
The Versatility of Collagen in Pharmacology: Targeting Collagen, Targeting with Collagen
 , , , and
, , , and 
Abstract
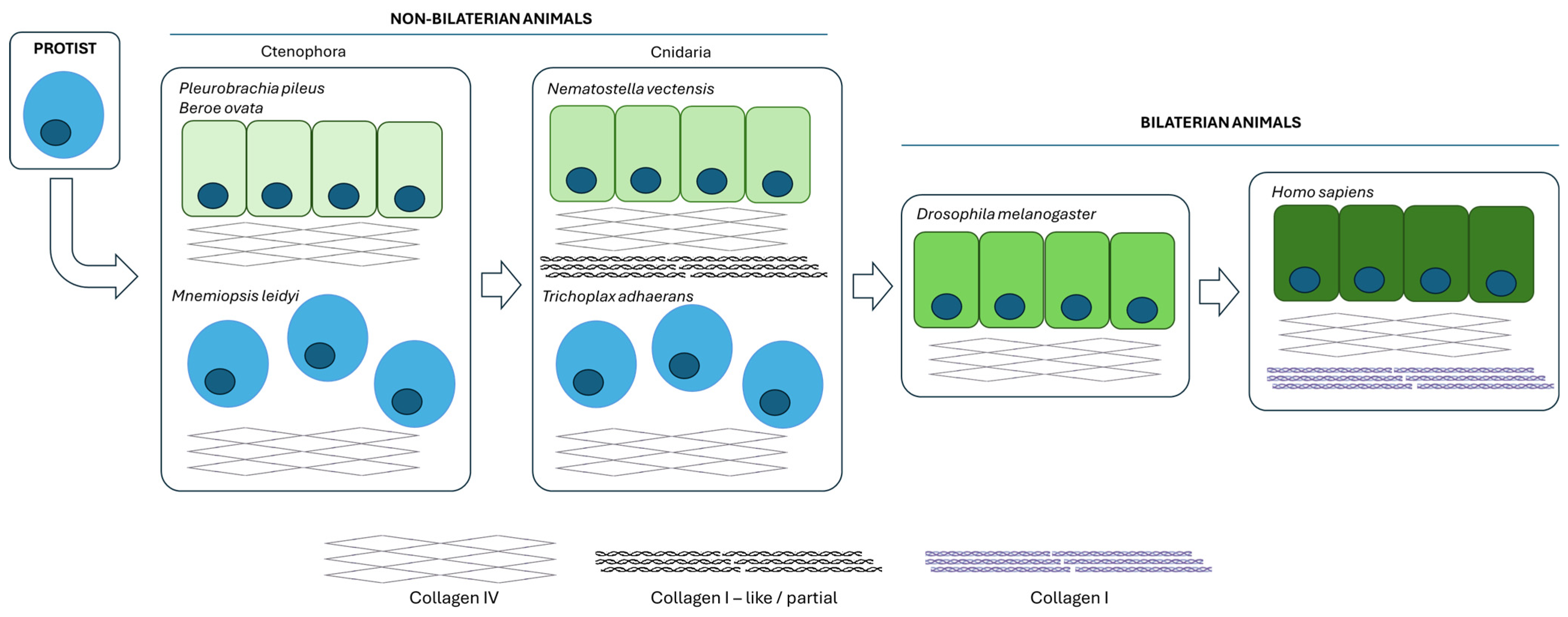
1. Introduction
2. Collagen-Related Diseases
2.1. Fibrosis and Wound Healing
2.2. Cancer
2.3. Hemostasis
2.4. Genetic Disorders
3. Direct Targeting
3.1. Collagenases
3.2. Targeting Collagen with Biding Proteins and Peptides
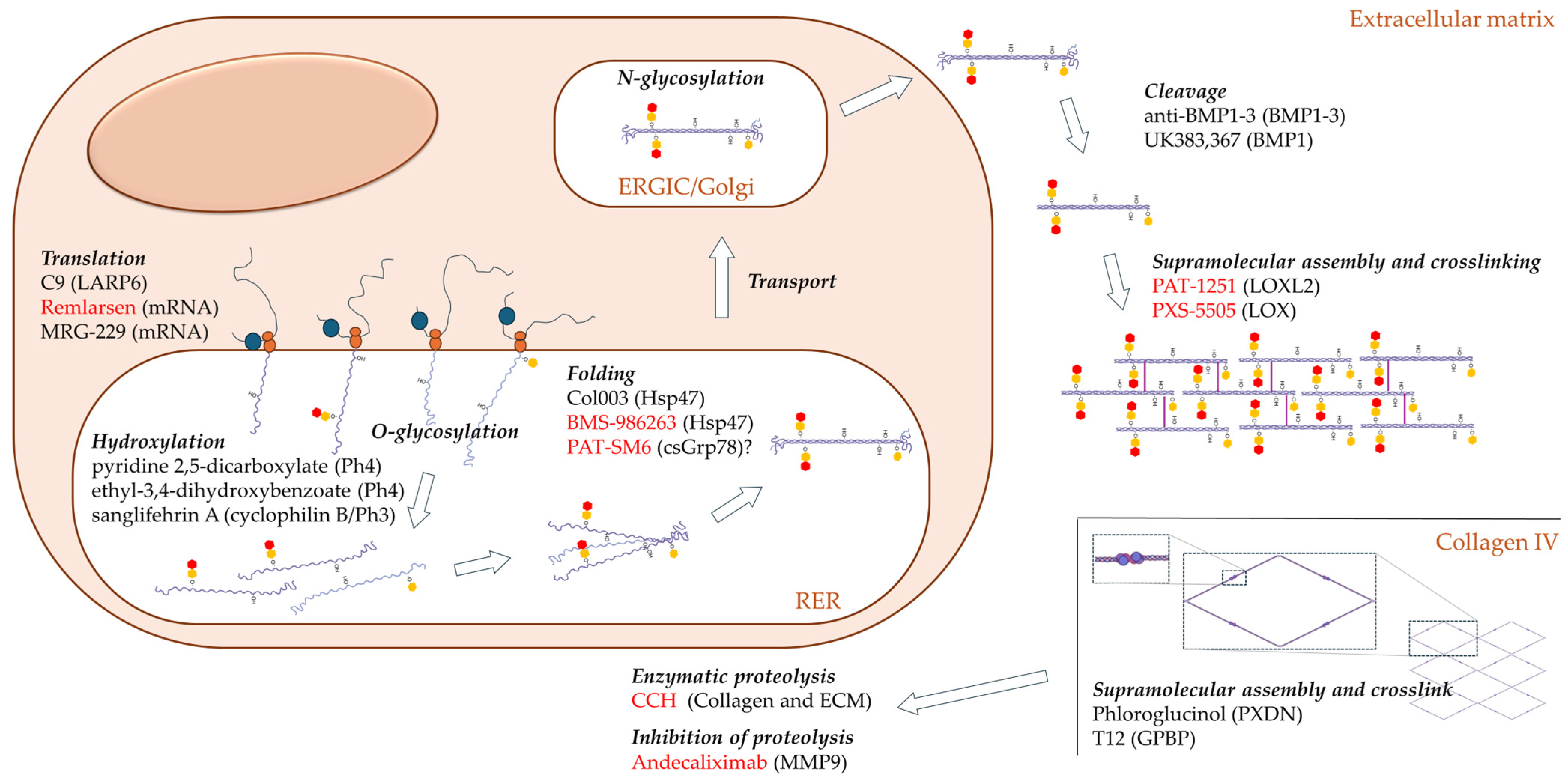
4. Indirect Targeting
4.1. Translation
4.2. Prolyl-Hydroxylation
4.3. Lysil-Hydroxylation
4.4. Glycosylation
4.5. Protein Folding
4.6. Protein Trafficking
4.7. Propeptide Cleavage
4.8. Supramolecular Assembly and Crosslinking of Fibrillar Collagens
5. Indirect Targeting of Collagen IV: Protein Folding and Supramolecular Assembly
6. Targeting with Collagen
6.1. Collagen I
6.2. Collagen II
6.3. Collagen IV
6.4. Collagen VIII
6.5. Collagen XVIII
6.6. Collagens XV and XIX
7. Epilogue: Gene Therapy
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ricard-Blum, S. The Collagen Family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, I.; Muragaki, Y.; Olsen, B.R. Tissue-Specific Forms of Type IX Collagen-Proteoglycan Arise from the Use of Two Widely Separated Promoters. J. Biol. Chem. 1989, 264, 20033–20041. [Google Scholar] [CrossRef]
- Heljasvaara, R.; Aikio, M.; Ruotsalainen, H.; Pihlajaniemi, T. Collagen XVIII in Tissue Homeostasis and Dysregulation—Lessons Learned from Model Organisms and Human Patients. Matrix Biol. 2017, 57–58, 55–75. [Google Scholar] [CrossRef]
- McAlinden, A.; Havlioglu, N.; Liang, L.; Davies, S.R.; Sandell, L.J. Alternative Splicing of Type II Procollagen Exon 2 Is Regulated by the Combination of a Weak 5′ Splice Site and an Adjacent Intronic Stem-Loop Cis Element. J. Biol. Chem. 2005, 280, 32700–32711. [Google Scholar] [CrossRef]
- Van Der Rest, M.; Mayne, R. Type IX Collagen Proteoglycan from Cartilage Is Covalently Cross-Linked to Type II Collagen. J. Biol. Chem. 1988, 263, 1615–1618. [Google Scholar] [CrossRef]
- Koch, M.; Bernasconi, C.; Chiquet, M. A Major Oligomeric Fibroblast Proteoglycan Identified as a Novel Large Form of Type-XII Collagen. Eur. J. Biochem. 1992, 207, 847–856. [Google Scholar] [CrossRef]
- Ehnis, T.; Dieterich, W.; Bauer, M.; Kresse, H.; Schuppan, D. Localization of a Binding Site for the Proteoglycan Decorin on Collagen XIV (Undulin). J. Biol. Chem. 1997, 272, 20414–20419. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.C.; Amenta, P.S.; Dion, A.S.; Sciancalepore, J.P.; Nagaswami, C.; Weisel, J.W.; Yurchenco, P.D. The Molecular Structure of Human Tissue Type XV Presents a Unique Conformation among the Collagens. Biochem. J. 2007, 404, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Halfter, W.; Dong, S.; Schurer, B.; Cole, G.J. Collagen XVIII Is a Basement Membrane Heparan Sulfate Proteoglycan. J. Biol. Chem. 1998, 273, 25404–25412. [Google Scholar] [CrossRef]
- Callebaut, I.; Mignotte, V.; Souchet, M.; Mornon, J.-P. EMI Domains Are Widespread and Reveal the Probable Orthologs of the Caenorhabditis Elegans CED-1 Protein. Biochem. Biophys. Res. Commun. 2003, 300, 619–623. [Google Scholar] [CrossRef]
- Ye, J.J.; Bian, X.; Lim, J.; Medzhitov, R. Adiponectin and Related C1q/TNF-Related Proteins Bind Selectively to Anionic Phospholipids and Sphingolipids. Proc. Natl. Acad. Sci. USA 2020, 117, 17381–17388. [Google Scholar] [CrossRef] [PubMed]
- Fidler, A.L.; Darris, C.E.; Chetyrkin, S.V.; Pedchenko, V.K.; Boudko, S.P.; Brown, K.L.; Gray Jerome, W.; Hudson, J.K.; Rokas, A.; Hudson, B.G. Collagen IV and Basement Membrane at the Evolutionary Dawn of Metazoan Tissues. eLife 2017, 6, e24176. [Google Scholar] [CrossRef] [PubMed]
- Fidler, A.L.; Boudko, S.P.; Rokas, A.; Hudson, B.G. The Triple Helix of Collagens—An Ancient Protein Structure That Enabled Animal Multicellularity and Tissue Evolution. J. Cell Sci. 2018, 131, jcs203950. [Google Scholar] [CrossRef] [PubMed]
- Calvete, J.J.; Revert, F.; Blanco, M.; Cervera, J.; Tárrega, C.; Sanz, L.; Revert-Ros, F.; Granero, F.; Pérez-Payá, E.; Hudson, B.G.; et al. Conformational Diversity of the Goodpasture Antigen, the Noncollagenous-1 Domain of the A3 Chain of Collagen IV. Proteomics 2006, 6, S237–S244. [Google Scholar] [CrossRef]
- Revert, F.; Revert-Ros, F.; Blasco, R.; Artigot, A.; López-Pascual, E.; Gozalbo-Rovira, R.; Ventura, I.; Gutiérrez-Carbonell, E.; Roda, N.; Ruíz-Sanchis, D.; et al. Selective Targeting of Collagen IV in the Cancer Cell Microenvironment Reduces Tumor Burden. Oncotarget 2018, 9, 11020. [Google Scholar] [CrossRef] [PubMed]
- Casino, P.; Gozalbo-Rovira, R.; Rodríguez-Díaz, J.; Banerjee, S.; Boutaud, A.; Rubio, V.; Hudson, B.G.; Saus, J.; Cervera, J.; Marina, A. Structures of Collagen IV Globular Domains: Insight into Associated Pathologies, Folding and Network Assembly. IUCrJ 2018, 5, 765–779. [Google Scholar] [CrossRef] [PubMed]
- Antar, S.A.; Ashour, N.A.; Marawan, M.E.; Al-Karmalawy, A.A. Fibrosis: Types, Effects, Markers, Mechanisms for Disease Progression, and Its Relation with Oxidative Stress, Immunity, and Inflammation. Int. J. Mol. Sci. 2023, 24, 4004. [Google Scholar] [CrossRef] [PubMed]
- Kasashima, H.; Duran, A.; Martinez-Ordoñez, A.; Nakanishi, Y.; Kinoshita, H.; Linares, J.F.; Reina-Campos, M.; Kudo, Y.; L’Hermitte, A.; Yashiro, M.; et al. Stromal SOX2 Upregulation Promotes Tumorigenesis through the Generation of a SFRP1/2-Expressing Cancer-Associated Fibroblast Population. Dev. Cell 2021, 56, 95–110.e10. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of Fibrosis: Therapeutic Translation for Fibrotic Disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Jessen, H.; Hoyer, N.; Prior, T.S.; Frederiksen, P.; Karsdal, M.A.; Leeming, D.J.; Bendstrup, E.; Sand, J.M.B.; Shaker, S.B. Turnover of Type I and III Collagen Predicts Progression of Idiopathic Pulmonary Fibrosis. Respir. Res. 2021, 22, 205. [Google Scholar] [CrossRef]
- Savin, I.A.; Zenkova, M.A.; Sen’kova, A.V. Pulmonary Fibrosis as a Result of Acute Lung Inflammation: Molecular Mechanisms, Relevant In Vivo Models, Prognostic and Therapeutic Approaches. Int. J. Mol. Sci. 2022, 23, 14959. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Jayasinghe, G.; Deore, S.; Wahed, K.; Bhan, K.; Bakti, N.; Singh, B. Dupuytren’s Contracture–Current Concepts. J. Clin. Orthop. Trauma 2020, 11, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.L.; Alom, M.; Trost, L. The Etiology of Peyronie’s Disease: Pathogenesis and Genetic Contributions. Sex. Med. Rev. 2020, 8, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Iovanna, J.; Santofimia-Castaño, P. Targeting Fibrosis: The Bridge That Connects Pancreatitis and Pancreatic Cancer. Int. J. Mol. Sci. 2021, 22, 4970. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac Fibrosis. Cardiovasc. Res. 2020, 117, 1450–1488. [Google Scholar] [CrossRef]
- Simões, F.C.; Cahill, T.J.; Kenyon, A.; Gavriouchkina, D.; Vieira, J.M.; Sun, X.; Pezzolla, D.; Ravaud, C.; Masmanian, E.; Weinberger, M.; et al. Macrophages Directly Contribute Collagen to Scar Formation during Zebrafish Heart Regeneration and Mouse Heart Repair. Nat. Commun. 2020, 11, 600. [Google Scholar] [CrossRef]
- Sobolewski, P.; Maślińska, M.; Wieczorek, M.; Łagun, Z.; Malewska, A.; Roszkiewicz, M.; Nitskovich, R.; Szymańska, E.; Walecka, I. Systemic Sclerosis–Multidisciplinary Disease: Clinical Features and Treatment. Reumatologia 2019, 57, 221–233. [Google Scholar] [CrossRef]
- Andrews, J.P.; Marttala, J.; Macarak, E.; Rosenbloom, J.; Uitto, J. Keloids: The Paradigm of Skin Fibrosis—Pathomechanisms and Treatment. Matrix Biol. 2016, 51, 37–46. [Google Scholar] [CrossRef]
- Revert, F.; Merino, R.; Monteagudo, C.; Macias, J.; Peydró, A.; Alcácer, J.; Muniesa, P.; Marquina, R.; Blanco, M.; Iglesias, M. Increased Goodpasture Antigen-Binding Protein Expression Induces Type IV Collagen Disorganization and Deposit of Immunoglobulin A in Glomerular Basement Membrane. Am. J. Pathol. 2007, 171, 1419–1430. [Google Scholar] [CrossRef]
- Bülow, R.D.; Boor, P. Extracellular Matrix in Kidney Fibrosis: More Than Just a Scaffold. J. Histochem. Cytochem. 2019, 67, 643–661. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, J.; Zhang, H.; Wang, J.; Hua, H.; Jiang, Y. The Role of Network-Forming Collagens in Cancer Progression. Int. J. Cancer 2022, 151, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Bourgot, I.; Primac, I.; Louis, T.; Noël, A.; Maquoi, E. Reciprocal Interplay Between Fibrillar Collagens and Collagen-Binding Integrins: Implications in Cancer Progression and Metastasis. Front. Oncol. 2020, 10, 1488. [Google Scholar] [CrossRef]
- De Martino, D.; Bravo-Cordero, J.J. Collagens in Cancer: Structural Regulators and Guardians of Cancer Progression. Cancer Res. 2023, 83, 1386–1392. [Google Scholar] [CrossRef]
- Brisson, B.K.; Mauldin, E.A.; Lei, W.; Vogel, L.K.; Power, A.M.; Lo, A.; Dopkin, D.; Khanna, C.; Wells, R.G.; Puré, E.; et al. Type III Collagen Directs Stromal Organization and Limits Metastasis in a Murine Model of Breast Cancer. Am. J. Pathol. 2015, 185, 1471–1486. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, J.S.; Nobre, A.R.; Mondal, C.; Taha, I.; Farias, E.F.; Fertig, E.J.; Naba, A.; Aguirre-Ghiso, J.A.; Bravo-Cordero, J.J. A Tumor-Derived Type III Collagen-Rich ECM Niche Regulates Tumor Cell Dormancy. Nat. Cancer 2022, 3, 90–107. [Google Scholar] [CrossRef] [PubMed]
- Slobodianuk, T.L.; Kochelek, C.; Foeckler, J.; Kalloway, S.; Weiler, H.; Flood, V.H. Defective Collagen Binding and Increased Bleeding in a Murine Model of von Willebrand Disease Affecting Collagen IV Binding. J. Thromb. Haemost. 2019, 17, 63–71. [Google Scholar] [CrossRef]
- Pareti, F.I.; Niiya, K.; McPherson, J.M.; Ruggeri, Z.M. Isolation and Characterization of Two Domains of Human von Willebrand Factor That Interact with Fibrillar Collagen Types I and III. J. Biol. Chem. 1987, 262, 13835–13841. [Google Scholar] [CrossRef]
- Lamandé, S.R.; Bateman, J.F. Genetic Disorders of the Extracellular Matrix. Anat. Rec. 2020, 303, 1527–1542. [Google Scholar] [CrossRef]
- Marom, R.; Rabenhorst, B.M.; Morello, R. Osteogenesis Imperfecta: An Update on Clinical Features and Therapies. Eur. J. Endocrinol. 2020, 183, R95–R106. [Google Scholar] [CrossRef]
- Has, C.; Bauer, J.W.; Bodemer, C.; Bolling, M.C.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.-D.; Heagerty, A.; Hovnanian, A.; Marinkovich, M.P.; et al. Consensus Reclassification of Inherited Epidermolysis Bullosa and Other Disorders with Skin Fragility. Br. J. Dermatol. 2020, 183, 614–627. [Google Scholar] [CrossRef] [PubMed]
- Badalamente, M.A.; Hurst, L.C.; Benhaim, P.; Cohen, B.M. Efficacy and Safety of Collagenase Clostridium Histolyticum in the Treatment of Proximal Interphalangeal Joints in Dupuytren Contracture: Combined Analysis of 4 Phase 3 Clinical Trials. J. Hand. Surg. Am. 2015, 40, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Cerveró, R.; Vazquez-Ferreiro, P.; Gómez-Herrero, D.; Carrera-Hueso, F.J.; Fikri-Banbrahim, N. Evolución al Año de Tratamiento Con CCH Para La Contractura de Dupuytren: Estudio Prospectivo. Rev. Esp. Cir. Ortop. Traumatol. 2018, 62, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Zarb, R.M.; Graf, A.R.; Talhelm, J.E.; Stehr, R.C.; Sanger, J.R.; Matloub, H.S.; Daley, R.A. Dupuytren’s Contracture Recurrence and Treatment Following Collagenase Clostridium Histolyticum Injection: A Longitudinal Assessment in a Veteran Population. Mil. Med. 2023, 188, e2975–e2981. [Google Scholar] [CrossRef] [PubMed]
- Teloken, P.; Katz, D. Medical Management of Peyronie’s Disease: Review of the Clinical Evidence. Med. Sci. 2019, 7, 96. [Google Scholar] [CrossRef] [PubMed]
- Xiapex | European Medicines Agency. Document: EMA/95504/2020. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/xiapex (accessed on 31 May 2024).
- Endo Pharmaceuticals. A Phase 2B, Open-Label Study to Explore Tissue Histopathology Following Subcutaneous Injection of Collagenase Clostridium Histolyticum Using an Abdominoplasty Model. clinicaltrials.gov, 2022. Available online: https://www.clinicaltrials.gov/search?term=NCT04236635 (accessed on 7 June 2024).
- Fitzpatrick, J.; Richardson, C.; Klaber, I.; Richardson, M.D. Clostridium Histolyticum (AA4500) for the Treatment of Adhesive Capsulitis of the Shoulder: A Randomised Double-Blind, Placebo-Controlled Study for the Safety and Efficacy of Collagenase-Single Site Report. Drug Des. Dev. Ther. 2020, 14, 2707–2713. [Google Scholar] [CrossRef] [PubMed]
- García-Olmo, D.; Villarejo Campos, P.; Barambio, J.; Gomez-Heras, S.G.; Vega-Clemente, L.; Olmedillas-Lopez, S.; Guadalajara, H.; Garcia-Arranz, M. Intraperitoneal Collagenase as a Novel Therapeutic Approach in an Experimental Model of Colorectal Peritoneal Carcinomatosis. Sci. Rep. 2021, 11, 503. [Google Scholar] [CrossRef] [PubMed]
- Dolor, A.; Szoka, F. Digesting a Path Forward: The Utility of Collagenase Tumor Treatment for Improved Drug Delivery. Mol. Pharm. 2018, 15, 2069–2083. [Google Scholar] [CrossRef] [PubMed]
- Sheets, A.R.; Demidova-Rice, T.N.; Shi, L.; Ronfard, V.; Grover, K.V.; Herman, I.M. Identification and Characterization of Novel Matrix-Derived Bioactive Peptides: A Role for Collagenase from Santyl® Ointment in Post-Debridement Wound Healing? PLoS ONE 2016, 11, e0159598. [Google Scholar] [CrossRef]
- Das, A.; Datta, S.; Roche, E.; Chaffee, S.; Jose, E.; Shi, L.; Grover, K.; Khanna, S.; Sen, C.K.; Roy, S. Novel Mechanisms of Collagenase Santyl Ointment (CSO) in Wound Macrophage Polarization and Resolution of Wound Inflammation. Sci. Rep. 2018, 8, 1696. [Google Scholar] [CrossRef]
- Evrosimovska Andonovska, B.; Boris, V.; Dimova, C.; Veleska, D. Matrix Metalloproteinases (with Accent to Collagenases). J. Cell Anim. Biol. 2011, 5, 113–120. [Google Scholar]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuña, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.P.; Hansch, C. Matrix Metalloproteinases (MMPs): Chemical-Biological Functions and (Q)SARs. Bioorg. Med. Chem. 2007, 15, 2223–2268. [Google Scholar] [CrossRef] [PubMed]
- Winer, A.; Adams, S.; Mignatti, P. Matrix Metalloproteinase Inhibitors in Cancer Therapy: Turning Past Failures into Future Successes. Mol. Cancer Ther. 2018, 17, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A.; Bodoky, G.; Starodub, A.; Cunningham, D.; Yip, D.; Wainberg, Z.A.; Bendell, J.; Thai, D.; He, J.; Bhargava, P.; et al. Phase III Study to Evaluate Efficacy and Safety of Andecaliximab With mFOLFOX6 as First-Line Treatment in Patients With Advanced Gastric or GEJ Adenocarcinoma (GAMMA-1). J. Clin. Oncol. 2021, 39, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A.; Cunningham, D.; Metges, J.-P.; Van Cutsem, E.; Wainberg, Z.; Elboudwarej, E.; Lin, K.-W.; Turner, S.; Zavodovskaya, M.; Inzunza, D.; et al. Randomized, Open-Label, Phase 2 Study of Andecaliximab plus Nivolumab versus Nivolumab Alone in Advanced Gastric Cancer Identifies Biomarkers Associated with Survival. J. Immunother. Cancer 2021, 9, e003580. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.; Siegel, C.A.; Friedenberg, K.A.; Younes, Z.H.; Seidler, U.; Bhandari, B.R.; Wang, K.; Wendt, E.; McKevitt, M.; Zhao, S.; et al. A Phase 2, Randomized, Placebo-Controlled Study Evaluating Matrix Metalloproteinase-9 Inhibitor, Andecaliximab, in Patients With Moderately to Severely Active Crohn’s Disease. J. Crohns Colitis 2018, 12, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Addi, C.; Murschel, F.; De Crescenzo, G. Design and Use of Chimeric Proteins Containing a Collagen-Binding Domain for Wound Healing and Bone Regeneration. Tissue Eng. Part B Rev. 2016, 23, 163–182. [Google Scholar] [CrossRef] [PubMed]
- Ingham, K.; Brew, S.; Isaacs, B. Interaction of Fibronectin and Its Gelatin-Binding Domains with Fluorescent-Labeled Chains of Type I Collagen. J. Biol. Chem. 1988, 263, 4624–4628. [Google Scholar] [CrossRef]
- An, B.; Abbonante, V.; Yigit, S.; Balduini, A.; Kaplan, D.L.; Brodsky, B. Definition of the Native and Denatured Type II Collagen Binding Site for Fibronectin Using a Recombinant Collagen System. J. Biol. Chem. 2014, 289, 4941–4951. [Google Scholar] [CrossRef]
- Patten, J.; Wang, K. Fibronectin in Development and Wound Healing. Adv. Drug Deliv. Rev. 2020, 170, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Yee, A.; Gildersleeve, R.; Gu, S.; Kretz, C.; McGee, B.; Carr, K.; Pipe, S.; Ginsburg, D. A von Willebrand Factor Fragment Containing the D’D3 Domains Is Sufficient to Stabilize Coagulation Factor VIII in Mice. Blood 2014, 124, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Bergmeier, W.; Hynes, R.O. Extracellular Matrix Proteins in Hemostasis and Thrombosis. Cold Spring Harb. Perspect. Biol. 2012, 4, a005132. [Google Scholar] [CrossRef]
- Tan, H.; Song, Y.; Jin, J.; Zhao, X.; Chen, J.; Qing, Z.; Yu, S.; Huang, L. vWF A3-GPI Modification of EPCs Accelerates Reendothelialization of Injured Vessels via Collagen Targeting in Mice. J. Drug Target. 2016, 24, 744–751. [Google Scholar] [CrossRef]
- Ishihara, J.; Ishihara, A.; Sasaki, K.; Lee, S.S.-Y.; Williford, J.-M.; Yasui, M.; Abe, H.; Potin, L.; Hosseinchi, P.; Fukunaga, K.; et al. Targeted Antibody and Cytokine Cancer Immunotherapies through Collagen Affinity. Sci. Transl. Med. 2019, 11, eaau3259. [Google Scholar] [CrossRef] [PubMed]
- Dewerchin, M.; Carmeliet, P. PlGF: A Multitasking Cytokine with Disease-Restricted Activity. Cold Spring Harb. Perspect. Med. 2012, 2, a011056. [Google Scholar] [CrossRef]
- Martino, M.; Briquez, P.; Guc, E.; Tortelli, F.; Kilarski, W.; Metzger, S.; Rice, J.; Kuhn, G.; Müller, R.; Swartz, M.; et al. Growth Factors Engineered for Super-Affinity to the Extracellular Matrix Enhance Tissue Healing. Science 2014, 343, 885–888. [Google Scholar] [CrossRef] [PubMed]
- de Souza, S.J.; Brentani, R. Collagen Binding Site in Collagenase Can Be Determined Using the Concept of Sense-Antisense Peptide Interactions. J. Biol. Chem. 1992, 267, 13763–13767. [Google Scholar] [CrossRef]
- Chen, B.; Lin, H.; Wang, J.; Zhao, Y.; Wang, B.; Zhao, W.; Sun, W.; Dai, J. Homogeneous Osteogenesis and Bone Regeneration by Demineralized Bone Matrix Loading with Collagen-Targeting Bone Morphogenetic Protein-2. Biomaterials 2007, 28, 1027–1035. [Google Scholar] [CrossRef]
- Yu, S.; Li, Y.; Kim, D. Collagen Mimetic Peptides: Progress Towards Functional Applications. Soft Matter. 2011, 7, 7927–7938. [Google Scholar] [CrossRef]
- Chakravarti, S.; Stallings, R.L.; SundarRaj, N.; Cornuet, P.K.; Hassell, J.R. Primary Structure of Human Lumican (Keratan Sulfate Proteoglycan) and Localization of the Gene (LUM) to Chromosome 12q21.3-Q22. Genomics 1995, 27, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, Y.; Cao, Y.; Wang, H.; Zhou, Y.; Gao, L.; Zeng, Z.; Cheng, M.; Jin, X.; Chen, J.; et al. Lumican Is Elevated in the Lung in Human and Experimental Acute Respiratory Distress Syndrome and Promotes Early Fibrotic Responses to Lung Injury. J. Transl. Med. 2022, 20, 392. [Google Scholar] [CrossRef] [PubMed]
- Momin, N.; Mehta, N.K.; Bennett, N.R.; Ma, L.; Palmeri, J.R.; Chinn, M.M.; Lutz, E.A.; Kang, B.; Irvine, D.J.; Spranger, S.; et al. Anchoring of Intratumorally Administered Cytokines to Collagen Safely Potentiates Systemic Cancer Immunotherapy. Sci. Transl. Med. 2019, 11, eaaw2614. [Google Scholar] [CrossRef] [PubMed]
- Avilés-Reyes, A.; Miller, J.H.; Lemos, J.A.; Abranches, J. Collagen-Binding Proteins of Streptococcus Mutans and Related Streptococci. Mol. Oral Microbiol. 2017, 32, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Hulme, J.T.; D’Souza, W.N.; McBride, H.J.; Yoon, B.-R.P.; Willee, A.M.; Duguay, A.; Thomas, M.; Fan, B.; Dayao, M.R.; Rottman, J.B.; et al. Novel Protein Therapeutic Joint Retention Strategy Based on Collagen-Binding Avimers. J. Orthop. Res. 2018, 36, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Stefanovic, B. mTORC1 Phosphorylates LARP6 to Stimulate Type I Collagen Expression. Sci. Rep. 2017, 7, 41173. [Google Scholar] [CrossRef] [PubMed]
- Stefanovic, B.; Manojlovic, Z.; Vied, C.; Badger, C.-D.; Stefanovic, L. Discovery and Evaluation of Inhibitor of LARP6 as Specific Antifibrotic Compound. Sci. Rep. 2019, 9, 326. [Google Scholar] [CrossRef] [PubMed]
- Szwed, A.; Kim, E.; Jacinto, E. Regulation and Metabolic Functions of mTORC1 and mTORC2. Physiol. Rev. 2021, 101, 1371–1426. [Google Scholar] [CrossRef] [PubMed]
- Verdura, S.; Cuyàs, E.; Ruiz-Torres, V.; Micol, V.; Joven, J.; Bosch-Barrera, J.; Menendez, J.A. Lung Cancer Management with Silibinin: A Historical and Translational Perspective. Pharmaceuticals 2021, 14, 559. [Google Scholar] [CrossRef]
- Choi, S.; Ham, S.; Lee, Y.I.; Kim, J.; Lee, W.J.; Lee, J.H. Silibinin Downregulates Types I and III Collagen Expression via Suppression of the mTOR Signaling Pathway. Int. J. Mol. Sci. 2023, 24, 14386. [Google Scholar] [CrossRef]
- Suzuki, H.I. Roles of MicroRNAs in Disease Biology. JMA J. 2023, 6, 104–113. [Google Scholar] [CrossRef]
- Cushing, L.; Kuang, P.P.; Qian, J.; Shao, F.; Wu, J.; Little, F.; Thannickal, V.J.; Cardoso, W.V.; Lü, J. miR-29 Is a Major Regulator of Genes Associated with Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2011, 45, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Liu, L.; Liu, S.; Song, S.; Li, H. MicroRNA-29a Inhibits Collagen Expression and Induces Apoptosis in Human Fetal Scleral Fibroblasts by Targeting the Hsp47/Smad3 Signaling Pathway. Exp. Eye Res. 2022, 225, 109275. [Google Scholar] [CrossRef]
- Cushing, L.; Kuang, P.; Lü, J. The Role of miR-29 in Pulmonary Fibrosis. Biochem. Cell Biol. 2015, 93, 109–118. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Itami, S.; Kuroda, M.; Yoshizato, K.; Kawada, N.; Murakami, Y. MiR-29a Assists in Preventing the Activation of Human Stellate Cells and Promotes Recovery From Liver Fibrosis in Mice. Mol. Ther. 2016, 24, 1848–1859. [Google Scholar] [CrossRef] [PubMed]
- Chioccioli, M.; Roy, S.; Newell, R.; Pestano, L.; Dickinson, B.; Rigby, K.; Herazo-Maya, J.; Jenkins, G.; Ian, S.; Saini, G.; et al. A Lung Targeted miR-29 Mimic as a Therapy for Pulmonary Fibrosis. eBioMedicine 2022, 85, 104304. [Google Scholar] [CrossRef]
- miRagen Therapeutics, Inc. A Phase 2, Double-Blind, Placebo-Controlled Study to Investigate the Efficacy, Safety and Tolerability of MRG-201 Following Intradermal Injection in Subjects With a History of Keloids. clinicaltrials.gov, 2021. Available online: https://www.clinicaltrials.gov/search?term=NCT03601052 (accessed on 7 June 2024).
- Devos, H.; Zoidakis, J.; Roubelakis, M.; Latosinska, A.; Vlahou, A. Reviewing the Regulators of COL1A1. Int. J. Mol. Sci. 2023, 24, 10004. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.-H.; Li, L.-C.; Yang, S.-F.; Tsai, C.-F.; Chuang, Y.-T.; Chu, H.-J.; Ueng, K.-C. MicroRNA Let-7a, -7e and -133a Attenuate Hypoxia-Induced Atrial Fibrosis via Targeting Collagen Expression and the JNK Pathway in HL1 Cardiomyocytes. Int. J. Mol. Sci. 2022, 23, 9636. [Google Scholar] [CrossRef]
- Koski, M.K.; Anantharajan, J.; Kursula, P.; Dhavala, P.; Murthy, A.V.; Bergmann, U.; Myllyharju, J.; Wierenga, R.K. Assembly of the Elongated Collagen Prolyl 4-Hydroxylase A2β2 Heterotetramer around a Central A2 Dimer. Biochem. J. 2017, 474, 751–769. [Google Scholar] [CrossRef]
- Rappu, P.; Salo, A.M.; Myllyharju, J.; Heino, J. Role of Prolyl Hydroxylation in the Molecular Interactions of Collagens. Essays Biochem. 2019, 63, 325–335. [Google Scholar] [CrossRef]
- Luo, Y.; Xu, W.; Chen, H.; Warburton, D.; Dong, R.; Qian, B.; Selman, M.; Gauldie, J.; Kolb, M.; Shi, W. A Novel Profibrotic Mechanism Mediated by TGF-β-Stimulated Collagen Prolyl Hydroxylase Expression in Fibrotic Lung Mesenchymal Cells. J. Pathol. 2015, 236, 384. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Cao, Y.; Zhao, H.; Wang, P.; Zhu, Z. Prolyl 4-Hydroxylase P4HA1 Mediates the Interplay Between Glucose Metabolism and Stemness in Pancreatic Cancer Cells. Curr. Stem Cell Res. Ther. 2023, 18, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Chaturvedi, P.; Bajpai, S.; Wong, C.C.; Wei, H.; Pitcairn, S.; Hubbi, M.E.; Wirtz, D.; Semenza, G.L. Collagen Prolyl Hydroxylases Are Essential for Breast Cancer Metastasis. Cancer Res. 2013, 73, 3285–3296. [Google Scholar] [CrossRef] [PubMed]
- Gjaltema, R.A.F.; Bank, R.A. Molecular Insights into Prolyl and Lysyl Hydroxylation of Fibrillar Collagens in Health and Disease. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 74–95. [Google Scholar] [CrossRef] [PubMed]
- Terajima, M.; Taga, Y.; Cabral, W.A.; Nagasawa, M.; Sumida, N.; Hattori, S.; Marini, J.C.; Yamauchi, M. Cyclophilin B Deficiency Causes Abnormal Dentin Collagen Matrix. J. Proteome Res. 2017, 16, 2914–2923. [Google Scholar] [CrossRef] [PubMed]
- Marini, J.C.; Cabral, W.A.; Barnes, A.M.; Chang, W. Components of the Collagen Prolyl 3-Hydroxylation Complex Are Crucial for Normal Bone Development. Cell Cycle 2007, 6, 1675–1681. [Google Scholar] [CrossRef] [PubMed]
- Flaxman, H.A.; Chrysovergi, M.-A.; Han, H.; Kabir, F.; Lister, R.T.; Chang, C.-F.; Black, K.E.; Lagares, D.; Woo, C.M. Sanglifehrin A Mitigates Multi-Organ Fibrosis in Vivo by Inducing Secretion of the Collagen Chaperone Cyclophilin B. bioRxiv 2023. [Google Scholar] [CrossRef] [PubMed]
- Pokidysheva, E.; Boudko, S.; Vranka, J.; Zientek, K.; Maddox, K.; Moser, M.; Fässler, R.; Ware, J.; Bächinger, H.P. Biological Role of Prolyl 3-Hydroxylation in Type IV Collagen. Proc. Natl. Acad. Sci. USA 2014, 111, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Pang, K.; Pang, H.; Zhang, J.; Zhang, Z.; He, H.; Zhou, R.; Shi, Z.; Han, C. Knockdown of P3H4 Inhibits Proliferation and Invasion of Bladder Cancer. Aging 2020, 12, 2156–2168. [Google Scholar] [CrossRef]
- Saito, T.; Terajima, M.; Taga, Y.; Hayashi, F.; Oshima, S.; Kasamatsu, A.; Okubo, Y.; Ito, C.; Toshimori, K.; Sunohara, M.; et al. Decrease of Lysyl Hydroxylase 2 Activity Causes Abnormal Collagen Molecular Phenotypes, Defective Mineralization and Compromised Mechanical Properties of Bone. Bone 2022, 154, 116242. [Google Scholar] [CrossRef]
- Xu, F.; Zhang, J.; Hu, G.; Liu, L.; Liang, W. Hypoxia and TGF-Β1 Induced PLOD2 Expression Improve the Migration and Invasion of Cervical Cancer Cells by Promoting Epithelial-to-Mesenchymal Transition (EMT) and Focal Adhesion Formation. Cancer Cell Int. 2017, 17, 54. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Parag-Sharma, K.; Terajima, M.; Musicant, A.M.; Murphy, R.M.; Ramsey, M.R.; Hibi, H.; Yamauchi, M.; Amelio, A.L. Lysyl Hydroxylase 2-Induced Collagen Cross-Link Switching Promotes Metastasis in Head and Neck Squamous Cell Carcinomas. Neoplasia 2021, 23, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Guo, H.; Wang, S.; Maghsoud, Y.; Vázquez-Montelongo, E.A.; Jing, Z.; Sammons, R.M.; Cho, E.J.; Ren, P.; Cisneros, G.A.; et al. Unleashing the Potential of 1,3-Diketone Analogues as Selective LH2 Inhibitors. ACS Med. Chem. Lett. 2023, 14, 1396–1403. [Google Scholar] [CrossRef] [PubMed]
- Baumann, S.; Hennet, T. Collagen Accumulation in Osteosarcoma Cells Lacking GLT25D1 Collagen Galactosyltransferase. J. Biol. Chem. 2016, 291, 18514–18524. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; He, L.; Xiao, F.; Gao, M.; Wei, H.; Yang, J.; Shu, Y.; Zhang, F.; Ye, X.; Li, P.; et al. Upregulation of GLT25D1 in Hepatic Stellate Cells Promotes Liver Fibrosis via the TGF-Β1/SMAD3 Pathway In Vivo and In Vitro. J. Clin. Transl. Hepatol. 2023, 11, 1–14. [Google Scholar] [CrossRef]
- Li, R.C.; Wong, M.Y.; DiChiara, A.S.; Hosseini, A.S.; Shoulders, M.D. Collagen’s Enigmatic, Highly Conserved N-Glycan Has an Essential Proteostatic Function. Proc. Natl. Acad. Sci. USA 2021, 118, e2026608118. [Google Scholar] [CrossRef]
- Kozlov, G.; Määttänen, P.; Thomas, D.Y.; Gehring, K. A Structural Overview of the PDI Family of Proteins. FEBS J. 2010, 277, 3924–3936. [Google Scholar] [CrossRef]
- Bekendam, R.H.; Flaumenhaft, R. Inhibition of Protein Disulfide Isomerase in Thrombosis. Basic. Clin. Pharmacol. Toxicol. 2016, 119, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Flaumenhaft, R.; Furie, B.; Zwicker, J.I. Therapeutic Implications of Protein Disulfide Isomerase Inhibition in Thrombotic Disease. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 16–23. [Google Scholar] [CrossRef]
- DiChiara, A.S.; Taylor, R.J.; Wong, M.Y.; Doan, N.D.; Del Rosario, A.M.; Shoulders, M.D. Mapping and Exploring the Collagen-I Proteostasis Network. ACS Chem. Biol. 2016, 11, 1408–1421. [Google Scholar] [CrossRef]
- Paglia, G.; Minacori, M.; Meschiari, G.; Fiorini, S.; Chichiarelli, S.; Eufemi, M.; Altieri, F. Protein Disulfide Isomerase A3 (PDIA3): A Pharmacological Target in Glioblastoma? Int. J. Mol. Sci. 2023, 24, 13279. [Google Scholar] [CrossRef] [PubMed]
- Revert, F.; Ventura, I.; Martínez-Martínez, P.; Granero-Moltó, F.; Revert-Ros, F.; Macías, J.; Saus, J. Goodpasture Antigen-Binding Protein Is a Soluble Exportable Protein That Interacts with Type IV Collagen. J. Biol. Chem. 2008, 283, 30246. [Google Scholar] [CrossRef] [PubMed]
- Rasche, L.; Duell, J.; Castro, I.C.; Dubljevic, V.; Chatterjee, M.; Knop, S.; Hensel, F.; Rosenwald, A.; Einsele, H.; Topp, M.S.; et al. GRP78-Directed Immunotherapy in Relapsed or Refractory Multiple Myeloma-Results from a Phase 1 Trial with the Monoclonal Immunoglobulin M Antibody PAT-SM6. Haematologica 2015, 100, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Elfiky, A.A.; Baghdady, A.M.; Ali, S.A.; Ahmed, M.I. GRP78 Targeting: Hitting Two Birds with a Stone. Life Sci. 2020, 260, 118317. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Cho, Y.; Lee, S. Cell Surface GRP94 as a Novel Emerging Therapeutic Target for Monoclonal Antibody Cancer Therapy. Cells 2021, 10, 670. [Google Scholar] [CrossRef] [PubMed]
- Siekierka, J.J.; Hung, S.H.; Poe, M.; Lin, C.S.; Sigal, N.H. A Cytosolic Binding Protein for the Immunosuppressant FK506 Has Peptidyl-Prolyl Isomerase Activity but Is Distinct from Cyclophilin. Nature 1989, 341, 755–757. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Vranka, J.; Wirz, J.; Nagata, K.; Bächinger, H.P. The Rough Endoplasmic Reticulum-Resident FK506-Binding Protein FKBP65 Is a Molecular Chaperone That Interacts with Collagens. J. Biol. Chem. 2008, 283, 31584–31590. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, U.; Cundy, T.; Pyott, S.M.; Christiansen, H.E.; Hegde, M.R.; Bank, R.A.; Pals, G.; Ankala, A.; Conneely, K.; Seaver, L.; et al. Mutations in FKBP10, Which Result in Bruck Syndrome and Recessive Forms of Osteogenesis Imperfecta, Inhibit the Hydroxylation of Telopeptide Lysines in Bone Collagen. Hum. Mol. Genet. 2013, 22, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Staab-Weijnitz, C.A.; Fernandez, I.E.; Knüppel, L.; Maul, J.; Heinzelmann, K.; Juan-Guardela, B.M.; Hennen, E.; Preissler, G.; Winter, H.; Neurohr, C.; et al. FK506-Binding Protein 10, a Potential Novel Drug Target for Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 455–467. [Google Scholar] [CrossRef]
- Xie, S.; Xing, Y.; Shi, W.; Zhang, M.; Chen, M.; Fang, W.; Liu, S.; Zhang, T.; Zeng, X.; Chen, S.; et al. Cardiac Fibroblast Heat Shock Protein 47 Aggravates Cardiac Fibrosis Post Myocardial Ischemia–Reperfusion Injury by Encouraging Ubiquitin Specific Peptidase 10 Dependent Smad4 Deubiquitination. Acta Pharm. Sin. B 2022, 12, 4138–4153. [Google Scholar] [CrossRef]
- Qosa, H.; de Oliveira, C.H.M.C.; Cizza, G.; Lawitz, E.J.; Colletti, N.; Wetherington, J.; Charles, E.D.; Tirucherai, G.S. Pharmacokinetics, Safety, and Tolerability of BMS-986263, a Lipid Nanoparticle Containing HSP47 siRNA, in Participants with Hepatic Impairment. Clin. Transl. Sci. 2023, 16, 1791–1802. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Goldberg, J. TANGO1/cTAGE5 Receptor as a Polyvalent Template for Assembly of Large COPII Coats. Proc. Natl. Acad. Sci. USA 2016, 113, 10061–10066. [Google Scholar] [CrossRef] [PubMed]
- Raote, I.; Ortega-Bellido, M.; Santos, A.J.; Foresti, O.; Zhang, C.; Garcia-Parajo, M.F.; Campelo, F.; Malhotra, V. TANGO1 Builds a Machine for Collagen Export by Recruiting and Spatially Organizing COPII, Tethers and Membranes. eLife 2018, 7, e32723. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.G.; Phamluong, K.; Li, L.; Sun, M.; Cao, T.C.; Liu, P.S.; Modrusan, Z.; Sandoval, W.N.; Rangell, L.; Carano, R.A.D.; et al. Global Defects in Collagen Secretion in a Mia3/TANGO1 Knockout Mouse. J. Cell Biol. 2011, 193, 935–951. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.J.; Nogueira, C.; Ortega-Bellido, M.; Malhotra, V. TANGO1 and Mia2/cTAGE5 (TALI) cooperate to export bulky pre-chylomicrons/VLDLs from the endoplasmic reticulum. J. Cell Biol. 2016, 213, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Man, J.; Zhou, W.; Zuo, S.; Zhao, X.; Wang, Q.; Ma, H.; Li, H.-Y. TANGO1 Interacts with NRTN to Promote Hepatocellular Carcinoma Progression by Regulating the PI3K/AKT/mTOR Signaling Pathway. Biochem. Pharmacol. 2023, 213, 115615. [Google Scholar] [CrossRef] [PubMed]
- Arnolds, O.; Stoll, R. Characterization of a Fold in TANGO1 Evolved from SH3 Domains for the Export of Bulky Cargos. Nat. Commun. 2023, 14, 2273. [Google Scholar] [CrossRef] [PubMed]
- Vadon-Le Goff, S.; Hulmes, D.J.S.; Moali, C. BMP-1/Tolloid-like Proteinases Synchronize Matrix Assembly with Growth Factor Activation to Promote Morphogenesis and Tissue Remodeling. Matrix Biol. 2015, 44–46, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, T.; Watabe, T. Bone Morphogenetic Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021899. [Google Scholar] [CrossRef]
- Grgurevic, L.; Erjavec, I.; Grgurevic, I.; Dumic-Cule, I.; Brkljacic, J.; Verbanac, D.; Matijasic, M.; Paljetak, H.C.; Novak, R.; Plecko, M.; et al. Systemic Inhibition of BMP1-3 Decreases Progression of CCl4-Induced Liver Fibrosis in Rats. Growth Factors 2017, 35, 201–215. [Google Scholar] [CrossRef]
- Talantikite, M.; Lécorché, P.; Beau, F.; Damour, O.; Becker-Pauly, C.; Ho, W.; Dive, V.; Vadon-Le Goff, S.; Moali, C. Inhibitors of BMP-1/Tolloid-like Proteinases: Efficacy, Selectivity and Cellular Toxicity. FEBS Open Bio 2018, 8, 2011–2021. [Google Scholar] [CrossRef]
- Bai, M.; Lei, J.; Wang, S.; Ding, D.; Yu, X.; Guo, Y.; Chen, S.; Du, Y.; Li, D.; Zhang, Y.; et al. BMP1 Inhibitor UK383,367 Attenuates Renal Fibrosis and Inflammation in CKD. Am. J. Physiol. Renal Physiol. 2019, 317, F1430–F1438. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.-Y.; N’Diaye, E.-N.; Caplazi, P.; Huang, Z.; Arlantico, A.; Jeet, S.; Wong, A.; Brightbill, H.D.; Li, Q.; Wong, W.R.; et al. BMP1 Is Not Required for Lung Fibrosis in Mice. Sci. Rep. 2022, 12, 5466. [Google Scholar] [CrossRef] [PubMed]
- Bekhouche, M.; Colige, A. The Procollagen N-Proteinases ADAMTS2, 3 and 14 in Pathophysiology. Matrix Biol. 2015, 44–46, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Kelwick, R.; Desanlis, I.; Wheeler, G.N.; Edwards, D.R. The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin Motifs) Family. Genome Biol. 2015, 16, 113. [Google Scholar] [CrossRef] [PubMed]
- Brebion, F.; Gosmini, R.; Deprez, P.; Varin, M.; Peixoto, C.; Alvey, L.; Jary, H.; Bienvenu, N.; Triballeau, N.; Blanque, R.; et al. Discovery of GLPG1972/S201086, a Potent, Selective, and Orally Bioavailable ADAMTS-5 Inhibitor for the Treatment of Osteoarthritis. J. Med. Chem. 2021, 64, 2937–2952. [Google Scholar] [CrossRef] [PubMed]
- Canty, E.G.; Lu, Y.; Meadows, R.S.; Shaw, M.K.; Holmes, D.F.; Kadler, K.E. Coalignment of Plasma Membrane Channels and Protrusions (Fibripositors) Specifies the Parallelism of Tendon. J. Cell Biol. 2004, 165, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Musiime, M.; Chang, J.; Hansen, U.; Kadler, K.E.; Zeltz, C.; Gullberg, D. Collagen Assembly at the Cell Surface: Dogmas Revisited. Cells 2021, 10, 662. [Google Scholar] [CrossRef] [PubMed]
- Revell, C.K.; Herrera, J.A.; Lawless, C.; Lu, Y.; Kadler, K.E.; Chang, J.; Jensen, O.E. Modeling Collagen Fibril Self-Assembly from Extracellular Medium in Embryonic Tendon. Biophys. J. 2023, 122, 3219–3237. [Google Scholar] [CrossRef]
- Lloyd, S.M.; He, Y. Exploring Extracellular Matrix Crosslinking as a Therapeutic Approach to Fibrosis. Cells 2024, 13, 438. [Google Scholar] [CrossRef]
- Eyre, D.R.; Weis, M.; Rai, J. Analyses of Lysine Aldehyde Cross-Linking in Collagen Reveal That the Mature Cross-Link Histidinohydroxylysinonorleucine Is an Artifact. J. Biol. Chem. 2019, 294, 6578–6590. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M.; Taga, Y.; Terajima, M. Analyses of Lysine Aldehyde Cross-Linking in Collagen Reveal That the Mature Cross-Link Histidinohydroxylysinonorleucine Is an Artifact. J. Biol. Chem. 2019, 294, 14163. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.S.; Sharma, M.; Kabekkodu, S.P.; Kumar, N.A.; Satyamoorthy, K.; Radhakrishnan, R. Understanding the Molecular Mechanism Associated with Reversal of Oral Submucous Fibrosis Targeting Hydroxylysine Aldehyde-Derived Collagen Cross-Links. J. Carcinog. 2021, 20, 9. [Google Scholar] [PubMed]
- Rosell-García, T.; Paradela, A.; Bravo, G.; Dupont, L.; Bekhouche, M.; Colige, A.; Rodriguez-Pascual, F. Differential Cleavage of Lysyl Oxidase by the Metalloproteinases BMP1 and ADAMTS2/14 Regulates Collagen Binding through a Tyrosine Sulfate Domain. J. Biol. Chem. 2019, 294, 11087–11100. [Google Scholar] [CrossRef] [PubMed]
- Barry, V.; Spangler, R.; Marshall, D.; McCauley, S.; Rodriguez, H.; Oyasu, M.; Mikels, A.; Vaysberg, M.; Ghermazien, H.; Wai, C.; et al. Allosteric Inhibition of Lysyl Oxidase-like-2 Impedes the Development of a Pathologic Microenvironment. Nat. Med. 2010, 16, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Brown, K.K.; Collard, H.R.; Cottin, V.; Gibson, K.F.; Kaner, R.J.; Lederer, D.J.; Martinez, F.J.; Noble, P.W.; Song, J.W.; et al. Efficacy of Simtuzumab versus Placebo in Patients with Idiopathic Pulmonary Fibrosis: A Randomised, Double-Blind, Controlled, Phase 2 Trial. Lancet Respir. Med. 2017, 5, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Muir, A.J.; Levy, C.; Janssen, H.L.A.; Montano-Loza, A.J.; Shiffman, M.L.; Caldwell, S.; Luketic, V.; Ding, D.; Jia, C.; McColgan, B.J.; et al. Simtuzumab for Primary Sclerosing Cholangitis: Phase 2 Study Results With Insights on the Natural History of the Disease. Hepatology 2019, 69, 684–698. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Savona, M.R.; Mesa, R.A.; Oh, S.; Dong, H.; Thai, D.; Gotlib, J. A Phase 2 Study to Evaluate the Efficacy and Safety of Simtuzumab in Adult Subjects with Primary, Post Polycythemia Vera (PV) or Post Essential Thrombocythemia (ET) Myelofibrosis. Blood 2015, 126, 2810. [Google Scholar] [CrossRef]
- Harrison, S.A.; Abdelmalek, M.F.; Caldwell, S.; Shiffman, M.L.; Diehl, A.M.; Ghalib, R.; Lawitz, E.J.; Rockey, D.C.; Schall, R.A.; Jia, C.; et al. Simtuzumab Is Ineffective for Patients With Bridging Fibrosis or Compensated Cirrhosis Caused by Nonalcoholic Steatohepatitis. Gastroenterology 2018, 155, 1140–1153. [Google Scholar] [CrossRef] [PubMed]
- Meissner, E.G.; McLaughlin, M.; Matthews, L.; Gharib, A.M.; Wood, B.J.; Levy, E.; Sinkus, R.; Virtaneva, K.; Sturdevant, D.; Martens, C.; et al. Simtuzumab Treatment of Advanced Liver Fibrosis in HIV and HCV-Infected Adults: Results of a 6-Month Open-Label Safety Trial. Liver Int. 2016, 36, 1783–1792. [Google Scholar] [CrossRef]
- Benson, A.B.; Wainberg, Z.A.; Hecht, J.R.; Vyushkov, D.; Dong, H.; Bendell, J.; Kudrik, F. A Phase II Randomized, Double-Blind, Placebo-Controlled Study of Simtuzumab or Placebo in Combination with Gemcitabine for the First-Line Treatment of Pancreatic Adenocarcinoma. Oncologist 2017, 22, 241-e15. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.R.; Benson, A.B.; Vyushkov, D.; Yang, Y.; Bendell, J.; Verma, U. A Phase II, Randomized, Double-Blind, Placebo-Controlled Study of Simtuzumab in Combination with FOLFIRI for the Second-Line Treatment of Metastatic KRAS Mutant Colorectal Adenocarcinoma. Oncologist 2017, 22, 243-e23. [Google Scholar] [CrossRef] [PubMed]
- Eraso, P.; Mazón, M.J.; Jiménez, V.; Pizarro-García, P.; Cuevas, E.P.; Majuelos-Melguizo, J.; Morillo-Bernal, J.; Cano, A.; Portillo, F. New Functions of Intracellular LOXL2: Modulation of RNA-Binding Proteins. Molecules 2023, 28, 4433. [Google Scholar] [CrossRef] [PubMed]
- M.D. Anderson Cancer Center. Open Label Phase 2 Single Agent Study of PAT-1251 in Patients With Primary Myelofibrosis (PMF), Post-Polycythemia Vera Myelofibrosis (Post-PV MF), or Post-Essential Thrombocytosis Myelofibrosis (Post-ET MF). clinicaltrials.gov, 2019. Available online: https://www.clinicaltrials.gov/search?term=NCT04054245 (accessed on 7 June 2024).
- Syntara. A Phase 1/2a Study to Evaluate Safety, Pharmacokinetic and Pharmacodynamic Dose Escalation and Expansion Study of PXS-5505 in Patients with Primary, Postpolycythemia Vera or Post-Essential Thrombocythemia Myelofibrosis. clinicaltrials.gov, 2024. Available online: https://www.clinicaltrials.gov/search?term=NCT04676529 (accessed on 7 June 2024).
- Vachhani, P.; Baskar, J.; Charlton, B.; Cheung, S.; Jarolimek, W.; Lee, S.-E.; Tan, P.; Watson, A.M.; Wu, S.-J. PXS5505-MF-101: A Phase 1/2a Study to Evaluate Safety, Pharmacokinetics and Pharmacodynamics of Pxs-5505 in Patients with Primary, Post-Polycythemia Vera or Post-Essential Thrombocythemia Myelofibrosis. Blood 2023, 142, 625. [Google Scholar] [CrossRef]
- Badri, N. A Phase 1b/2 Trial of PXS-5505 Combined with First Line Atezolizumab Plus Bevacizumab for Treating Patients with Unresectable Hepatocellular Carcinoma. clinicaltrials.gov, 2023. Available online: https://www.clinicaltrials.gov/search?term=NCT05109052 (accessed on 7 June 2024).
- Vanacore, R.; Friedman, D.; Ham, A.; Sundaramoorthy, M.; Hudson, B. Identification of S-Hydroxylysyl-Methionine as the Covalent Cross-Link of the Noncollagenous (NC1) Hexamer of the A1α1α2 Collagen IV Network. J. Biol. Chem. 2005, 280, 29300–29310. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Shi, R. Mammalian Peroxidasin (PXDN): From Physiology to Pathology. Free Radic. Biol. Med. 2022, 182, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.E.; Fessler, L.I.; Takagi, Y.; Blumberg, B.; Keene, D.R.; Olson, P.F.; Parker, C.G.; Fessler, J.H. Peroxidasin: A Novel Enzyme-matrix Protein of Drosophila Development. EMBO J. 1994, 13, 3438–3447. [Google Scholar] [CrossRef] [PubMed]
- Bhave, G.; Cummings, C.F.; Vanacore, R.M.; Kumagai-Cresse, C.; Ero-Tolliver, I.A.; Rafi, M.; Kang, J.-S.; Pedchenko, V.P.; Fessler, L.I.; Fessler, J.H.; et al. Peroxidasin Forms Sulfilimine Chemical Bonds Using Hypohalous Acids In Tissue Genesis. Nat. Chem. Biol. 2012, 8, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Colon, S.; Luan, H.; Liu, Y.; Meyer, C.; Gewin, L.; Bhave, G. Peroxidasin and Eosinophil Peroxidase, but Not Myeloperoxidase, Contribute to Renal Fibrosis in the Murine Unilateral Ureteral Obstruction Model. Am. J. Physiol. Renal Physiol. 2019, 316, F360–F371. [Google Scholar] [CrossRef]
- Paumann-Page, M.; Obinger, C.; Winterbourn, C.C.; Furtmüller, P.G. Peroxidasin Inhibition by Phloroglucinol and Other Peroxidase Inhibitors. Antioxidants 2024, 13, 23. [Google Scholar] [CrossRef]
- Kumar, V.A.; Taylor, N.L.; Jalan, A.A.; Hwang, L.K.; Wang, B.K.; Hartgerink, J.D. A Nanostructured Synthetic Collagen Mimic for Hemostasis. Biomacromolecules 2014, 15, 1484–1490. [Google Scholar] [CrossRef] [PubMed]
- Maquart, F.X.; Siméon, A.; Pasco, S.; Monboisse, J.C. Regulation of cell activity by the extracellular matrix: The concept of matrikines. J. Soc. Biol. 1999, 193, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Davis, G.E.; Bayless, K.J.; Davis, M.J.; Meininger, G.A. Regulation of Tissue Injury Responses by the Exposure of Matricryptic Sites within Extracellular Matrix Molecules. Am. J. Pathol. 2000, 156, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, M.L.; Iyer, R.P.; Zamilpa, R.; Yabluchanskiy, A.; DeLeon-Pennell, K.Y.; Hall, M.E.; Kaplan, A.; Zouein, F.A.; Bratton, D.; Flynn, E.R.; et al. A Novel Collagen Matricryptin Reduces Left Ventricular Dilation Post-Myocardial Infarction by Promoting Scar Formation and Angiogenesis. J. Am. Coll. Cardiol. 2015, 66, 1364–1374. [Google Scholar] [CrossRef] [PubMed]
- Grilo, G.A.; Cakir, S.N.; Shaver, P.R.; Iyer, R.P.; Whitehead, K.; McClung, J.M.; Vahdati, A.; de Castro Brás, L.E. Collagen Matricryptin Promotes Cardiac Function by Mediating Scar Formation. Life Sci. 2023, 321, 121598. [Google Scholar] [CrossRef] [PubMed]
- Pozzo, C.F.S.D.; Sielski, M.S.; de Campos Vidal, B.; Werneck, C.C.; Vicente, C.P. A Collagen I Derived Matricryptin Increases Aorta Vascular Wall Remodeling after Induced Thrombosis in Mouse. Thromb. Res. 2022, 209, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Gauci, S.J.; Golub, S.B.; Tatarczuch, L.; Lee, E.; Chan, D.; Walsh, N.C.; Little, C.B.; Stanton, H.; Lokmic, Z.; Sims, N.A.; et al. Disrupted Type II Collagenolysis Impairs Angiogenesis, Delays Endochondral Ossification and Initiates Aberrant Ossification in Mouse Limbs. Matrix Biol. 2019, 83, 77–96. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Guo, Z.; Zhang, J.; Yang, Y.; Liu, C.; Zhang, L.; Gu, Z.; Li, Y.; Ding, Z.; Shi, G. Recombinant Human Arresten and Canstatin Inhibit Angiogenic Behaviors of HUVECs via Inhibiting the PI3K/Akt Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 8995. [Google Scholar] [CrossRef] [PubMed]
- Kamphaus, G.D.; Colorado, P.C.; Panka, D.J.; Hopfer, H.; Ramchandran, R.; Torre, A.; Maeshima, Y.; Mier, J.W.; Sukhatme, V.P.; Kalluri, R. Canstatin, a Novel Matrix-Derived Inhibitor of Angiogenesis and Tumor Growth. J. Biol. Chem. 2000, 275, 1209–1215. [Google Scholar] [CrossRef]
- Colorado, P.C.; Torre, A.; Kamphaus, G.; Maeshima, Y.; Hopfer, H.; Takahashi, K.; Volk, R.; Zamborsky, E.D.; Herman, S.; Sarkar, P.K.; et al. Anti-Angiogenic Cues from Vascular Basement Membrane Collagen. Cancer Res. 2000, 60, 2520–2526. [Google Scholar]
- Aikio, M.; Alahuhta, I.; Nurmenniemi, S.; Suojanen, J.; Palovuori, R.; Teppo, S.; Sorsa, T.; López-Otín, C.; Pihlajaniemi, T.; Salo, T.; et al. Arresten, a Collagen-Derived Angiogenesis Inhibitor, Suppresses Invasion of Squamous Cell Carcinoma. PLoS ONE 2012, 7, e51044. [Google Scholar] [CrossRef]
- Sugiyama, A.; Shimizu, Y.; Okada, M.; Otani, K.; Yamawaki, H. Preventive Effect of Canstatin against Ventricular Arrhythmia Induced by Ischemia/Reperfusion Injury: A Pilot Study. Int. J. Mol. Sci. 2021, 22, 1004. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, A.; Kaisho, M.; Okada, M.; Otani, K.; Yamawaki, H. Decreased Expression of Canstatin in Rat Model of Monocrotaline-Induced Pulmonary Arterial Hypertension: Protective Effect of Canstatin on Right Ventricular Remodeling. Int. J. Mol. Sci. 2020, 21, 6797. [Google Scholar] [CrossRef] [PubMed]
- Sudhakar, A.; Sugimoto, H.; Yang, C.; Lively, J.; Zeisberg, M.; Kalluri, R. Human Tumstatin and Human Endostatin Exhibit Distinct Antiangiogenic Activities Mediated by Alpha v Beta 3 and Alpha 5 Beta 1 Integrins. Proc. Natl. Acad. Sci. USA 2003, 100, 4766–4771. [Google Scholar] [CrossRef] [PubMed]
- Maeshima, Y.; Yerramalla, U.L.; Dhanabal, M.; Holthaus, K.A.; Barbashov, S.; Kharbanda, S.; Reimer, C.; Manfredi, M.; Dickerson, W.M.; Kalluri, R. Extracellular Matrix-Derived Peptide Binds to Alpha(v)Beta(3) Integrin and Inhibits Angiogenesis. J. Biol. Chem. 2001, 276, 31959–31968. [Google Scholar] [CrossRef]
- Liu, F.; Wang, F.; Dong, X.; Xiu, P.; Sun, P.; Li, Z.; Shi, X.; Zhong, J. T7 Peptide Cytotoxicity in Human Hepatocellular Carcinoma Cells Is Mediated by Suppression of Autophagy. Int. J. Mol. Med. 2019, 44, 523–534. [Google Scholar] [CrossRef]
- Riaz, M.K.; Zhang, X.; Wong, K.H.; Chen, H.; Liu, Q.; Chen, X.; Zhang, G.; Lu, A.; Yang, Z. Pulmonary Delivery of Transferrin Receptors Targeting Peptide Surface-Functionalized Liposomes Augments the Chemotherapeutic Effect of Quercetin in Lung Cancer Therapy. Int. J. Nanomed. 2019, 14, 2879–2902. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Zuo, Z.; Xu, C.; Ji, M.; He, J.; Li, J. Tumstatin (69–88) Alleviates Heart Failure via Attenuating Oxidative Stress in Rats with Myocardial Infarction. Heliyon 2022, 8, e10582. [Google Scholar] [CrossRef]
- Yasuda, J.; Okada, M.; Yamawaki, H. T3 Peptide, an Active Fragment of Tumstatin, Inhibits H2O2-Induced Apoptosis in H9c2 Cardiomyoblasts. Eur. J. Pharmacol. 2017, 807, 64–70. [Google Scholar] [CrossRef]
- Ocugen. A Phase 1 Study to Assess the Safety and Efficacy Of OCU200 For Center-Involved Diabetic Macular Edema. clinicaltrials.gov, 2023. Available online: https://www.clinicaltrials.gov/search?term=NCT05802329 (accessed on 7 June 2024).
- Upadhyay, A.K.; Arumugham, R.; Naha, P.; Singh, D. OCU200 (Transferrin-Tumstatin Fusion Protein): A Potential Therapeutic for DME, DR, and Wet-AMD. Investig. Ophthalmol. Vis. Sci. 2021, 62, 992. [Google Scholar]
- Petitclerc, E.; Boutaud, A.; Prestayko, A.; Xu, J.; Sado, Y.; Ninomiya, Y.; Sarras, M.P.; Hudson, B.G.; Brooks, P.C. New Functions for Non-Collagenous Domains of Human Collagen Type IV. Novel Integrin Ligands Inhibiting Angiogenesis and Tumor Growth in Vivo. J. Biol. Chem. 2000, 275, 8051–8061. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, E.D.; Popel, A.S. A Systematic Methodology for Proteome-Wide Identification of Peptides Inhibiting the Proliferation and Migration of Endothelial Cells. Proc. Natl. Acad. Sci. USA 2008, 105, 13775–13780. [Google Scholar] [CrossRef] [PubMed]
- Brassart-Pasco, S.; Sénéchal, K.; Thevenard, J.; Ramont, L.; Devy, J.; Di Stefano, L.; Dupont-Deshorgue, A.; Brézillon, S.; Feru, J.; Jazeron, J.-F.; et al. Tetrastatin, the NC1 Domain of the A4(IV) Collagen Chain: A Novel Potent Anti-Tumor Matrikine. PLoS ONE 2012, 7, e29587. [Google Scholar] [CrossRef] [PubMed]
- Lambert, E.; Fuselier, E.; Ramont, L.; Brassart, B.; Dukic, S.; Oudart, J.-B.; Dupont-Deshorgue, A.; Sellier, C.; Machado, C.; Dauchez, M.; et al. Conformation-Dependent Binding of a Tetrastatin Peptide to Avβ3 Integrin Decreases Melanoma Progression through FAK/PI3K/Akt Pathway Inhibition. Sci. Rep. 2018, 8, 9837. [Google Scholar] [CrossRef] [PubMed]
- Vautrin-Glabik, A.; Devy, J.; Bour, C.; Baud, S.; Choulier, L.; Hoarau, A.; Dupont-Deshorgue, A.; Sellier, C.; Brassart, B.; Oudart, J.-B.; et al. Angiogenesis Inhibition by a Short 13 Amino Acid Peptide Sequence of Tetrastatin, the A4(IV) NC1 Domain of Collagen IV. Front. Cell Dev. Biol. 2020, 8, 775. [Google Scholar] [CrossRef] [PubMed]
- Koskimaki, J.E.; Karagiannis, E.D.; Tang, B.C.; Hammers, H.; Watkins, D.N.; Pili, R.; Popel, A.S. Pentastatin-1, a Collagen IV Derived 20-Mer Peptide, Suppresses Tumor Growth in a Small Cell Lung Cancer Xenograft Model. BMC Cancer 2010, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Mutgan, A.C.; Jandl, K.; Radic, N.; Valzano, F.; Kolb, D.; Hoffmann, J.; Foris, V.; Wilhelm, J.; Boehm, P.M.; Hoetzenecker, K.; et al. Pentastatin, a Matrikine of the Collagen IVα5, Is a Novel Endogenous Mediator of Pulmonary Endothelial Dysfunction. Am. J. Physiol. Cell Physiol. 2023, 325, C1294–C1312. [Google Scholar] [CrossRef] [PubMed]
- Weckmann, M.; Moir, L.M.; Heckman, C.A.; Black, J.L.; Oliver, B.G.; Burgess, J.K. Lamstatin-a Novel Inhibitor of Lymphangiogenesis Derived from Collagen IV. J. Cell. Mol. Med. 2012, 16, 3062–3073. [Google Scholar] [CrossRef] [PubMed]
- Mundel, T.M.; Yliniemi, A.-M.; Maeshima, Y.; Sugimoto, H.; Kieran, M.; Kalluri, R. Type IV Collagen Alpha6 Chain-Derived Noncollagenous Domain 1 (Alpha6(IV)NC1) Inhibits Angiogenesis and Tumor Growth. Int. J. Cancer 2008, 122, 1738–1744. [Google Scholar] [CrossRef]
- Gunda, V.; Verma, R.K.; Sudhakar, Y.A. Inhibition of Elastin Peptide-Mediated Angiogenic Signaling Mechanism(s) in Choroidal Endothelial Cells by the A6(IV)NC1 Collagen Fragment. Investig. Ophthalmol. Vis. Sci. 2013, 54, 7828–7835. [Google Scholar] [CrossRef]
- Xu, R.; Yao, Z.Y.; Xin, L.; Zhang, Q.; Li, T.P.; Gan, R.B. NC1 Domain of Human Type VIII Collagen (Alpha 1) Inhibits Bovine Aortic Endothelial Cell Proliferation and Causes Cell Apoptosis. Biochem. Biophys. Res. Commun. 2001, 289, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Yao, C.; Wang, Z.; Yue, L.; Fang, Z.; Yao, H.; Lin, F.; Zhao, H.; Sun, Y.-J.; Bian, X.-W.; et al. Vastatin, an Endogenous Antiangiogenesis Polypeptide That Is Lost in Hepatocellular Carcinoma, Effectively Inhibits Tumor Metastasis. Mol. Ther. 2016, 24, 1358–1368. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, J.; Woo, Y.M.; Shen, Z.; Yao, H.; Cai, Y.; Lin, M.C.-M.; Poon, W.S. Enhanced Expression of Vastatin Inhibits Angiogenesis and Prolongs Survival in Murine Orthotopic Glioblastoma Model. BMC Cancer 2017, 17, 126. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.S.; Boehm, T.; Shing, Y.; Fukai, N.; Vasios, G.; Lane, W.S.; Flynn, E.; Birkhead, J.R.; Olsen, B.R.; Folkman, J. Endostatin: An Endogenous Inhibitor of Angiogenesis and Tumor Growth. Cell 1997, 88, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-M.; Hwang, S.; Kim, Y.-M.; Pyun, B.-J.; Kim, T.-Y.; Lee, S.-T.; Gho, Y.S.; Kwon, Y.-G. Endostatin Blocks Vascular Endothelial Growth Factor-Mediated Signaling via Direct Interaction with KDR/Flk-1. J. Biol. Chem. 2002, 277, 27872–27879. [Google Scholar] [CrossRef] [PubMed]
- Rehn, M.; Veikkola, T.; Kukk-Valdre, E.; Nakamura, H.; Ilmonen, M.; Lombardo, C.; Pihlajaniemi, T.; Alitalo, K.; Vuori, K. Interaction of Endostatin with Integrins Implicated in Angiogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 1024–1029. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Huang, Y.; Zhou, H.; Song, X.; Yuan, S.; Fu, Y.; Luo, Y. Nucleolin Is a Receptor That Mediates Antiangiogenic and Antitumor Activity of Endostatin. Blood 2007, 110, 2899–2906. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Zhong, K.; Tian, H.; Gao, X.; Xu, X.; Yin, X.; Yao, W. Characterization of a monoPEG20000-Endostar. Int. J. Biol. Macromol. 2010, 46, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Hua, L.; Hu, B.; Wang, J. Pre-Clinical Efficacy and Safety Pharmacology of PEGylated Recombinant Human Endostatin. Curr. Mol. Med. 2024, 24, 389–396. [Google Scholar] [CrossRef]
- Chen, Y.; Du, Y.; Li, P.; Wu, F.; Fu, Y.; Li, Z.; Luo, Y. Phase I Trial of M2ES, a Novel Polyethylene Glycosylated Recombinant Human Endostatin, plus Gemcitabine in Advanced Pancreatic Cancer. Mol. Clin. Oncol. 2014, 2, 586–590. [Google Scholar] [CrossRef]
- Cattaneo, M.G.; Pola, S.; Francescato, P.; Chillemi, F.; Vicentini, L.M. Human Endostatin-Derived Synthetic Peptides Possess Potent Antiangiogenic Properties in Vitro and in Vivo. Exp. Cell Res. 2003, 283, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Wickström, S.A.; Alitalo, K.; Keski-Oja, J. An Endostatin-Derived Peptide Interacts with Integrins and Regulates Actin Cytoskeleton and Migration of Endothelial Cells. J. Biol. Chem. 2004, 279, 20178–20185. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.-M.; Yin, R.; Chen, L.; Siraj, S.; Huang, X.; Wang, M.; Fang, H.; Wang, Y. An RGD-Modified Endostatin-Derived Synthetic Peptide Shows Antitumor Activity in Vivo. Bioconjug. Chem. 2008, 19, 1980–1986. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Takihara, T.; Chambers, R.A.; Veraldi, K.L.; Larregina, A.T.; Feghali-Bostwick, C.A. A Peptide Derived from Endostatin Ameliorates Organ Fibrosis. Sci. Transl. Med. 2012, 4, 136ra71. [Google Scholar] [CrossRef] [PubMed]
- Mlakar, L.; Garrett, S.M.; Watanabe, T.; Sanderson, M.; Nishimoto, T.; Heywood, J.; Helke, K.L.; Pilewski, J.M.; Herzog, E.L.; Feghali-Bostwick, C. Ameliorating Fibrosis in Murine and Human Tissues with END55, an Endostatin-Derived Fusion Protein Made in Plants. Biomedicines 2022, 10, 2861. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Nieto, G.; Heljasvaara, R.; Heikkinen, A.; Kaski, H.-K.; Devarajan, R.; Rinne, O.; Henriksson, C.; Thomson, E.; von Hertzen, C.; Miinalainen, I.; et al. Deletion of Col15a1 Modulates the Tumour Extracellular Matrix and Leads to Increased Tumour Growth in the MMTV-PyMT Mouse Mammary Carcinoma Model. Int. J. Mol. Sci. 2021, 22, 9978. [Google Scholar] [CrossRef] [PubMed]
- Ramchandran, R.; Dhanabal, M.; Volk, R.; Waterman, M.J.; Segal, M.; Lu, H.; Knebelmann, B.; Sukhatme, V.P. Antiangiogenic Activity of Restin, NC10 Domain of Human Collagen XV: Comparison to Endostatin. Biochem. Biophys. Res. Commun. 1999, 255, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Mutolo, M.J.; Morris, K.J.; Leir, S.-H.; Caffrey, T.C.; Lewandowska, M.A.; Hollingsworth, M.A.; Harris, A. Tumor Suppression by Collagen XV Is Independent of the Restin Domain. Matrix Biol. 2012, 31, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Ramont, L.; Brassart-Pasco, S.; Thevenard, J.; Deshorgue, A.; Venteo, L.; Laronze, J.Y.; Pluot, M.; Monboisse, J.-C.; Maquart, F.-X. The NC1 Domain of Type XIX Collagen Inhibits in Vivo Melanoma Growth. Mol. Cancer Ther. 2007, 6, 506–514. [Google Scholar] [CrossRef]
- Oudart, J.-B.; Brassart-Pasco, S.; Vautrin, A.; Sellier, C.; Machado, C.; Dupont-Deshorgue, A.; Brassart, B.; Baud, S.; Dauchez, M.; Monboisse, J.-C.; et al. Plasmin Releases the Anti-Tumor Peptide from the NC1 Domain of Collagen XIX. Oncotarget 2015, 6, 3656–3668. [Google Scholar] [CrossRef]
- Oudart, J.-B.; Villemin, M.; Brassart, B.; Sellier, C.; Terryn, C.; Dupont-Deshorgue, A.; Monboisse, J.C.; Maquart, F.-X.; Ramont, L.; Brassart-Pasco, S. F4, a Collagen XIX-Derived Peptide, Inhibits Tumor Angiogenesis through Avβ3 and A5β1 Integrin Interaction. Cell Adh. Migr. 2021, 15, 215–223. [Google Scholar] [CrossRef]
- Guide, S.V.; Gonzalez, M.E.; Bağcı, I.S.; Agostini, B.; Chen, H.; Feeney, G.; Steimer, M.; Kapadia, B.; Sridhar, K.; Quesada Sanchez, L.; et al. Trial of Beremagene Geperpavec (B-VEC) for Dystrophic Epidermolysis Bullosa. N. Engl. J. Med. 2022, 387, 2211–2219. [Google Scholar] [CrossRef] [PubMed]
- Tovar Vetencourt, A.; Sayed-Ahmed, I.; Gomez, J.; Chen, H.; Agostini, B.; Carroll, K.; Parry, T.; Krishnan, S.; Sabater, A.L. Ocular Gene Therapy in a Patient with Dystrophic Epidermolysis Bullosa. N. Engl. J. Med. 2024, 390, 530–535. [Google Scholar] [CrossRef]
- Abeona Therapeutics, Inc. VIITAL: A Phase 3 Study of EB-101 for the Treatment of Recessive Dystrophic Epidermolysis Bullosa (RDEB). clinicaltrials.gov, 2022. Available online: https://www.clinicaltrials.gov/search?term=NCT04227106 (accessed on 7 June 2024).
- Siprashvili, Z.; Nguyen, N.T.; Gorell, E.S.; Loutit, K.; Khuu, P.; Furukawa, L.K.; Lorenz, H.P.; Leung, T.H.; Keene, D.R.; Rieger, K.E.; et al. Safety and Wound Outcomes Following Genetically Corrected Autologous Epidermal Grafts in Patients With Recessive Dystrophic Epidermolysis Bullosa. JAMA 2016, 316, 1808–1817. [Google Scholar] [CrossRef] [PubMed]
- So, J.Y.; Nazaroff, J.; Iwummadu, C.V.; Harris, N.; Gorell, E.S.; Fulchand, S.; Bailey, I.; McCarthy, D.; Siprashvili, Z.; Marinkovich, M.P.; et al. Long-Term Safety and Efficacy of Gene-Corrected Autologous Keratinocyte Grafts for Recessive Dystrophic Epidermolysis Bullosa. Orphanet. J. Rare Dis. 2022, 17, 377. [Google Scholar] [CrossRef] [PubMed]
- Takashima, S.; Shinkuma, S.; Fujita, Y.; Nomura, T.; Ujiie, H.; Natsuga, K.; Iwata, H.; Nakamura, H.; Vorobyev, A.; Abe, R.; et al. Efficient Gene Reframing Therapy for Recessive Dystrophic Epidermolysis Bullosa with CRISPR/Cas9. J. Investig. Dermatol. 2019, 139, 1711–1721.e4. [Google Scholar] [CrossRef] [PubMed]
- Bonafont, J.; Mencía, Á.; García, M.; Torres, R.; Rodríguez, S.; Carretero, M.; Chacón-Solano, E.; Modamio-Høybjør, S.; Marinas, L.; León, C.; et al. Clinically Relevant Correction of Recessive Dystrophic Epidermolysis Bullosa by Dual sgRNA CRISPR/Cas9-Mediated Gene Editing. Mol. Ther. 2019, 27, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-Replace Genome Editing without Double-Strand Breaks or Donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-A.; Kim, S.-E.; Lee, A.-Y.; Hwang, G.-H.; Kim, J.H.; Iwata, H.; Kim, S.-C.; Bae, S.; Lee, S.E. Therapeutic Base Editing and Prime Editing of COL7A1 Mutations in Recessive Dystrophic Epidermolysis Bullosa. Mol. Ther. 2022, 30, 2664–2679. [Google Scholar] [CrossRef]
- Ablinger, M.; Lettner, T.; Friedl, N.; Potocki, H.; Palmetzhofer, T.; Koller, U.; Illmer, J.; Liemberger, B.; Hainzl, S.; Klausegger, A.; et al. Personalized Development of Antisense Oligonucleotides for Exon Skipping Restores Type XVII Collagen Expression in Junctional Epidermolysis Bullosa. Int. J. Mol. Sci. 2021, 22, 3326. [Google Scholar] [CrossRef]
- Holostem Terapie Avanzate s.r.l. Prospective, Open-Label, Uncontrolled Clinical Trial to Assess the Safety and Efficacy of Autologous Cultured Epidermal Grafts Containing Epidermal Stem Cells Genetically Modified With a Gamma-Retroviral (Rv) Vector Carrying COL17A1 cDNA for Restoration of Epidermis in Patients With Junctional Epidermolysis Bullosa. clinicaltrials.gov, 2022. Available online: https://www.clinicaltrials.gov/search?term=NCT03490331 (accessed on 7 June 2024).
- Petković, I.; Bischof, J.; Kocher, T.; March, O.P.; Liemberger, B.; Hainzl, S.; Strunk, D.; Raninger, A.M.; Binder, H.-M.; Reichelt, J.; et al. COL17A1 Editing via Homology-Directed Repair in Junctional Epidermolysis Bullosa. Front. Med. 2022, 9, 976604. [Google Scholar] [CrossRef] [PubMed]
- Bischof, J.; March, O.P.; Liemberger, B.; Haas, S.A.; Hainzl, S.; Petković, I.; Leb-Reichl, V.; Illmer, J.; Korotchenko, E.; Klausegger, A.; et al. Paired Nicking-Mediated COL17A1 Reframing for Junctional Epidermolysis Bullosa. Mol. Ther. 2022, 30, 2680–2692. [Google Scholar] [CrossRef] [PubMed]
- Klermund, J.; Rhiel, M.; Kocher, T.; Chmielewski, K.O.; Bischof, J.; Andrieux, G.; El Gaz, M.; Hainzl, S.; Boerries, M.; Cornu, T.I.; et al. On- and off-Target Effects of Paired CRISPR-Cas Nickase in Primary Human Cells. Mol. Ther. 2024, 32, 1298–1310. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-S.; Sato, T.; Chaugule, S.; Ma, H.; Xie, J.; Gao, G.; Shim, J.-H. AAV-Based Gene Editing of Type 1 Collagen Mutation to Treat Osteogenesis Imperfecta. Mol. Ther. Nucleic Acids 2024, 35, 102111. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, S.; Tavormina, J.; Tampe, D.; Zeisberg, M.; Wang, H.; Mahadevan, K.K.; Wu, C.-J.; Sugimoto, H.; Chang, C.-C.; et al. Oncogenic Collagen I Homotrimers from Cancer Cells Bind to A3β1 Integrin and Impact Tumor Microbiome and Immunity to Promote Pancreatic Cancer. Cancer Cell 2022, 40, 818–834.e9. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Organization | Type (+GAG) a | Chains (with alt. Splicing Isoforms; Autoantigen) b | Triple Helical Combinations | Phenotype of Human Mutations (Affected Chain) |
|---|---|---|---|---|
| Basement membrane network | IV | α1(IV) α2(IV) α3(IV) α4(IV) α5(IV) α6(IV) | [α1(IV)]2 α2(IV) | Angiopathies, nephropathy (α1, α2). Kidney disease, hematuria, loss hearing, eye abnormalities (α3, α4, α5). Deafness (α6)?. |
| α3(IV) α4(IV) α5(IV) | ||||
| [α5(IV)]2 α6(IV) | ||||
| Basketweave like network | VI | α1(VI) α2(VI) α3(VI) α4(VI) c α5(VI) d α6(IV) | α1(VI) α2(VI) α3(VI) | Muscular dystrophy, myopathy (α1, α2, α3). Involuntary movements, dystonia (α3). Chronic neuropathic itch (α5)? |
| α1(VI) α2(VI) α5(VI) ? | ||||
| α1(VI) α2(VI) α6(VI) ? | ||||
| Hexagonal networks | VIII | α1(VIII) α2(VIII) | [α1(VIII)]2 α2(VIII) | Corneal dystrophy (α2). |
| α1(VIII) [α2(VIII)]2 | ||||
| [α1(VIII)]3 | ||||
| [α2(VIII)]3 | ||||
| X | α1(X) | [α1(X)]3 | Chondrodysplasia. | |
| Fibrillar | I | α1(I) α2(I) | [α1(I)]2 α2(I) | Aberrant osteogenesis, osteoporosis, overly flexible joints, stretchy-fragile skin (α1, α2). |
| [α1(I)]3 | ||||
| II | α1(II) | [α1(II)]3 | Hipochondrogenosis, spondyloepiphyseal dysplasia, retinal detachment. | |
| III | α1(III) | [α1(III)]3 | Joint laxity and stretchy-fragile skin, vascular problems and aortic dissection. | |
| V | α1(V) α2(V) α3(V) | [α1(V)]2 α2(V) | Corneal problems, fibromuscular dysplasia, skin hyperextensibility, dystrophic scarring, and joint hypermobility (α1, α2). | |
| [α1(V)]3 | ||||
| α1(XI) α1(V) α1(II) e | ||||
| XI | α1(XI) α2(XI) | α1(XI) α2(XI) α1(II) e | Ophthalmologic, deafness, skeletal abnormalities, fibrochondrogenesis (α1, α2). | |
| α1(XI) α1(V) α1(II) e | ||||
| XXIV | α1(XXIV) | [α1(XXIV)]3 | ||
| XXVII | α1(XXVII) | [α1(XXVII)]3 | Short stature, bilateral congenital hip dislocation, radial head dislocation, carpal coalition, scoliosis, dysmorphic face. | |
| Fibrillar-associated collagens with interrupted triple helices (FACIT) | IX | α1(IX) α2(IX) α3(IX) | α1(IX) α2(IX) α3(IX) | Epiphyseal dysplasia, arthro-ophthalmodystrophy (α1, α2, α3). |
| XII | α1(XII) | [α1(XII)]3 | Muscular dystrophy, myopathy. | |
| XIV | α1(XIV) | [α1(XIV)]3 | Punctate abnormal thickening of the stratum corneum of the palms and soles? | |
| XVI | α1(XVI) | [α1(XVI)]3 | ||
| XIX | α1(XIX) | [α1(XIX)]3 | ||
| XX | α1(XX) | [α1(XX)]3 | Striate abnormal thickening of the stratum corneum of the palms and soles? | |
| XXI | α1(XXI) | [α1(XXI)]3 | ||
| XXII | α1(XXII) | [α1(XXII)]3 | ||
| Anchoring fibrils | VII | α1(VII) | [α1(VII)]3 | Cutaneous and mucosal fragility resulting in blisters and superficial ulcerations. |
| Membrane bound | XIII | α1(XIII) | [α1(XIII)]3 | Skeletal muscle weakness. |
| XVII | α1(XVII) | [α1(XVII)]3 | Atrophy of the skin and nonscarring blistering, epithelial dystrophy. | |
| XXIII | α1(XXIII) | [α1(XXIII)]3 | ||
| XXV | α1(XXV) | [α1(XXV)]3 | Fibrosis of extraocular muscles, ophthalmoplegia. | |
| Multiplexins | XV | α1(XV) | [α1(XV)]3 | |
| XVIII | α1(XVIII) | [α1(XVIII)]3 | High myopia, vitreoretinal degeneration and occipital skull defects | |
| Other | XXVI | α1(XXVI) | [α1(XXVI)]3 | |
| XXVIII | α1(XXVIII) | [α1(XXVIII)]3 |
| Collagen | Chain | Matrikine/Matricryptin Registered in ClinicalTrials.org | Activity |
|---|---|---|---|
| I | α1 | p1158/59 | Potentiates the remodeling of aorta wall after thrombosis |
| IV | α1 | Arresten | Antiangiogenic |
| α2 | Canstatin | Antiangiogenic. Inhibits the production of ROS and the elevation of intracellular Ca2+ levels. Antifibrotic | |
| α3 | Tumstatin | Antiangiogenic | |
| Tumstatin-peptide T7 | Cytotoxic | ||
| Tumstatin-peptide T3 | Improves heart function and reduces heart hypertrophy, fibrosis, and oxidative stress after myocardial infarction | ||
| Tumstatin-transferrin fusion polypeptide: OCU200 | Inhibits endothelial cell proliferation and damage in an oxygen-induced retinopathy | ||
| α4 | Tetrastatin-2 | Antiangiogenic | |
| Tetrastatin peptide QS-13 | Antiangiogenic. Antimigratory | ||
| α5 | Pentastatin-1 | Antiangiogenic. Antitumoral | |
| Lamstatin | Inhibits lymphangiogenesis | ||
| Lamstatin peptide CP17 | |||
| α6 | Hexastatin | Antiangiogenic. Antitumoral | |
| VIII | α1 | Vastatin | Antiangiogenic. Antitumoral |
| XV | α1 | Restin | Antiangiogenic. Antitumoral |
| XVIII | α1 | Endostatin | Antiangiogenic. Antitumoral |
| PEGylated endostatin M2ES | |||
| Endostar™ (recombinant endostatin approved in China) | |||
| Endostatin-derived E3 peptide | Antifibrotic | ||
| E3 analogue END55 | |||
| XIX | α1 | Peptide F4 | Antiangiogenic. Antimetastatic |
| Group | Drug (Type) | Clinical Trial | Disease | Refs |
|---|---|---|---|---|
| Direct | Collagenase from Clostridium histolyticum | Marketed | Fibrosis: Dupuytren’s contracture | [42,43,44] |
| Fibrosis: Peyronie’s disease | [45] | |||
| Debriding chronic dermal ulcers and severely burned areas | [51,52] | |||
| Phase II: efficacy not proven | Adhesive capsulitis of the shoulder | [46] | ||
| Indirect | MMPs inhibitors (12) | Phase I–III: efficacy not proven | Cancer | [55,56] |
| Macular degeneration | ||||
| Arthritis | ||||
| Inflammation | ||||
| MMP-9 inhibitor | Phase II and III: efficacy not proven | Gastric Cancer, Advanced Gastric, or GEJ Adenocarcinoma | [57,58] | |
| Phase II: efficacy not proven | Crohn’s Disease | [59] | ||
| mimic of miR-29 | Phase II: efficacy not proven | Keloid formation | [88,89] | |
| anti-Grp78 | Phase I: favorable safety profile | Refractory multiple myeloma | [116] | |
| HSP47 siRNA | Phase II: promising results | Hepatic fibrosis secondary to HCV infection | [124] | |
| Anti-LOXL2 | Phase II: efficacy not proven | Idiopathic pulmonary fibrosis | [149] | |
| Primary sclerosing cholangitis | [150] | |||
| Primary myelofibrosis | [151] | |||
| Myelofibrosis secondary to polycythemia vera | [151] | |||
| Essential thrombocythemia | [151] | |||
| Bridging Fibrosis or Compensated Cirrhosis Caused by Nonalcoholic Steatohepatitis | [152] | |||
| Liver fibrosis in HIV and HCV-infected adults | [153] | |||
| Steatohepatitis pancreatic adenocarcinoma | [154] | |||
| Metastatic KRAS mutant colorectal cancer | [155] | |||
| Broad-spectrum LOX inhibitors | Phase I/II: promising results | Myelofibrosis | [159] | |
| Collagen Peptides | Tumstatin based drugs [α3(IV)] | Phase I: | Diabetic macular edema | [186] |
| Endostatin, PEGylated endostatin [α1(XVIII)] | Phase III—marketed | Cancer | [205] | |
| Gene therapy | Recombinant COL7A1 expression | Marketed | Dystrophic epidermolysis bullosa | [219,220] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Revert-Ros, F.; Ventura, I.; Prieto-Ruiz, J.A.; Hernández-Andreu, J.M.; Revert, F. The Versatility of Collagen in Pharmacology: Targeting Collagen, Targeting with Collagen. Int. J. Mol. Sci. 2024, 25, 6523. https://doi.org/10.3390/ijms25126523
Revert-Ros F, Ventura I, Prieto-Ruiz JA, Hernández-Andreu JM, Revert F. The Versatility of Collagen in Pharmacology: Targeting Collagen, Targeting with Collagen. International Journal of Molecular Sciences. 2024; 25(12):6523. https://doi.org/10.3390/ijms25126523
Chicago/Turabian StyleRevert-Ros, Francisco, Ignacio Ventura, Jesús A. Prieto-Ruiz, José Miguel Hernández-Andreu, and Fernando Revert. 2024. "The Versatility of Collagen in Pharmacology: Targeting Collagen, Targeting with Collagen" International Journal of Molecular Sciences 25, no. 12: 6523. https://doi.org/10.3390/ijms25126523
APA StyleRevert-Ros, F., Ventura, I., Prieto-Ruiz, J. A., Hernández-Andreu, J. M., & Revert, F. (2024). The Versatility of Collagen in Pharmacology: Targeting Collagen, Targeting with Collagen. International Journal of Molecular Sciences, 25(12), 6523. https://doi.org/10.3390/ijms25126523