Guan, W.; Spafford, J.D. Cav3 T-type channels: Regulators for gating, membrane expression, and cation selectivity. Pflug. Arch. 2014, 466, 645–660. [Google Scholar] [CrossRef]
- Le Quang, K.; Naud, P.; Qi, X.; Duval, F.; Shi, Y.; Gillis, M.; Comtois, P.; Tardif, J.; Li, D.; Levesque, P.; et al. Role of T-type calcium channel subunits in post-myocardial infarction remodelling probed with genetically engineered mice. Cardiovasc. Res. 2011, 91, 420–428. [Google Scholar] [CrossRef]
- Ono, K.; Iijima, T. Cardiac T-type Ca2+ channels in the heart. J. Mol. Cell. Cardiol. 2010, 48, 65–70. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, Q.; Hu, H.; Yang, C.; Zhao, Q. Esketamine alleviates hypoxia/reoxygenation injury of cardiomyocytes by regulating TRPV1 expression and inhibiting intracellular Ca2+ concentration. Clinics 2024, 79, 100363. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zhou, X.; Zhao, X.; Wang, Z.; Huang, A.; Huang, Y.; Sun, H.; Guan, F.; Jiang, W. Tetrandrine downregulates TRPV2 expression to ameliorate myocardial ischemia/reperfusion injury in rats via regulation of cardiomyocyte apoptosis, calcium homeostasis and mitochondrial function. Eur. J. Pharmacol. 2024, 964, 176246. [Google Scholar] [CrossRef] [PubMed]
- MacKinnon, R. Potassium channels. FEBS Lett. 2003, 555, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, S.; Kidd, J.; Law, R.; Franks, C.; Sattelle, D. Structure and function of two-pore-domain K+ channels: Contributions from genetic model organisms. Trends Pharmacol. Sci. 2005, 26, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Q.; Purhonen, P.; Hebert, H. Structure of potassium channels. Cell. Mol. Life Sci. 2015, 72, 3677–3693. [Google Scholar] [CrossRef] [PubMed]
- Grandi, E.; Sanguinetti, M.; Bartos, D.; Bers, D.; Chen-Izu, Y.; Chiamvimonvat, N.; Colecraft, H.; Delisle, B.; Heijman, J.; Navedo, M.; et al. Potassium channels in the heart: Structure, function and regulation. J. Physiol. 2017, 595, 2209–2228. [Google Scholar] [CrossRef] [PubMed]
- Isidoro Tavares, N.; Philip-Couderc, P.; Papageorgiou, I.; Baertschi, A.; Lerch, R.; Montessuit, C. Expression and function of ATP-dependent potassium channels in late post-infarction remodeling. J. Mol. Cell. Cardiol. 2007, 42, 1016–1025. [Google Scholar] [CrossRef]
- Kamatham, S.; Waters, C.M.; Schwingshackl, A.; Mancarella, S. TREK-1 protects the heart against ischemia-reperfusion-induced injury and from adverse remodeling after myocardial infarction. Pflug. Arch. 2019, 471, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Qiao, X.; Zhang, L.; Wang, D.; Zhang, L.; Feng, Q.; Wu, B.; Cao, J.; Liu, Q. IK1 channel agonist zacopride suppresses ventricular arrhythmias in conscious rats with healing myocardial infarction. Life Sci. 2019, 239, 117075. [Google Scholar] [CrossRef]
- Kaprielian, R.; Wickenden, A.D.; Kassiri, Z.; Parker, T.G.; Liu, P.P.; Backx, P.H. Relationship between K+ channel down-regulation and [Ca2+]i in rat ventricular myocytes following myocardial infarction. J. Physiol. 1999, 517 Pt 1, 229–245. [Google Scholar] [CrossRef]
- Elasoru, S.E.; Rhana, P.; de Oliveira Barreto, T.; Naves de Souza, D.L.; Menezes-Filho, J.E.R.; Souza, D.S.; Loes Moreira, M.V.; Gomes Campos, M.T.; Adedosu, O.T.; Roman-Campos, D.; et al. Andrographolide protects against isoproterenol-induced myocardial infarction in rats through inhibition of L-type Ca2+ and increase of cardiac transient outward K+ currents. Eur. J. Pharmacol. 2021, 906, 174194. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; van der Heyden, M.A.G. The network of cardiac KIR2.1: Its function, cellular regulation, electrical signaling, diseases and new drug avenues. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2024. [Google Scholar] [CrossRef]
- Song, W.; Shou, W. Cardiac Sodium Channel Nav1.5 Mutations and Cardiac Arrhythmia. Pediatr. Cardiol. 2012, 33, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Sara, N.; Sandra, B.G.; Jesse, B.Y.; Lakshmi, S.; Richard, W.A.; Gordon, F.T.; Manu, B.-J.; L Mario, A. Structural basis of cytoplasmic NaV1.5 and NaV1.4 regulation. J. Gen. Physiol. 2020, 153, e202012722. [Google Scholar] [CrossRef] [PubMed]
- Bers, D. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Shi, H.; Tonggu, L.; Gamal El-Din, T.; Lenaeus, M.; Zhao, Y.; Yoshioka, C.; Zheng, N.; Catterall, W. Structure of the Cardiac Sodium Channel. Cell 2020, 180, 122–134.e10. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.; Wang, L.; Zhong, J. Sodium channels, cardiac arrhythmia, and therapeutic strategy. Adv. Pharmacol. 2014, 70, 367–392. [Google Scholar] [CrossRef] [PubMed]
- Coronel, R.; Lau, D.; Sosunov, E.; Janse, M.; Danilo, P.; Anyukhovsky, E.; Wilms-Schopman, F.; Opthof, T.; Shlapakova, I.; Ozgen, N.; et al. Cardiac expression of skeletal muscle sodium channels increases longitudinal conduction velocity in the canine 1-week myocardial infarction. Heart Rhythm 2010, 7, 1104–1110. [Google Scholar] [CrossRef]
- Kang, G.; Xie, A.; Liu, H.; Dudley, S. MIR448 antagomir reduces arrhythmic risk after myocardial infarction by upregulating the cardiac sodium channel. JCI Insight 2020, 5, e140759. [Google Scholar] [CrossRef]
- Ahern, C.A.; Zhang, J.F.; Wookalis, M.J.; Horn, R. Modulation of the cardiac sodium channel NaV1.5 by Fyn, a Src family tyrosine kinase. Circ. Res. 2005, 96, 991–998. [Google Scholar] [CrossRef]
- Remme, C.A.; Bezzina, C.R. Sodium channel (dys)function and cardiac arrhythmias. Cardiovasc. Ther. 2010, 28, 287–294. [Google Scholar] [CrossRef]
- Dhar Malhotra, J.; Chen, C.; Rivolta, I.; Abriel, H.; Malhotra, R.; Mattei, L.N.; Brosius, F.C.; Kass, R.S.; Isom, L.L. Characterization of sodium channel alpha- and beta-subunits in rat and mouse cardiac myocytes. Circulation 2001, 103, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.H.; Lenkowski, P.W.; Lee, H.C.; Mounsey, J.P.; Patel, M.K. Modulation of Na(v)1.5 by beta1-- and beta3-subunit co-expression in mammalian cells. Pflug. Arch. 2005, 449, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Medeiros-Domingo, A.; Kaku, T.; Tester, D.J.; Iturralde-Torres, P.; Itty, A.; Ye, B.; Valdivia, C.; Ueda, K.; Canizales-Quinteros, S.; Tusié-Luna, M.T.; et al. SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome. Circulation 2007, 116, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Abriel, H.; Kass, R.S. Regulation of the voltage-gated cardiac sodium channel Nav1.5 by interacting proteins. Trends Cardiovasc. Med. 2005, 15, 35–40. [Google Scholar] [CrossRef] [PubMed]
- London, B.; Michalec, M.; Mehdi, H.; Zhu, X.; Kerchner, L.; Sanyal, S.; Viswanathan, P.C.; Pfahnl, A.E.; Shang, L.L.; Madhusudanan, M.; et al. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation 2007, 116, 2260–2268. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Yong, S.L.; Fan, C.; Ni, Y.; Yoo, S.; Zhang, T.; Zhang, X.; Obejero-Paz, C.A.; Rho, H.J.; Ke, T.; et al. Identification of a new co-factor, MOG1, required for the full function of cardiac sodium channel Nav 1.5. J. Biol. Chem. 2008, 283, 6968–6978. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.S.; Verkerk, A.O.; Bhuiyan, Z.A.; Wilde, A.A.; Tan, H.L. Novel Brugada syndrome-causing mutation in ion-conducting pore of cardiac Na+ channel does not affect ion selectivity properties. Acta Physiol. Scand. 2005, 185, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.N.; Yang, P.C.; Behrens, S.B.; Moreno, J.D.; Clancy, C.E. Computational approaches to understand cardiac electrophysiology and arrhythmias. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H766–H783. [Google Scholar] [CrossRef]
- Hale, S.L.; Shryock, J.C.; Belardinelli, L.; Sweeney, M.; Kloner, R.A. Late sodium current inhibition as a new cardioprotective approach. J. Mol. Cell. Cardiol. 2008, 44, 954–967. [Google Scholar] [CrossRef]
- Chu, S.; Wang, W.; Zhang, N.; Liu, T.; Li, J.; Chu, X.; Zuo, S.; Ma, Z.; Ma, D.; Chu, L. Protective effects of 18β-Glycyrrhetinic acid against myocardial infarction: Involvement of PI3K/Akt pathway activation and inhibiting Ca2+ influx via L-type Ca2+ channels. Food Sci. Nutr. 2021, 9, 6831–6843. [Google Scholar] [CrossRef]
- Huang, K.; Huang, D.; Fu, S.; Yang, C.; Liao, Y. Abnormal calcium “sparks” in cardiomyocytes of post-myocardial infarction heart. J. Huazhong Univ. Sci. Technol. Med. Sci. 2008, 28, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.X.; Joshi, A.; Guo, D.; Hlaing, T.; Martin, J.; Xu, X.; Kowey, P.R. Phase 2 reentry as a trigger to initiate ventricular fibrillation during early acute myocardial ischemia. Circulation 2004, 110, 1036–1041. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.F.; Luo, Y.; Ding, Y.J.; Chen, Y.; Pu, S.X.; Wu, H.J.; Wang, Z.F.; Tao, B.B.; Wang, W.W.; Zhu, Y.C. Hydrogen Sulfide Targets the Cys320/Cys529 Motif in Kv4.2 to Inhibit the Ito Potassium Channels in Cardiomyocytes and Regularizes Fatal Arrhythmia in Myocardial Infarction. Antioxid. Redox Signal. 2015, 23, 129–147. [Google Scholar] [CrossRef] [PubMed]
- Balderas, E.; Zhang, J.; Stefani, E.; Toro, L. Mitochondrial BKCa channel. Front. Physiol. 2015, 6, 104. [Google Scholar] [CrossRef] [PubMed]
- Goswami, S.K.; Ponnalagu, D.; Hussain, A.T.; Shah, K.; Karekar, P.; Gururaja Rao, S.; Meredith, A.L.; Khan, M.; Singh, H. Expression and Activation of BK(Ca) Channels in Mice Protects Against Ischemia-Reperfusion Injury of Isolated Hearts by Modulating Mitochondrial Function. Front. Cardiovasc. Med. 2018, 5, 194. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Asp. Med. 2019, 65, 70–99. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, C.; Dries, E.; Popovic, N.; Singh, A.; Haemers, P.; Roderick, H.; Claus, P.; Sipido, K.; Driesen, R. Global fibroblast activation throughout the left ventricle but localized fibrosis after myocardial infarction. Sci. Rep. 2017, 7, 10801. [Google Scholar] [CrossRef] [PubMed]
- Gwanyanya, A.; Mubagwa, K. Emerging role of transient receptor potential (TRP) ion channels in cardiac fibroblast pathophysiology. Front. Physiol. 2022, 13, 968393. [Google Scholar] [CrossRef] [PubMed]
- Fomovsky, G.; Rouillard, A.; Holmes, J. Regional mechanics determine collagen fiber structure in healing myocardial infarcts. J. Mol. Cell. Cardiol. 2012, 52, 1083–1090. [Google Scholar] [CrossRef]
- Pilla, J.; Koomalsingh, K.; McGarvey, J.; Witschey, W.; Dougherty, L.; Gorman, J.; Gorman, R. Regional myocardial three-dimensional principal strains during postinfarction remodeling. Ann. Thorac. Surg. 2015, 99, 770–778. [Google Scholar] [CrossRef]
- Stewart, L.; Turner, N. Channelling the Force to Reprogram the Matrix: Mechanosensitive Ion Channels in Cardiac Fibroblasts. Cells 2021, 10, 990. [Google Scholar] [CrossRef] [PubMed]
- Abraham, D.M.; Lee, T.E.; Watson, L.J.; Mao, L.; Chandok, G.; Wang, H.G.; Frangakis, S.; Pitt, G.S.; Shah, S.H.; Wolf, M.J.; et al. The two-pore domain potassium channel TREK-1 mediates cardiac fibrosis and diastolic dysfunction. J. Clin. Investig. 2018, 128, 4843–4855. [Google Scholar] [CrossRef] [PubMed]
- Pertiwi, K.R.; Hillman, R.M.; Scott, C.A.; Chilton, E.L. Ischemia Reperfusion Injury Produces, and Ischemic Preconditioning Prevents, Rat Cardiac Fibroblast Differentiation: Role of K(ATP) Channels. J. Cardiovasc. Dev. Dis. 2019, 6, 22. [Google Scholar] [CrossRef]
- Davis, J.; Burr, A.R.; Davis, G.F.; Birnbaumer, L.; Molkentin, J.D. A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev. Cell 2012, 23, 705–715. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Li, D.; Zhang, Y.; Tang, B.; Li, G.; Yang, Y.; Yang, D. Transgenic overexpression of transient receptor potential vanilloid subtype 1 attenuates isoproterenol-induced myocardial fibrosis in mice. Int. J. Mol. Med. 2016, 38, 601–609. [Google Scholar] [CrossRef]
- Adapala, R.K.; Thoppil, R.J.; Luther, D.J.; Paruchuri, S.; Meszaros, J.G.; Chilian, W.M.; Thodeti, C.K. TRPV4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J. Mol. Cell. Cardiol. 2013, 54, 45–52. [Google Scholar] [CrossRef]
- Wulff, H.; Köhler, R. Endothelial small-conductance and intermediate-conductance KCa channels: An update on their pharmacology and usefulness as cardiovascular targets. J. Cardiovasc. Pharmacol. 2013, 61, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Stanley, N. Electrical coupling between cardiomyocytes and fibroblasts: Experimental testing of a challenging and important concept. Cardiovasc. Res. 2018, 114, 349–352. [Google Scholar] [CrossRef]
- Camelliti, P.; Devlin, G.P.; Matthews, K.G.; Kohl, P.; Green, C.R. Spatially and temporally distinct expression of fibroblast connexins after sheep ventricular infarction. Cardiovasc. Res. 2004, 62, 415–425. [Google Scholar] [CrossRef]
- Rubart, M.; Tao, W.; Lu, X.-L.; Conway, S.J.; Reuter, S.P.; Lin, S.-F.; Soonpaa, M.H. Electrical coupling between ventricular myocytes and myofibroblasts in the infarcted mouse heart. Cardiovasc. Res. 2018, 114, 389–400. [Google Scholar] [CrossRef]
- Dhanjal, T.S.; Lellouche, N.; von Ruhland, C.J.; Abehsira, G.; Edwards, D.H.; Dubois-Randé, J.-L.; Moschonas, K.; Teiger, E.; Williams, A.J.; George, C.H. Massive Accumulation of Myofibroblasts in the Critical Isthmus Is Associated with Ventricular Tachycardia Inducibility in Post-Infarct Swine Heart. JACC Clin. Electrophysiol. 2017, 3, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Feske, S.; Wulff, H.; Skolnik, E.Y. Ion channels in innate and adaptive immunity. Annu. Rev. Immunol. 2015, 33, 291–353. [Google Scholar] [CrossRef] [PubMed]
- Boukenna, M.; Rougier, J.S.; Aghagolzadeh, P.; Pradervand, S.; Guichard, S.; Hämmerli, A.F.; Pedrazzini, T.; Abriel, H. Multiomics uncover the proinflammatory role of Trpm4 deletion after myocardial infarction in mice. Am. J. Physiol. Heart Circ. Physiol. 2023, 324, H504–H518. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Lee, K.Y.; Chang, K. The Protective Role of TREM2 in the Heterogenous Population of Macrophages during Post-Myocardial Infarction Inflammation. Int. J. Mol. Sci. 2023, 24, 5556. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K.; Nahrendorf, M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science 2013, 339, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Leuschner, F.; Rauch, P.J.; Ueno, T.; Gorbatov, R.; Marinelli, B.; Lee, W.W.; Dutta, P.; Wei, Y.; Robbins, C.; Iwamoto, Y.; et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J. Exp. Med. 2012, 209, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wülfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 2017, 169, 510–522.e20. [Google Scholar] [CrossRef] [PubMed]
- Fei, Y.-D.; Wang, Q.; Hou, J.-W.; Li, W.; Cai, X.-X.; Yang, Y.-L.; Zhang, L.-H.; Wei, Z.-X.; Chen, T.-Z.; Wang, Y.-P.; et al. Macrophages facilitate post myocardial infarction arrhythmias: Roles of gap junction and KCa3.1. Theranostics 2019, 9, 6396–6411. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Bevan, M.J. CD8+ T cells: Foot soldiers of the immune system. Immunity 2011, 35, 161–168. [Google Scholar] [CrossRef]
- Acharya, T.K.; Tiwari, A.; Majhi, R.K.; Goswami, C. TRPM8 channel augments T-cell activation and proliferation. Cell Biol. Int. 2021, 45, 198–210. [Google Scholar] [CrossRef]
- Jairaman, A.; Othy, S.; Dynes, J.L.; Yeromin, A.V.; Zavala, A.; Greenberg, M.L.; Nourse, J.L.; Holt, J.R.; Cahalan, S.M.; Marangoni, F.; et al. Piezo1 channels restrain regulatory T cells but are dispensable for effector CD4+ T cell responses. Sci. Adv. 2021, 7, eabg5859. [Google Scholar] [CrossRef] [PubMed]
- Bertin, S.; Aoki-Nonaka, Y.; Lee, J.; de Jong, P.R.; Kim, P.; Han, T.; Yu, T.; To, K.; Takahashi, N.; Boland, B.S.; et al. The TRPA1 ion channel is expressed in CD4+ T cells and restrains T-cell-mediated colitis through inhibition of TRPV1. Gut 2017, 66, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Fung-Leung, W.P.; Edwards, W.; Liu, Y.; Ngo, K.; Angsana, J.; Castro, G.; Wu, N.; Liu, X.; Swanson, R.V.; Wickenden, A.D. T Cell Subset and Stimulation Strength-Dependent Modulation of T Cell Activation by Kv1.3 Blockers. PLoS ONE 2017, 12, e0170102. [Google Scholar] [CrossRef]
- Ruck, T.; Bock, S.; Pfeuffer, S.; Schroeter, C.B.; Cengiz, D.; Marciniak, P.; Lindner, M.; Herrmann, A.; Liebmann, M.; Kovac, S.; et al. K(2P)18.1 translates T cell receptor signals into thymic regulatory T cell development. Cell Res. 2022, 32, 72–88. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Khan, M.; Mohsin, S. The Regulatory Role of T Cell Responses in Cardiac Remodeling Following Myocardial Infarction. Int. J. Mol. Sci. 2020, 21, 5013. [Google Scholar] [CrossRef] [PubMed]
- Kologrivova, I.; Shtatolkina, M.; Suslova, T.; Ryabov, V. Cells of the Immune System in Cardiac Remodeling: Main Players in Resolution of Inflammation and Repair after Myocardial Infarction. Front. Immunol. 2021, 12, 664457. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Tan, M.; Meng, X.; Dong, J.; Zhang, Y. Effects of potassium channel knockdown on peripheral blood T lymphocytes and NFAT signaling pathway in Xinjiang Kazak patients with hypertension. Clin. Exp. Hypertens. 2023, 45, 2169449. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhao, M.; Liu, X.; Li, Y.; Xu, B.; Zhou, L.; Sun, X.; Sun, W.; Kang, N.; Ji, Z.; et al. Ion channel TRPV2 is critical in enhancing B cell activation and function. J. Exp. Med. 2024, 221, e20221042. [Google Scholar] [CrossRef]
- Mahtani, T.; Sheth, H.; Smith, L.K.; Benedict, L.; Brecier, A.; Ghasemlou, N.; Treanor, B. The ion channel TRPV5 regulates B-cell signaling and activation. Front. Immunol. 2024, 15, 1386719. [Google Scholar] [CrossRef]
- Daverkausen-Fischer, L.; Pröls, F. Regulation of calcium homeostasis and flux between the endoplasmic reticulum and the cytosol. J. Biol. Chem. 2022, 298, 102061. [Google Scholar] [CrossRef]
- Porsch, F.; Mallat, Z.; Binder, C.J. Humoral immunity in atherosclerosis and myocardial infarction: From B cells to antibodies. Cardiovasc. Res. 2021, 117, 2544–2562. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.S.; Alicot, E.M.; Schuerpf, F.; Chiu, I.; Li, J.; Moore, F.D.; Carroll, M.C. Blockade of self-reactive IgM significantly reduces injury in a murine model of acute myocardial infarction. Cardiovasc. Res. 2010, 87, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Elliott, W.J.; Ram, C.V. Calcium channel blockers. J. Clin. Hypertens. 2011, 13, 687–689. [Google Scholar] [CrossRef] [PubMed]
- Frishman, W.H. Calcium channel blockers: Differences between subclasses. Am. J. Cardiovasc. Drugs 2007, 7 (Suppl. S1), 17–23. [Google Scholar] [CrossRef] [PubMed]
- Furberg, C.D.; Psaty, B.M.; Meyer, J.V. Nifedipine. Dose-related increase in mortality in patients with coronary heart disease. Circulation 1995, 92, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Opie, L.H.; Messerli, F.H.; Stason, W.B.; Schmid, C.H.; Niedzwiecki, D.; Whiting, G.W.; Luo, D.; Ross, S.D.; Chalmers, T.C.; Saseen, J.J.; et al. Nifedipine and mortality. Grave defects in the dossier. Circulation 1995, 92, 1068–1073. [Google Scholar] [CrossRef] [PubMed]
- Jamerson, K.; Weber, M.A.; Bakris, G.L.; Dahlöf, B.; Pitt, B.; Shi, V.; Hester, A.; Gupte, J.; Gatlin, M.; Velazquez, E.J. Benazepril plus amlodipine or hydrochlorothiazide for hypertension in high-risk patients. N. Engl. J. Med. 2008, 359, 2417–2428. [Google Scholar] [CrossRef] [PubMed]
- Qiang, S.; Tun Swe, N.; Lang, L. Adenosine and verapamil for no-reflow during primary percutaneous coronary intervention in people with acute myocardial infarction. Cochrane Database Syst. Rev. 2015, 2015, CD009503. [Google Scholar] [CrossRef] [PubMed]
- Sueta, D.; Tabata, N.; Hokimoto, S. Clinical roles of calcium channel blockers in ischemic heart diseases. Hypertens. Res. 2017, 40, 423–428. [Google Scholar] [CrossRef]
- Levitan, B.M.; Ahern, B.M.; Aloysius, A.; Brown, L.; Wen, Y.; Andres, D.A.; Satin, J. Rad-GTPase contributes to heart rate via L-type calcium channel regulation. J. Mol. Cell. Cardiol. 2021, 154, 60–69. [Google Scholar] [CrossRef]
- Viola, H.M.; Jordan, M.C.; Roos, K.P.; Hool, L.C. Decreased myocardial injury and improved contractility after administration of a peptide derived against the alpha-interacting domain of the L-type calcium channel. J. Am. Heart Assoc. 2014, 3, e000961. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Li, M.; Zhao, Z.; Zhang, Y.; Zhang, J.; Zhang, X.; Zhang, Y.; Guan, S.; Chu, L. Mechanisms underlying the cardio-protection of total ginsenosides against myocardial ischemia in rats in vivo and in vitro: Possible involvement of L-type Ca2+ channels, contractility and Ca2+ homeostasis. J. Pharmacol. Sci. 2019, 139, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Chu, X.; Zhang, X.; Bao, Y.; Zhang, Y.; Guo, H.; Liu, Y.; Liu, H.; Zhang, J.; Zhang, Y.; et al. Mechanisms underlying the cardioprotective effect of Salvianic acid A against isoproterenol-induced myocardial ischemia injury in rats: Possible involvement of L-type calcium channels and myocardial contractility. J. Ethnopharmacol. 2016, 189, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Zhang, M.; Zheng, B.; Zhang, Y.; Chu, X.; Liu, Y.; Li, Z.; Han, X.; Chu, L. [8]-Gingerol exerts anti-myocardial ischemic effects in rats via modulation of the MAPK signaling pathway and L-type Ca2+ channels. Pharmacol. Res. Perspect. 2021, 9, e00852. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wen, Z.; Xue, Y.; Han, X.; Ma, D.; Ma, Z.; Wu, Z.; Guan, S.; Sun, S.; Chu, L. Cardioprotective effects of glycyrrhizic acid involve inhibition of calcium influx via L-type calcium channels and myocardial contraction in rats. Naunyn Schmiedebergs Arch. Pharmacol. 2020, 393, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhang, Y.; Liu, M.; Han, X.; Zhang, J.; Zhang, X.; Chu, L. Protective effect of quercetin against myocardial ischemia as a Ca2+ channel inhibitor: Involvement of inhibiting contractility and Ca2+ influx via L-type Ca2+ channels. Arch. Pharm. Res. 2020, 43, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Grover, G.J. Protective effects of ATP-sensitive potassium-channel openers in experimental myocardial ischemia. J. Cardiovasc. Pharmacol. 1994, 24 (Suppl. S4), S18–S27. [Google Scholar] [PubMed]
- Gomma, A.H.; Purcell, H.J.; Fox, K.M. Potassium channel openers in myocardial ischaemia: Therapeutic potential of nicorandil. Drugs 2001, 61, 1705–1710. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; Seetharaman, S.; Paucek, P.; Garlid, K.D. Bioenergetic consequences of opening the ATP-sensitive K+ channel of heart mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H649–H657. [Google Scholar] [CrossRef]
- Maslov, L.N.; Popov, S.V.; Naryzhnaya, N.V.; Mukhomedzyanov, A.V.; Kurbatov, B.K.; Derkachev, I.A.; Boshchenko, A.A.; Prasad, N.R.; Ma, H.; Zhang, Y.; et al. K(ATP) channels are regulators of programmed cell death and targets for the creation of novel drugs against ischemia/reperfusion cardiac injury. Fundam. Clin. Pharmacol. 2023, 37, 1020–1049. [Google Scholar] [CrossRef]
- Xia, Z.; Chen, B.; Zhou, C.; Wang, Y.; Ren, J.; Yao, X.; Yang, Y.; Wan, Q.; Lian, Z. Protective effect of ischaemic postconditioning combined with nicorandil on myocardial ischaemia–reperfusion injury in diabetic rats. BMC Cardiovasc. Disord. 2022, 22, 518. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.F.; Zou, J.; Wang, N.; Ma, H.; Zhu, L.L.; Liu, K.; Liu, M.D.; Wang, K.K.; Xiao, X.Z. Nicorandil alleviates cardiac remodeling and dysfunction post-infarction by up-regulating the nucleolin/autophagy axis. Cell. Signal. 2022, 92, 110272. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.T.; Xiong, Y.Y.; Ning, Y.; Tang, R.J.; Xu, J.Y.; Jiang, W.Y.; Li, X.S.; Zhang, L.L.; Chen, C.; Pan, Q.; et al. Nicorandil-Pretreated Mesenchymal Stem Cell-Derived Exosomes Facilitate Cardiac Repair after Myocardial Infarction via Promoting Macrophage M2 Polarization by Targeting miR-125a-5p/TRAF6/IRF5 Signaling Pathway. Int. J. Nanomed. 2024, 19, 2005–2024. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Chen, X.; Li, K.; Li, H.; Qi, C. Myocardial Perfusion in ST-Segment Elevation Myocardial Infarction Patients after Percutaneous Coronary Intervention: Influencing Factors and Intervention Strategies. Cureus 2023, 15, e42841. [Google Scholar] [CrossRef] [PubMed]
- Ilyas, M.; Noor, M.; Khan, H.S.; Haroon, S.; Farhat, K.; Ali, S. Cardio protective effect of nicorandil in reperfusion injury among patients undergoing primary percutaneous coronary intervention. Pak. J. Med. Sci. 2023, 39, 177–181. [Google Scholar] [CrossRef]
- Shen, L.; Qiu, L.; Liu, J.; Li, N.; Shu, H.; Zhou, N. Clinical Implications of Nicorandil Combined with Trimetazidine in Patients with Coronary Heart Disease: A Real-World Observational Study. Adv. Ther. 2022, 39, 655–673. [Google Scholar] [CrossRef] [PubMed]
- Babic, V.; Petitpain, N.; Guy, C.; Trechot, P.; Bursztejn, A.C.; Faillie, J.L.; Vial, T.; Schmutz, J.L.; Gillet, P. Nicorandil-induced ulcerations: A 10-year observational study of all cases spontaneously reported to the French pharmacovigilance network. Int. Wound J. 2018, 15, 508–518. [Google Scholar] [CrossRef]
- Salloum, F.N.; Takenoshita, Y.; Ockaili, R.A.; Daoud, V.P.; Chou, E.; Yoshida, K.; Kukreja, R.C. Sildenafil and vardenafil but not nitroglycerin limit myocardial infarction through opening of mitochondrial K(ATP) channels when administered at reperfusion following ischemia in rabbits. J. Mol. Cell. Cardiol. 2007, 42, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Hutchings, D.C.; Anderson, S.G.; Caldwell, J.L.; Trafford, A.W. Phosphodiesterase-5 inhibitors and the heart: Compound cardioprotection? Heart 2018, 104, 1244–1250. [Google Scholar] [CrossRef]
- Fox, K.; Thadani, U.; Ma, P.; Nash, S.; Keating, Z.; Czorniak, M.; Gillies, H.; Keltai, M. Sildenafil citrate does not reduce exercise tolerance in men with erectile dysfunction and chronic stable angina. Eur. Heart J. 2003, 24, 2206–2212. [Google Scholar] [CrossRef]
- Mehrotra, N.; Gupta, M.; Kovar, A.; Meibohm, B. The role of pharmacokinetics and pharmacodynamics in phosphodiesterase-5 inhibitor therapy. Int. J. Impot. Res. 2007, 19, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Muirhead, G.J.; Wilner, K.; Colburn, W.; Haug-Pihale, G.; Rouviex, B. The effects of age and renal and hepatic impairment on the pharmacokinetics of sildenafil. Br. J. Clin. Pharmacol. 2002, 53 (Suppl. S1), 21s–30s. [Google Scholar] [CrossRef] [PubMed]
- McCormack, J.; Barr, R.; Wolff, A.; Lopaschuk, G. Ranolazine stimulates glucose oxidation in normoxic, ischemic, and reperfused ischemic rat hearts. Circulation 1996, 93, 135–142. [Google Scholar] [CrossRef] [PubMed]
- MacInnes, A.; Fairman, D.; Binding, P.; Rhodes, J.; Wyatt, M.; Phelan, A.; Haddock, P.; Karran, E. The antianginal agent trimetazidine does not exert its functional benefit via inhibition of mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ. Res. 2003, 93, e26–e32. [Google Scholar] [CrossRef] [PubMed]
- Antzelevitch, C.; Belardinelli, L.; Wu, L.; Fraser, H.; Zygmunt, A.C.; Burashnikov, A.; Di Diego, J.M.; Fish, J.M.; Cordeiro, J.M.; Goodrow, R.J., Jr.; et al. Electrophysiologic properties and antiarrhythmic actions of a novel antianginal agent. J. Cardiovasc. Pharmacol. Ther. 2004, 9 (Suppl. S1), S65–S83. [Google Scholar] [CrossRef] [PubMed]
- Rupprecht, H.J.; vom Dahl, J.; Terres, W.; Seyfarth, K.M.; Richardt, G.; Schultheibeta, H.P.; Buerke, M.; Sheehan, F.H.; Drexler, H. Cardioprotective effects of the Na+/H+ exchange inhibitor cariporide in patients with acute anterior myocardial infarction undergoing direct PTCA. Circulation 2000, 101, 2902–2908. [Google Scholar] [CrossRef] [PubMed]
- Zeymer, U.; Suryapranata, H.; Monassier, J.P.; Opolski, G.; Davies, J.; Rasmanis, G.; Linssen, G.; Tebbe, U.; Schröder, R.; Tiemann, R.; et al. The Na+/H+ exchange inhibitor eniporide as an adjunct to early reperfusion therapy for acute myocardial infarction. Results of the evaluation of the safety and cardioprotective effects of eniporide in acute myocardial infarction (ESCAMI) trial. J. Am. Coll. Cardiol. 2001, 38, 1644–1650. [Google Scholar] [CrossRef] [PubMed]
- Pickell, Z.; Williams, A.M.; Alam, H.B.; Hsu, C.H. Histone Deacetylase Inhibitors: A Novel Strategy for Neuroprotection and Cardioprotection Following Ischemia/Reperfusion Injury. J. Am. Heart Assoc. 2020, 9, e016349. [Google Scholar] [CrossRef]
- Lecour, S.; Andreadou, I.; Bøtker, H.E.; Davidson, S.M.; Heusch, G.; Ruiz-Meana, M.; Schulz, R.; Zuurbier, C.J.; Ferdinandy, P.; Hausenloy, D.J. IMproving Preclinical Assessment of Cardioprotective Therapies (IMPACT) criteria: Guidelines of the EU-CARDIOPROTECTION COST Action. Basic Res. Cardiol. 2021, 116, 52. [Google Scholar] [CrossRef]
- Ferdinandy, P.; Hausenloy, D.J.; Heusch, G.; Baxter, G.F.; Schulz, R. Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol. Rev. 2014, 66, 1142–1174. [Google Scholar] [CrossRef]
- Ferdinandy, P.; Andreadou, I.; Baxter, G.F.; Bøtker, H.E.; Davidson, S.M.; Dobrev, D.; Gersh, B.J.; Heusch, G.; Lecour, S.; Ruiz-Meana, M.; et al. Interaction of Cardiovascular Nonmodifiable Risk Factors, Comorbidities and Comedications with Ischemia/Reperfusion Injury and Cardioprotection by Pharmacological Treatments and Ischemic Conditioning. Pharmacol. Rev. 2023, 75, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, N.; Matsuda, R.; Hata, Y.; Shimamoto, K. Pharmacological characteristics and clinical applications of K201. Curr. Clin. Pharmacol. 2009, 4, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, N. New 1,4-benzothiazepine derivative, K201, demonstrates cardioprotective effects against sudden cardiac cell death and intracellular calcium blocking action. Drug Dev. Res. 1994, 33, 429–438. [Google Scholar] [CrossRef]
- Kong, W.; Huang, W.; Peng, C.; Zhang, B.; Duan, G.; Ma, W.; Huang, Z. Multiple machine learning methods aided virtual screening of NaV1.5 inhibitors. J. Cell. Mol. Med. 2023, 27, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Terashi, G.; Wang, X.; Kihara, D. Protein model refinement for cryo-EM maps using AlphaFold2 and the DAQ score. Acta Crystallogr. D Struct. Biol. 2023, 79, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Samways, D.S.K. Applications for Mass Spectrometry in the Study of Ion Channel Structure and Function. Adv. Exp. Med. Biol. 2019, 1140, 359–375. [Google Scholar] [CrossRef] [PubMed]
- Pliushcheuskaya, P.; Künze, G. Recent Advances in Computer-Aided Structure-Based Drug Design on Ion Channels. Int. J. Mol. Sci. 2023, 24, 9226. [Google Scholar] [CrossRef] [PubMed]
- Imbrici, P.; Nicolotti, O.; Leonetti, F.; Conte, D.; Liantonio, A. Ion Channels in Drug Discovery and Safety Pharmacology. Methods Mol. Biol. 2018, 1800, 313–326. [Google Scholar] [CrossRef]
- Amin, A.S.; Tan, H.L.; Wilde, A.A. Cardiac ion channels in health and disease. Heart Rhythm 2010, 7, 117–126. [Google Scholar] [CrossRef]
- Cao, Y.; Redd, M.A.; Fang, C.; Mizikovsky, D.; Li, X.; Macdonald, P.S.; King, G.F.; Palpant, N.J. New Drug Targets and Preclinical Modelling Recommendations for Treating Acute Myocardial Infarction. Heart Lung Circ. 2023, 32, 852–869. [Google Scholar] [CrossRef]
- Wang, H.W.; Wang, J.W. How cryo-electron microscopy and X-ray crystallography complement each other. Protein Sci. 2017, 26, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.F.; Yuan, C.; Du, Y.M.; Sun, K.L.; Zhang, X.K.; Vogel, H.; Jia, X.D.; Gao, Y.Z.; Zhang, Q.F.; Wang, D.P.; et al. Applications and prospects of cryo-EM in drug discovery. Mil. Med. Res. 2023, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Kramer, R.; Cohen, D. Functional genomics to new drug targets. Nat. Rev. Drug Discov. 2004, 3, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chiu, J.-F.; He, Q.-Y. Chapter 20—Genomics and Proteomics in Drug Design and Discovery. In Pharmacology; Hacker, M., Messer, W., Bachmann, K., Eds.; Academic Press: San Diego, CA, USA, 2009; pp. 561–573. [Google Scholar]
- Liu, J.; Cui, Y.; Tao, Y.; Zhang, H. Study of simulation technology for myocardial ion channels on pharmacological effects. In Proceedings of the 2011 Computing in Cardiology, Hangzhou, China, 18–21 September 2011; pp. 81–84. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, T.; Hui, W.; Huang, M.; Guo, Y.; Yu, M.; Yang, X.; Liu, Y.; Chen, X. Dynamic Changes in Ion Channels during Myocardial Infarction and Therapeutic Challenges. Int. J. Mol. Sci. 2024, 25, 6467. https://doi.org/10.3390/ijms25126467
Song T, Hui W, Huang M, Guo Y, Yu M, Yang X, Liu Y, Chen X. Dynamic Changes in Ion Channels during Myocardial Infarction and Therapeutic Challenges. International Journal of Molecular Sciences. 2024; 25(12):6467. https://doi.org/10.3390/ijms25126467
Chicago/Turabian StyleSong, Tongtong, Wenting Hui, Min Huang, Yan Guo, Meiyi Yu, Xiaoyu Yang, Yanqing Liu, and Xia Chen. 2024. "Dynamic Changes in Ion Channels during Myocardial Infarction and Therapeutic Challenges" International Journal of Molecular Sciences 25, no. 12: 6467. https://doi.org/10.3390/ijms25126467
APA StyleSong, T., Hui, W., Huang, M., Guo, Y., Yu, M., Yang, X., Liu, Y., & Chen, X. (2024). Dynamic Changes in Ion Channels during Myocardial Infarction and Therapeutic Challenges. International Journal of Molecular Sciences, 25(12), 6467. https://doi.org/10.3390/ijms25126467