
A-Syn(ful) MAM: A Fresh Perspective on a Converging Domain in Parkinson’s Disease


Abstract
:1. Introduction
Parkinson’s Disease
2. Overview of Alpha-Synuclein
2.1. Structure and Conformations of αSyn
2.2. Localization of αSyn
3. Mitochondria-Associated ER Membranes (MAMs)
3.1. Overview of the MAM
3.2. Investigating the MAM
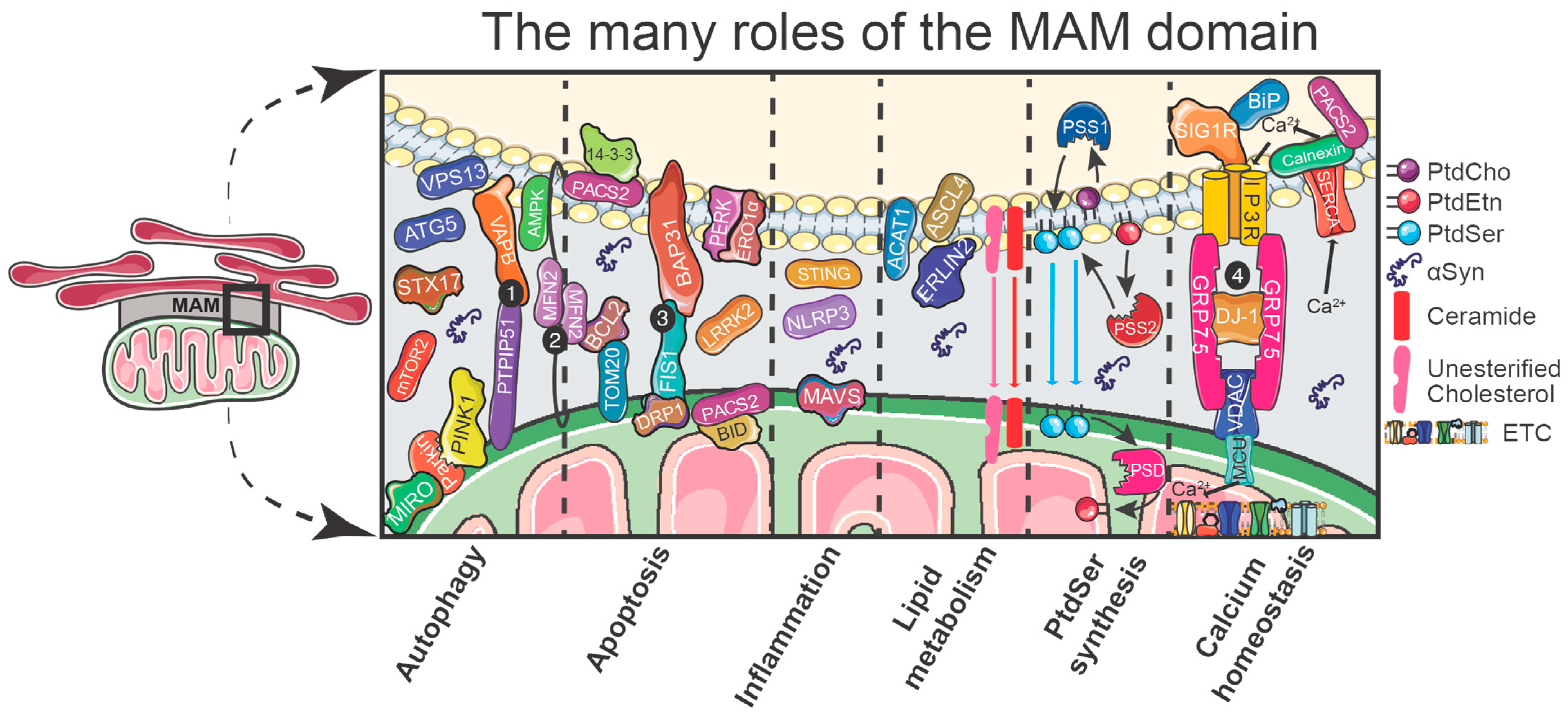
3.3. The Functions of the MAM
3.4. Autophagy
3.5. Apoptosis
3.6. Inflammation
3.7. Lipid Metabolism
3.8. Phosphatidylserine Synthesis
3.9. Calcium Homeostasis
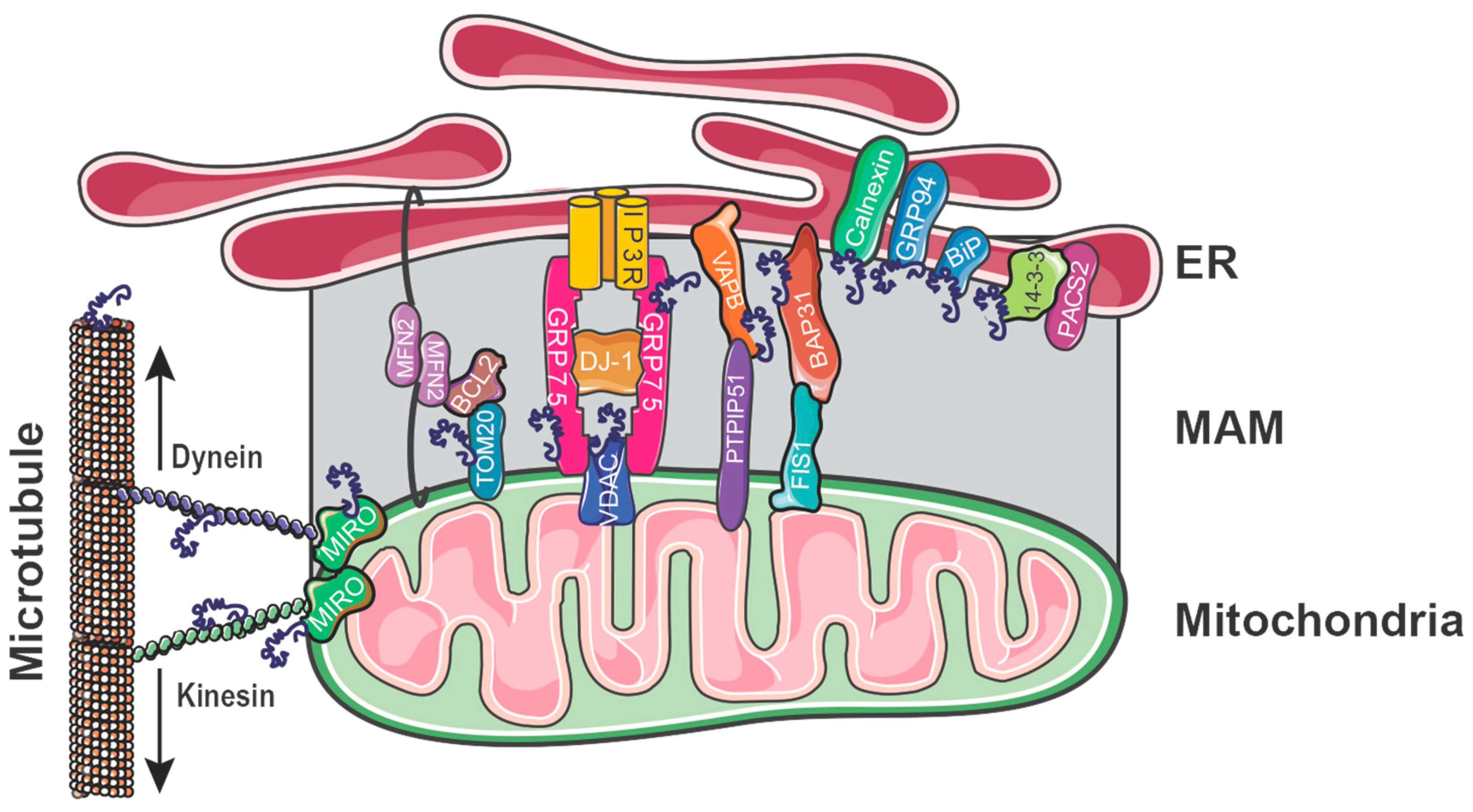
4. Regulating ER–Mitochondrial Apposition
4.1. PACS2
4.2. MAM Tethers
4.2.1. FIS1-BAP31
4.2.2. PTPIP51-VAPB
4.2.3. IP3R-GRP75-VDAC1
4.2.4. MFN2
5. Direct Association of αSyn with Proteins at the MAM
5.1. Association of αSyn Interacting Proteins at the MAM
5.2. Altering αSyn-Interacting Proteins at the MAM
5.3. αSyn as a MAM Regulator?
6. Conclusions and Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Roos, D.S.; Klein, M.; Deeg, D.J.H.; Doty, R.L.; Berendse, H.W. Prevalence of Prodromal Symptoms of Parkinson’s Disease in the Late Middle-Aged Population. J. Park. Dis. 2022, 12, 967–974. [Google Scholar] [CrossRef]
- Borghammer, P.; Horsager, J.; Andersen, K.; Van Den Berge, N.; Raunio, A.; Murayama, S.; Parkkinen, L.; Myllykangas, L. Neuropathological Evidence of Body-First vs. Brain-First Lewy Body Disease. Neurobiol. Dis. 2021, 161, 105557. [Google Scholar] [CrossRef]
- Hamza, T.H.; Zabetian, C.P.; Tenesa, A.; Laederach, A.; Montimurro, J.; Yearout, D.; Kay, D.M.; Doheny, K.F.; Paschall, J.; Pugh, E.; et al. Common Genetic Variation in the HLA Region Is Associated with Late-Onset Sporadic Parkinson’s Disease. Nat. Genet. 2010, 42, 781–785. [Google Scholar] [CrossRef]
- Tan, M.M.X.; Lawton, M.A.; Jabbari, E.; Reynolds, R.H.; Iwaki, H.; Blauwendraat, C.; Kanavou, S.; Pollard, M.I.; Hubbard, L.; Malek, N.; et al. Genome-Wide Association Studies of Cognitive and Motor Progression in Parkinson’s Disease. Mov. Disord. 2021, 36, 424–433. [Google Scholar] [CrossRef]
- Dong-Chen, X.; Yong, C.; Yang, X.; Chen-Yu, S.; Li-Hua, P. Signaling Pathways in Parkinson’s Disease: Molecular Mechanisms and Therapeutic Interventions. Sig. Transduct. Target. Ther. 2023, 8, 73. [Google Scholar] [CrossRef]
- Chiba-Falek, O.; Lopez, G.J.; Nussbaum, R.L. Levels of Alpha-Synuclein mRNA in Sporadic Parkinson Disease Patients. Mov. Disord. 2006, 21, 1703–1708. [Google Scholar] [CrossRef]
- Soldner, F.; Stelzer, Y.; Shivalila, C.S.; Abraham, B.J.; Latourelle, J.C.; Barrasa, M.I.; Goldmann, J.; Myers, R.H.; Young, R.A.; Jaenisch, R. Parkinson-Associated Risk Variant in Distal Enhancer of α-Synuclein Modulates Target Gene Expression. Nature 2016, 533, 95–99. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the Alpha-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schöls, L.; Riess, O. Ala30Pro Mutation in the Gene Encoding Alpha-Synuclein in Parkinson’s Disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Daida, K.; Shimonaka, S.; Shiba-Fukushima, K.; Ogata, J.; Yoshino, H.; Okuzumi, A.; Hatano, T.; Motoi, Y.; Hirunagi, T.; Katsuno, M.; et al. α-Synuclein V15A Variant in Familial Parkinson’s Disease Exhibits a Weaker Lipid-Binding Property. Mov. Disord. 2022, 37, 2075–2085. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The New Mutation, E46K, of α-Synuclein Causes Parkinson and Lewy Body Dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-Synuclein p.H50Q, a Novel Pathogenic Mutation for Parkinson’s Disease. Mov. Disord. 2013, 28, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Koros, C.; Strohäker, T.; Schulte, C.; Bozi, M.; Varvaresos, S.; Ibáñez de Opakua, A.; Simitsi, A.M.; Bougea, A.; Voumvourakis, K.; et al. A Novel SNCA A30G Mutation Causes Familial Parkinson’s Disease. Mov. Disord. 2021, 36, 1624–1633. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Hirano, M.; Stoessl, A.J.; Imamichi, Y.; Ikeda, A.; Li, Y.; Funayama, M.; Yamada, I.; Nakamura, Y.; Sossi, V.; et al. Homozygous Alpha-Synuclein p.A53V in Familial Parkinson’s Disease. Neurobiol. Aging 2017, 57, 248.e7–248.e12. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. Alpha-Synuclein Locus Triplication Causes Parkinson’s Disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.-C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-Synuclein Locus Duplication as a Cause of Familial Parkinson’s Disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, P.; Bonnet, A.-M.; Débarges, B.; Lohmann, E.; Tison, F.; Agid, Y.; Dürr, A.; Brice, A.; Pollak, P. Causal Relation between α-Synuclein Locus Duplication as a Cause of Familial Parkinson’s Disease. Lancet 2004, 364, 1169–1171. [Google Scholar] [CrossRef]
- Nishioka, K.; Ross, O.A.; Hattori, N. SNCA Gene Multiplication: A Model Mechanism of Parkinson Disease; Ross, O.A., Ed.; IntechOpen: Rijeka, Croatia, 2011; p. Ch. 20. [Google Scholar]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. α-Synuclein in Filamentous Inclusions of Lewy Bodies from Parkinson’s Disease and Dementia with Lewy Bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [PubMed]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy Pathology in Parkinson’s Disease Consists of Crowded Organelles and Lipid Membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Makasewicz, K.; Linse, S.; Sparr, E. Interplay of α-Synuclein with Lipid Membranes: Cooperative Adsorption, Membrane Remodeling and Coaggregation. JACS Au 2024, 4, 1250–1262. [Google Scholar] [CrossRef]
- Galvagnion, C. The Role of Lipids Interacting with α-Synuclein in the Pathogenesis of Parkinson’s Disease. J. Park. Dis. 2017, 7, 433–450. [Google Scholar] [CrossRef] [PubMed]
- Griggs, A.M.; Ysselstein, D.; Rochet, J.-C. Chapter 39—Role of Aberrant α-Synuclein–Membrane Interactions in Parkinson’s Disease. In Bio-Nanoimaging; Uversky, V.N., Lyubchenko, Y.L., Eds.; Academic Press: Boston, MA, USA, 2014; pp. 443–452. ISBN 978-0-12-394431-3. [Google Scholar]
- Sarchione, A.; Marchand, A.; Taymans, J.-M.; Chartier-Harlin, M.-C. Alpha-Synuclein and Lipids: The Elephant in the Room? Cells 2021, 10, 2452. [Google Scholar] [CrossRef] [PubMed]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. α-Synuclein Occurs Physiologically as a Helically Folded Tetramer That Resists Aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef]
- Lautenschläger, J.; Stephens, A.D.; Fusco, G.; Ströhl, F.; Curry, N.; Zacharopoulou, M.; Michel, C.H.; Laine, R.; Nespovitaya, N.; Fantham, M.; et al. C-Terminal Calcium Binding of α-Synuclein Modulates Synaptic Vesicle Interaction. Nat. Commun. 2018, 9, 712. [Google Scholar] [CrossRef] [PubMed]
- Moons, R.; Konijnenberg, A.; Mensch, C.; Van Elzen, R.; Johannessen, C.; Maudsley, S.; Lambeir, A.-M.; Sobott, F. Metal Ions Shape α-Synuclein. Sci. Rep. 2020, 10, 16293. [Google Scholar] [CrossRef] [PubMed]
- Bisi, N.; Feni, L.; Peqini, K.; Pérez-Peña, H.; Ongeri, S.; Pieraccini, S.; Pellegrino, S. α-Synuclein: An All-Inclusive Trip Around Its Structure, Influencing Factors and Applied Techniques. Front. Chem. 2021, 9, 666585. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.; McLaurin, J.; Yip, C.M.; George-Hyslop, P.S.; Fraser, P.E. α-Synuclein Membrane Interactions and Lipid Specificity. J. Biol. Chem. 2000, 275, 34328–34334. [Google Scholar] [CrossRef] [PubMed]
- Luäcke, C.; Gantz, D.L.; Klimtchuk, E.; Hamilton, J.A. Interactions between Fatty Acids and α-Synuclein. J. Lipid Res. 2006, 47, 1714–1724. [Google Scholar] [CrossRef] [PubMed]
- Fusco, G.; Pape, T.; Stephens, A.D.; Mahou, P.; Costa, A.R.; Kaminski, C.F.; Kaminski Schierle, G.S.; Vendruscolo, M.; Veglia, G.; Dobson, C.M.; et al. Structural Basis of Synaptic Vesicle Assembly Promoted by α-Synuclein. Nat. Commun. 2016, 7, 12563. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Carlus, D.; Yahi, N. The Fusogenic Tilted Peptide (67–78) of α-Synuclein Is a Cholesterol Binding Domain. Biochim. Biophys. Acta (BBA)-Biomembr. 2011, 1808, 2343–2351. [Google Scholar] [CrossRef]
- Fantini, J.; Yahi, N. Molecular Basis for the Glycosphingolipid-Binding Specificity of α-Synuclein: Key Role of Tyrosine 39 in Membrane Insertion. J. Mol. Biol. 2011, 408, 654–669. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhao, Y.; Xi, H.; Jiang, J.; Yu, Y.; Dong, W. The Membrane Interaction of Alpha-Synuclein. Front. Cell. Neurosci. 2021, 15, 633727. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, X.; Li, J.-D. The Roles of Post-Translational Modifications on α-Synuclein in the Pathogenesis of Parkinson’s Diseases. Front. Neurosci. 2019, 13, 381. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. Alpha-Synuclein Is Phosphorylated in Synucleinopathy Lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 Is the Dominant Pathological Modification of Alpha-Synuclein in Familial and Sporadic Lewy Body Disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Arawaka, S.; Hara, S.; Fukushima, S.; Koga, K.; Koyama, S.; Kato, T. Authentically Phosphorylated α-Synuclein at Ser129 Accelerates Neurodegeneration in a Rat Model of Familial Parkinson’s Disease. J. Neurosci. 2011, 31, 16884–16894. [Google Scholar] [CrossRef]
- Parra-Rivas, L.A.; Madhivanan, K.; Aulston, B.D.; Wang, L.; Prakashchand, D.D.; Boyer, N.P.; Saia-Cereda, V.M.; Branes-Guerrero, K.; Pizzo, D.P.; Bagchi, P.; et al. Serine-129 Phosphorylation of α-Synuclein Is an Activity-Dependent Trigger for Physiologic Protein-Protein Interactions and Synaptic Function. Neuron 2023, 111, 4006–4023.e10. [Google Scholar] [CrossRef]
- Sorrentino, Z.A.; Giasson, B.I. The Emerging Role of α-Synuclein Truncation in Aggregation and Disease. J. Biol. Chem. 2020, 295, 10224–10244. [Google Scholar] [CrossRef]
- Li, W.; West, N.; Colla, E.; Pletnikova, O.; Troncoso, J.C.; Marsh, L.; Dawson, T.M.; Jäkälä, P.; Hartmann, T.; Price, D.L.; et al. Aggregation Promoting C-Terminal Truncation of α-Synuclein Is a Normal Cellular Process and Is Enhanced by the Familial Parkinson’s Disease-Linked Mutations. Proc. Natl. Acad. Sci. USA 2005, 102, 2162–2167. [Google Scholar] [CrossRef]
- Cascella, R.; Bigi, A.; Cremades, N.; Cecchi, C. Effects of Oligomer Toxicity, Fibril Toxicity and Fibril Spreading in Synucleinopathies. Cell Mol. Life Sci. 2022, 79, 174. [Google Scholar] [CrossRef]
- Wang, W.; Perovic, I.; Chittuluru, J.; Kaganovich, A.; Nguyen, L.T.T.; Liao, J.; Auclair, J.R.; Johnson, D.; Landeru, A.; Simorellis, A.K.; et al. A Soluble α-Synuclein Construct Forms a Dynamic Tetramer. Proc. Natl. Acad. Sci. USA 2011, 108, 17797–17802. [Google Scholar] [CrossRef] [PubMed]
- Killinger, B.A.; Melki, R.; Brundin, P.; Kordower, J.H. Endogenous Alpha-Synuclein Monomers, Oligomers and Resulting Pathology: Let’s Talk about the Lipids in the Room. NPJ Park. Dis. 2019, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Vivona, S.; Diao, J.; Sharma, M.; Brunger, A.T.; Südhof, T.C. Properties of Native Brain α-Synuclein. Nature 2013, 498, E4–E6. [Google Scholar] [CrossRef] [PubMed]
- Espay, A.J.; Lees, A.J. Loss of Monomeric Alpha-Synuclein (Synucleinopenia) and the Origin of Parkinson’s Disease. Park. Relat. Disord. 2024, 112, 106077. [Google Scholar] [CrossRef] [PubMed]
- van Steenoven, I.; Majbour, N.K.; Vaikath, N.N.; Berendse, H.W.; van der Flier, W.M.; van de Berg, W.D.J.; Teunissen, C.E.; Lemstra, A.W.; El-Agnaf, O.M.A. α-Synuclein Species as Potential Cerebrospinal Fluid Biomarkers for Dementia with Lewy Bodies. Mov. Disord. 2018, 33, 1724–1733. [Google Scholar] [CrossRef] [PubMed]
- Wennström, M.; Surova, Y.; Hall, S.; Nilsson, C.; Minthon, L.; Boström, F.; Hansson, O.; Nielsen, H.M. Low CSF Levels of Both α-Synuclein and the α-Synuclein Cleaving Enzyme Neurosin in Patients with Synucleinopathy. PLoS ONE 2013, 8, e53250. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, A.; Schmitz, Y.; Fariñas, I.; Choi-Lundberg, D.; Ho, W.-H.; Castillo, P.E.; Shinsky, N.; Verdugo, J.M.G.; Armanini, M.; Ryan, A.; et al. Mice Lacking α-Synuclein Display Functional Deficits in the Nigrostriatal Dopamine System. Neuron 2000, 25, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Burré, J. The Synaptic Function of α-Synuclein. J. Park. Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial Import and Accumulation of Alpha-Synuclein Impair Complex I in Human Dopaminergic Neuronal Cultures and Parkinson Disease Brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. α-Synuclein Binds to the ER-Mitochondria Tethering Protein VAPB to Disrupt Ca2+ homeostasis and Mitochondrial ATP Production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef]
- Ramezani, M.; Wagenknecht-Wiesner, A.; Wang, T.; Holowka, D.A.; Eliezer, D.; Baird, B.A. Alpha Synuclein Modulates Mitochondrial Ca2+ Uptake from ER during Cell Stimulation and under Stress Conditions. NPJ Park. Dis. 2023, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Erustes, A.G.; D’Eletto, M.; Guarache, G.C.; Ureshino, R.P.; Bincoletto, C.; da Silva Pereira, G.J.; Piacentini, M.; Smaili, S.S. Overexpression of α-Synuclein Inhibits Mitochondrial Ca2+ Trafficking between the Endoplasmic Reticulum and Mitochondria through MAMs by Altering the GRP75–IP3R Interaction. J. Neurosci. Res. 2021, 99, 2932–2947. [Google Scholar] [CrossRef] [PubMed]
- Rosencrans, W.M.; Aguilella, V.M.; Rostovtseva, T.K.; Bezrukov, S.M. α-Synuclein Emerges as a Potent Regulator of VDAC-Facilitated Calcium Transport. Cell Calcium 2021, 95, 102355. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Vergnes, L.; Franich, N.R.; Reue, K.; Chesselet, M.-F. Region Specific Mitochondrial Impairment in Mice with Widespread Overexpression of Alpha-Synuclein. Neurobiol. Dis. 2014, 70, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Vicario, M.; Cieri, D.; Vallese, F.; Catoni, C.; Barazzuol, L.; Berto, P.; Grinzato, A.; Barbieri, L.; Brini, M.; Calì, T. A Split-GFP Tool Reveals Differences in the Sub-Mitochondrial Distribution of Wt and Mutant Alpha-Synuclein. Cell Death Dis. 2019, 10, 857. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. α-Synuclein Oligomers Interact with ATP Synthase and Open the Permeability Transition Pore in Parkinson’s Disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Becker, K.; Levine, N.; Zhang, M.; Lieberman, A.P.; Moore, D.J.; Ma, J. Pathogenic Alpha-Synuclein Aggregates Preferentially Bind to Mitochondria and Affect Cellular Respiration. Acta Neuropathol. Commun. 2019, 7, 41. [Google Scholar] [CrossRef]
- Jo, E.; Fuller, N.; Rand, R.P.; St George-Hyslop, P.; Fraser, P.E. Defective Membrane Interactions of Familial Parkinson’s Disease Mutant A30P Alpha-Synuclein. J. Mol. Biol. 2002, 315, 799–807. [Google Scholar] [CrossRef]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rub, C.; Liu, Y.; Magrane, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. A-Synuclein Is Localized to Mitochondria-Associated ER Membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef]
- Colla, E.; Coune, P.; Liu, Y.; Pletnikova, O.; Troncoso, J.C.; Iwatsubo, T.; Schneider, B.L.; Lee, M.K. Endoplasmic Reticulum Stress Is Important for the Manifestations of α-Synucleinopathy in vivo. J. Neurosci. 2012, 32, 3306–3320. [Google Scholar] [CrossRef]
- Colla, E.; Jensen, P.H.; Pletnikova, O.; Troncoso, J.C.; Glabe, C.; Lee, M.K. Accumulation of Toxic α-Synuclein Oligomer within Endoplasmic Reticulum Occurs in α-Synucleinopathy In Vivo. J. Neurosci. 2012, 32, 3301–3305. [Google Scholar] [CrossRef] [PubMed]
- Colla, E. Linking the Endoplasmic Reticulum to Parkinson’s Disease and Alpha-Synucleinopathy. Front. Neurosci. 2019, 13, 560. [Google Scholar] [CrossRef] [PubMed]
- Stojkovska, I.; Wani, W.Y.; Zunke, F.; Belur, N.R.; Pavlenko, E.A.; Mwenda, N.; Sharma, K.; Francelle, L.; Mazzulli, J.R. Rescue of α-Synuclein Aggregation in Parkinson’s Patient Neurons by Synergistic Enhancement of ER Proteostasis and Protein Trafficking. Neuron 2022, 110, 436–451.e11. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. α-Synuclein Blocks ER-Golgi Traffic and Rab1 Rescues Neuron Loss in Parkinson’s Models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Zunke, F.; Isacson, O.; Studer, L.; Krainc, D. α-Synuclein–Induced Lysosomal Dysfunction Occurs through Disruptions in Protein Trafficking in Human Midbrain Synucleinopathy Models. Proc. Natl. Acad. Sci. USA 2016, 113, 1931–1936. [Google Scholar] [CrossRef] [PubMed]
- Koss, D.J.; Erskine, D.; Porter, A.; Palmoski, P.; Menon, H.; Todd, O.G.J.; Leite, M.; Attems, J.; Outeiro, T.F. Nuclear Alpha-Synuclein Is Present in the Human Brain and Is Modified in Dementia with Lewy Bodies. Acta Neuropathol. Commun. 2022, 10, 98. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.; Moncalvo, M.; Tringali, D.; Tagliafierro, L.; Shriskanda, A.; Ilich, E.; Dong, W.; Kantor, B.; Chiba-Falek, O. The Mechanistic Role of Alpha-Synuclein in the Nucleus: Impaired Nuclear Function Caused by Familial Parkinson’s Disease SNCA Mutations. Hum. Mol. Genet. 2020, 29, 3107–3121. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.-L.; Song, L.-K.; Yuan, Y.-H.; Zhang, Y.; Han, N.; Gao, K.; Chen, N.-H. The Nuclear Accumulation of Alpha-Synuclein Is Mediated by Importin Alpha and Promotes Neurotoxicity by Accelerating the Cell Cycle. Neuropharmacology 2014, 82, 132–142. [Google Scholar] [CrossRef] [PubMed]
- English, A.R.; Voeltz, G.K. Endoplasmic Reticulum Structure and Interconnections with Other Organelles. Cold Spring Harb. Perspect. Biol. 2013, 5, a013227. [Google Scholar] [CrossRef]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef]
- Booms, A.; Coetzee, G.A. Functions of Intracellular Alpha-Synuclein in Microglia: Implications for Parkinson’s Disease Risk. Front. Cell. Neurosci. 2021, 15, 759571. [Google Scholar] [CrossRef] [PubMed]
- Djelloul, M.; Holmqvist, S.; Boza-Serrano, A.; Azevedo, C.; Yeung, M.S.; Goldwurm, S.; Frisén, J.; Deierborg, T.; Roybon, L. Alpha-Synuclein Expression in the Oligodendrocyte Lineage: An In Vitro and In Vivo Study Using Rodent and Human Models. Stem Cell Rep. 2015, 5, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.-Y.; Liu, G.-L.; Wang, D.-X.; Zhang, M.-M.; Kou, W.-Y.; Feng, T. Alpha-Synuclein in Peripheral Tissues in Parkinson’s Disease. ACS Chem. Neurosci. 2019, 10, 812–823. [Google Scholar] [CrossRef]
- Barbour, R.; Kling, K.; Anderson, J.P.; Banducci, K.; Cole, T.; Diep, L.; Fox, M.; Goldstein, J.M.; Soriano, F.; Seubert, P.; et al. Red Blood Cells Are the Major Source of Alpha-Synuclein in Blood. Neurodegener. Dis. 2008, 5, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Somayaji, M.; Cataldi, S.; Choi, S.J.; Edwards, R.H.; Mosharov, E.V.; Sulzer, D. A Dual Role for α-Synuclein in Facilitation and Depression of Dopamine Release from Substantia Nigra Neurons in Vivo. Proc. Natl. Acad. Sci. USA 2020, 117, 32701–32710. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.M.; Yang, D.; Li, X.-Q.; Liu, J.; Back, T.C.; Trivett, A.; Karim, B.; Barbut, D.; Zasloff, M.; Oppenheim, J.J. Alpha Synuclein, the Culprit in Parkinson Disease, Is Required for Normal Immune Function. Cell Rep. 2022, 38, 110090. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Autophagy and Alpha-Synuclein: Relevance to Parkinson’s Disease and Related Synucleopathies. Mov. Disord. 2016, 31, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Shaltouki, A.; Hsieh, C.-H.; Kim, M.J.; Wang, X. Alpha-Synuclein Delays Mitophagy and Targeting Miro Rescues Neuron Loss in Parkinson’s Models. Acta Neuropathol. 2018, 136, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Kanthasamy, A.; Ghosh, A.; Yang, Y.; Anantharam, V.; Kanthasamy, A.G. α-Synuclein Negatively Regulates Protein Kinase Cδ Expression to Suppress Apoptosis in Dopaminergic Neurons by Reducing P300 Histone Acetyltransferase Activity. J. Neurosci. 2011, 31, 2035–2051. [Google Scholar] [CrossRef]
- Calì, T.; Ottolini, D.; Negro, A.; Brini, M. α-Synuclein Controls Mitochondrial Calcium Homeostasis by Enhancing Endoplasmic Reticulum-Mitochondria Interactions. J. Biol. Chem. 2012, 287, 17914–17929. [Google Scholar] [CrossRef]
- Vance, J.E. Phospholipid Synthesis in a Membrane Fraction Associated with Mitochondria. J. Biol. Chem. 1990, 265, 7248–7256. [Google Scholar] [CrossRef]
- Rusiñol, A.E.; Cui, Z.; Chen, M.H.; Vance, J.E. A Unique Mitochondria-Associated Membrane Fraction from Rat Liver Has a High Capacity for Lipid Synthesis and Contains Pre-Golgi Secretory Proteins Including Nascent Lipoproteins. J. Biol. Chem. 1994, 269, 27494–27502. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Csordás, G.; Renken, C.; Várnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnóczky, G. Structural and Functional Features and Significance of the Physical Linkage between ER and Mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. MAM (Mitochondria-Associated Membranes) in Mammalian Cells: Lipids and Beyond. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2014, 1841, 595–609. [Google Scholar] [CrossRef]
- Brown, D.A.; London, E. Structure and Function of Sphingolipid- and Cholesterol-Rich Membrane Rafts. J. Biol. Chem. 2000, 275, 17221–17224. [Google Scholar] [CrossRef]
- Montesinos, J.; Area-Gomez, E. Chapter 2—Isolation of Mitochondria-Associated ER Membranes. In Methods in Cell Biology; Pon, L.A., Schon, E.A., Eds.; Mitochondria, 3rd ed.; Academic Press: Cambridge, MA, USA, 2020; Volume 155, pp. 33–44. [Google Scholar]
- MacDonald, L.; Baldini, G.; Storrie, B. Does Super Resolution Fluorescence Microscopy Obsolete Previous Microscopic Approaches to Protein Co-Localization? Methods Mol. Biol. 2015, 1270, 255–275. [Google Scholar] [CrossRef] [PubMed]
- Vallese, F.; Catoni, C.; Cieri, D.; Barazzuol, L.; Ramirez, O.; Calore, V.; Bonora, M.; Giamogante, F.; Pinton, P.; Brini, M.; et al. An Expanded Palette of Improved SPLICS Reporters Detects Multiple Organelle Contacts in Vitro and in vivo. Nat. Commun. 2020, 11, 6069. [Google Scholar] [CrossRef]
- Hertlein, V.; Flores-Romero, H.; Das, K.K.; Fischer, S.; Heunemann, M.; Calleja-Felipe, M.; Knafo, S.; Hipp, K.; Harter, K.; Fitzgerald, J.C.; et al. MERLIN: A Novel BRET-Based Proximity Biosensor for Studying Mitochondria–ER Contact Sites. Life Sci. Alliance 2019, 3, e201900600. [Google Scholar] [CrossRef]
- Chang, C.C.Y.; Lee Gregory, C.-Y.; Chang, E.T.; Cruz, J.C.; Levesque, M.C.; Chang, T.-Y. Recombinant Acyl-coA:Cholesterol Acyltransferase-1 (ACAT-1) Purified to Essential Homogeneity Utilizes Cholesterol in Mixed Micelles or in Vesicles in a Highly Cooperative Manner. J. Biol. Chem. 1998, 273, 35132–35141. [Google Scholar] [CrossRef]
- Area-Gomez, E. Chapter Eleven—Assessing the Function of Mitochondria-Associated ER Membranes. In Mitochondrial Function; Murphy, A.N., Chan, D.C.B.T.-M., Eds.; Academic Press: Cambridge, MA, USA, 2014; Volume 547, pp. 181–197. ISBN 0076-6879. [Google Scholar]
- Patergnani, S.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. Calcium Signaling around Mitochondria Associated Membranes (MAMs). Cell Commun. Signal 2011, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Thestrup, T.; Litzlbauer, J.; Bartholomäus, I.; Mues, M.; Russo, L.; Dana, H.; Kovalchuk, Y.; Liang, Y.; Kalamakis, G.; Laukat, Y.; et al. Optimized Ratiometric Calcium Sensors for Functional in Vivo Imaging of Neurons and T Lymphocytes. Nat. Methods 2014, 11, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cumberbatch, D.; Centanni, S.; Shi, S.-Q.; Winder, D.; Webb, D.; Johnson, C.H. Coupling Optogenetic Stimulation with NanoLuc-Based Luminescence (BRET) Ca++ Sensing. Nat. Commun. 2016, 7, 13268. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Woo, Y.; Suh, Y.; Suh, B.K.; Kim, S.J.; Nhung, T.T.M.; Yoo, J.Y.; Nghi, T.D.; Lee, S.B.; Mun, D.J.; et al. Ratiometric Measurement of MAM Ca2+ Dynamics Using a Modified CalfluxVTN. Nat. Commun. 2023, 14, 3586. [Google Scholar] [CrossRef] [PubMed]
- Pekkurnaz, G.; Wang, X. Mitochondrial Heterogeneity and Homeostasis through the Lens of a Neuron. Nat. Metab. 2022, 4, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.H.; Shen, S.; Wang, J.J.; He, Z.; Poon, A.; Li, J.; Qu, J.; Zhang, S.X. Comparative Proteomic Analysis of the Mitochondria-Associated ER Membrane (MAM) in a Long-Term Type 2 Diabetic Rodent Model. Sci. Rep. 2017, 7, 2062. [Google Scholar] [CrossRef] [PubMed]
- Poston, C.N.; Krishnan, S.C.; Bazemore-Walker, C.R. In-Depth Proteomic Analysis of Mammalian Mitochondria-Associated Membranes (MAM). J. Proteom. 2013, 79, 219–230. [Google Scholar] [CrossRef]
- Scorrano, L.; De Matteis, M.A.; Emr, S.; Giordano, F.; Hajnóczky, G.; Kornmann, B.; Lackner, L.L.; Levine, T.P.; Pellegrini, L.; Reinisch, K.; et al. Coming Together to Define Membrane Contact Sites. Nat. Commun. 2019, 10, 1287. [Google Scholar] [CrossRef] [PubMed]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in Healthy Aging and Disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, H.; Zhang, L.; Lin, X.; Li, X.; Zhuang, H.; Fan, H.; Meng, T.; He, Z.; Huang, H.; et al. The AMPK-MFN2 Axis Regulates MAM Dynamics and Autophagy Induced by Energy Stresses. Autophagy 2021, 17, 1142–1156. [Google Scholar] [CrossRef]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. mTOR Complex 2-Akt Signaling at Mitochondria-Associated Endoplasmic Reticulum Membranes (MAM) Regulates Mitochondrial Physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR Signaling Transduction Pathway and Targeted Therapies in Cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes Form at ER–Mitochondria Contact Sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, T.; Matarrese, P.; Manganelli, V.; Marconi, M.; Tinari, A.; Gambardella, L.; Faggioni, A.; Misasi, R.; Sorice, M.; Malorni, W. Evidence for the Involvement of Lipid Rafts Localized at the ER-Mitochondria Associated Membranes in Autophagosome Formation. Autophagy 2016, 12, 917–935. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The Ubiquitin Kinase PINK1 Recruits Autophagy Receptors to Induce Mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin Is Phosphorylated by PINK1 to Activate Parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Kuruvilla, J.; Tan, E.-K. Mitophagy and Reactive Oxygen Species Interplay in Parkinson’s Disease. NPJ Park. Dis. 2022, 8, 135. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 Stabilized by Mitochondrial Depolarization Recruits Parkin to Damaged Mitochondria and Activates Latent Parkin for Mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the Parkin Gene Cause Autosomal Recessive Juvenile Parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Gelmetti, V.; De Rosa, P.; Torosantucci, L.; Marini, E.S.; Romagnoli, A.; Di Rienzo, M.; Arena, G.; Vignone, D.; Fimia, G.M.; Valente, E.M. PINK1 and BECN1 Relocalize at Mitochondria-Associated Membranes during Mitophagy and Promote ER-Mitochondria Tethering and Autophagosome Formation. Autophagy 2017, 13, 654–669. [Google Scholar] [CrossRef] [PubMed]
- Choubey, V.; Cagalinec, M.; Liiv, J.; Safiulina, D.; Hickey, M.A.; Kuum, M.; Liiv, M.; Anwar, T.; Eskelinen, E.-L.; Kaasik, A. BECN1 Is Involved in the Initiation of Mitophagy. Autophagy 2014, 10, 1105–1119. [Google Scholar] [CrossRef] [PubMed]
- Guardia-Laguarta, C.; Liu, Y.; Lauritzen, K.H.; Erdjument-Bromage, H.; Martin, B.; Swayne, T.C.; Jiang, X.; Przedborski, S. PINK1 Content in Mitochondria Is Regulated by ER-Associated Degradation. J. Neurosci. 2019, 39, 7074–7085. [Google Scholar] [CrossRef]
- Fu, M.; St-Pierre, P.; Shankar, J.; Wang, P.T.C.; Joshi, B.; Nabi, I.R. Regulation of Mitophagy by the Gp78 E3 Ubiquitin Ligase. Mol. Biol. Cell 2013, 24, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Li, W.; Chen, H.; Jiang, L.; Zhu, R.; Feng, D. FUNDC1 Is a Novel Mitochondrial-Associated-Membrane (MAM) Protein Required for Hypoxia-Induced Mitochondrial Fission and Mitophagy. Autophagy 2016, 12, 1675–1676. [Google Scholar] [CrossRef] [PubMed]
- Marinković, M.; Novak, I. A Brief Overview of BNIP3L/NIX Receptor-Mediated Mitophagy. FEBS Open Bio 2021, 11, 3230–3236. [Google Scholar] [CrossRef] [PubMed]
- Prasad, M.; Kaur, J.; Pawlak, K.J.; Bose, M.; Whittal, R.M.; Bose, H.S. Mitochondria-Associated Endoplasmic Reticulum Membrane (MAM) Regulates Steroidogenic Activity via Steroidogenic Acute Regulatory Protein (StAR)-Voltage-Dependent Anion Channel 2 (VDAC2) Interaction. J. Biol. Chem. 2015, 290, 2604–2616. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER Tubules Mark Sites of Mitochondrial Division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 Mediate Drp1 Recruitment in Mitochondrial Fission. MBoC 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The Role of Dynamin-Related Protein 1, a Mediator of Mitochondrial Fission, in Apoptosis. Dev. Cell 2001, 1, 515–525. [Google Scholar] [CrossRef]
- Xian, H.; Yang, Q.; Xiao, L.; Shen, H.-M.; Liou, Y.-C. STX17 Dynamically Regulated by Fis1 Induces Mitophagy via Hierarchical Macroautophagic Mechanism. Nat. Commun. 2019, 10, 2059. [Google Scholar] [CrossRef]
- Iwasawa, R.; Mahul-Mellier, A.-L.; Datler, C.; Pazarentzos, E.; Grimm, S. Fis1 and Bap31 Bridge the Mitochondria-ER Interface to Establish a Platform for Apoptosis Induction. EMBO J. 2011, 30, 556–568. [Google Scholar] [CrossRef]
- Ihenacho, U.K.; Meacham, K.A.; Harwig, M.C.; Widlansky, M.E.; Hill, R.B. Mitochondrial Fission Protein 1: Emerging Roles in Organellar Form and Function in Health and Disease. Front. Endocrinol. 2021, 12, 660095. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Nguyen, M.; Chang, N.C.; Shore, G.C. Fis1, Bap31 and the Kiss of Death between Mitochondria and Endoplasmic Reticulum. EMBO J. 2011, 30, 451–452. [Google Scholar] [CrossRef]
- Dadsena, S.; Bockelmann, S.; Mina, J.G.M.; Hassan, D.G.; Korneev, S.; Razzera, G.; Jahn, H.; Niekamp, P.; Müller, D.; Schneider, M.; et al. Ceramides Bind VDAC2 to Trigger Mitochondrial Apoptosis. Nat. Commun. 2019, 10, 1832. [Google Scholar] [CrossRef]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.-P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK Is Required at the ER-Mitochondrial Contact Sites to Convey Apoptosis after ROS-Based ER Stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef]
- Bassot, A.; Chen, J.; Takahashi-Yamashiro, K.; Yap, M.C.; Gibhardt, C.S.; Le, G.N.T.; Hario, S.; Nasu, Y.; Moore, J.; Gutiérrez, T.; et al. The Endoplasmic Reticulum Kinase PERK Interacts with the Oxidoreductase ERO1 to Metabolically Adapt Mitochondria. Cell Rep. 2023, 42, 111899. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, J.C.; Arnoult, D.; Youle, R.J. Control of Mitochondrial Permeability by Bcl-2 Family Members. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2004, 1644, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Scheiblich, H.; Schlütter, A.; Golenbock, D.T.; Latz, E.; Martinez-Martinez, P.; Heneka, M.T. Activation of the NLRP3 Inflammasome in Microglia: The Role of Ceramide. J. Neurochem. 2017, 143, 534–550. [Google Scholar] [CrossRef]
- de la Roche, M.; Hamilton, C.; Mortensen, R.; Jeyaprakash, A.A.; Ghosh, S.; Anand, P.K. Trafficking of Cholesterol to the ER Is Required for NLRP3 Inflammasome Activation. J. Cell Biol. 2018, 217, 3560–3576. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.-K.; Chen, Z.J. Identification and Characterization of MAVS, a Mitochondrial Antiviral Signaling Protein That Activates NF-κB and IRF3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.L.; Zhu, J.; Sarkar, S.N.; Coyne, C.B. Regulation of Mitochondrial Antiviral Signaling (MAVS) Expression and Signaling by the Mitochondria-Associated Endoplasmic Reticulum Membrane (MAM) Protein Gp78. J. Biol. Chem. 2014, 289, 1604–1616. [Google Scholar] [CrossRef]
- Xue, C.; Dong, N.; Shan, A. Putative Role of STING-Mitochondria Associated Membrane Crosstalk in Immunity. Trends Immunol. 2022, 43, 513–522. [Google Scholar] [CrossRef]
- Szego, E.M.; Malz, L.; Bernhardt, N.; Rösen-Wolff, A.; Falkenburger, B.H.; Luksch, H. Constitutively Active STING Causes Neuroinflammation and Degeneration of Dopaminergic Neurons in Mice. eLife 2022, 11, e81943. [Google Scholar] [CrossRef] [PubMed]
- Hinkle, J.T.; Patel, J.; Panicker, N.; Karuppagounder, S.S.; Biswas, D.; Belingon, B.; Chen, R.; Brahmachari, S.; Pletnikova, O.; Troncoso, J.C.; et al. STING Mediates Neurodegeneration and Neuroinflammation in Nigrostriatal α-Synucleinopathy. Proc. Natl. Acad. Sci. USA 2022, 119, e2118819119. [Google Scholar] [CrossRef] [PubMed]
- Manganelli, V.; Longo, A.; Mattei, V.; Recalchi, S.; Riitano, G.; Caissutti, D.; Capozzi, A.; Sorice, M.; Misasi, R.; Garofalo, T. Role of ERLINs in the Control of Cell Fate through Lipid Rafts. Cells 2021, 10, 2408. [Google Scholar] [CrossRef]
- Stone, S.J.; Levin, M.C.; Zhou, P.; Han, J.; Walther, T.C.; Farese, R.V. The Endoplasmic Reticulum Enzyme DGAT2 Is Found in Mitochondria-Associated Membranes and Has a Mitochondrial Targeting Signal That Promotes Its Association with Mitochondria. J. Biol. Chem. 2009, 284, 5352–5361. [Google Scholar] [CrossRef]
- Aaltonen, M.J.; Alecu, I.; König, T.; Bennett, S.A.; Shoubridge, E.A. Serine Palmitoyltransferase Assembles at ER–Mitochondria Contact Sites. Life Sci. Alliance 2021, 5, e202101278. [Google Scholar] [CrossRef]
- Planas-Serra, L.; Launay, N.; Goicoechea, L.; Heron, B.; Jou, C.; Juliá-Palacios, N.; Ruiz, M.; Fourcade, S.; Casasnovas, C.; Torre, C.D.L.; et al. Sphingolipid Desaturase DEGS1 Is Essential for Mitochondria-Associated Membrane Integrity. J. Clin. Investig. 2023, 133, e162957. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Non-Apoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Mahoney-Sanchez, L.; Bouchaoui, H.; Boussaad, I.; Jonneaux, A.; Timmerman, K.; Berdeaux, O.; Ayton, S.; Krüger, R.; Duce, J.A.; Devos, D.; et al. Alpha Synuclein Determines Ferroptosis Sensitivity in Dopaminergic Neurons via Modulation of Ether-Phospholipid Membrane Composition. Cell Rep. 2022, 40, 111231. [Google Scholar] [CrossRef] [PubMed]
- Man, W.C.; Miyazaki, M.; Chu, K.; Ntambi, J. Colocalization of SCD1 and DGAT2: Implying Preference for Endogenous Monounsaturated Fatty Acids in Triglyceride Synthesis. J. Lipid Res. 2006, 47, 1928–1939. [Google Scholar] [CrossRef] [PubMed]
- Ganji, R.; Paulo, J.A.; Xi, Y.; Kline, I.; Zhu, J.; Clemen, C.S.; Weihl, C.C.; Purdy, J.G.; Gygi, S.P.; Raman, M. The P97-UBXD8 Complex Regulates ER-Mitochondria Contact Sites by Altering Membrane Lipid Saturation and Composition. Nat. Commun. 2023, 14, 638. [Google Scholar] [CrossRef] [PubMed]
- Lewin, T.M.; Kim, J.-H.; Granger, D.A.; Vance, J.E.; Coleman, R.A. Acyl-CoA Synthetase Isoforms 1, 4, and 5 Are Present in Different Subcellular Membranes in Rat Liver and Can Be Inhibited Independently. J. Biol. Chem. 2001, 276, 24674–24679. [Google Scholar] [CrossRef] [PubMed]
- Harayama, T.; Riezman, H. Understanding the Diversity of Membrane Lipid Composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.J.; Vance, J.E. Phosphatidylserine Synthase-1 and -2 Are Localized to Mitochondria-Associated Membranes. J. Biol. Chem. 2000, 275, 34534–34540. [Google Scholar] [CrossRef]
- Kennedy, E.P.; Weiss, S.B. The Function of Cytidine Coenzymes in the Biosynthesis of Phospholipides. J. Biol. Chem. 1956, 222, 193–214. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.K.; Kimura, T. Phosphatidylserine Biosynthesis Pathways in Lipid Homeostasis: Toward Resolution of the Pending Central Issue for Decades. FASEB J. 2021, 35, e21177. [Google Scholar] [CrossRef]
- Voelker, D.R. Chapter 15—Lipid Assembly into Cell Membranes. In New Comprehensive Biochemistry; Vance, D.E., Vance, J.E., Eds.; Biochemistry of Lipids, Lipoproteins and Membranes; Elsevier: Amsterdam, The Netherlands, 1996; Volume 31, pp. 391–423. [Google Scholar]
- Perrin, R.J.; Woods, W.S.; Clayton, D.F.; George, J.M. Interaction of Human α-Synuclein and Parkinson’s Disease Variants with Phospholipids: Structural analysis using site-directed mutagenesis. J. Biol. Chem. 2000, 275, 34393–34398. [Google Scholar] [CrossRef]
- Hannestad, J.K.; Rocha, S.; Agnarsson, B.; Zhdanov, V.P.; Wittung-Stafshede, P.; Höök, F. Single-Vesicle Imaging Reveals Lipid-Selective and Stepwise Membrane Disruption by Monomeric α-Synuclein. Proc. Natl. Acad. Sci. USA 2020, 117, 14178–14186. [Google Scholar] [CrossRef] [PubMed]
- Kay, J.G.; Fairn, G.D. Distribution, Dynamics and Functional Roles of Phosphatidylserine within the Cell. Cell Commun. Signal. 2019, 17, 126. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, C.; Yang, S.; Xiao, Y.; Xiong, X.; Chen, W.; Zhao, H.; Zhang, Q.; Han, Y.; Sun, L. Mitochondria-Associated ER Membranes—The Origin Site of Autophagy. Front. Cell Dev. Biol. 2020, 8, 595. [Google Scholar] [CrossRef]
- Brown, G.C.; Neher, J.J. Microglial Phagocytosis of Live Neurons. Nat. Rev. Neurosci. 2014, 15, 209–216. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive Microglia Are Positive for HLA-DR in the Substantia Nigra of Parkinson’s and Alzheimer’s Disease Brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Hishikawa, N.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Distribution of Major Histocompatibility Complex Class II-Positive Microglia and Cytokine Profile of Parkinson’s Disease Brains. Acta Neuropathol. 2003, 106, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Lynes, E.M.; Raturi, A.; Shenkman, M.; Sandoval, C.O.; Yap, M.C.; Wu, J.; Janowicz, A.; Myhill, N.; Benson, M.D.; Campbell, R.E.; et al. Palmitoylation Is the Switch That Assigns Calnexin to Quality Control or ER Ca2+ Signaling. J. Cell Sci. 2013, 126, 3893–3903. [Google Scholar] [CrossRef]
- Bartok, A.; Weaver, D.; Golenár, T.; Nichtova, Z.; Katona, M.; Bánsághi, S.; Alzayady, K.J.; Thomas, V.K.; Ando, H.; Mikoshiba, K.; et al. IP3 Receptor Isoforms Differently Regulate ER-Mitochondrial Contacts and Local Calcium Transfer. Nat. Commun. 2019, 10, 3726. [Google Scholar] [CrossRef]
- Hayashi, T.; Su, T.-P. Sigma-1 Receptor Chaperones at the ER-Mitochondrion Interface Regulate Ca(2+) Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef]
- Myhill, N.; Lynes, E.M.; Nanji, J.A.; Blagoveshchenskaya, A.D.; Fei, H.; Carmine Simmen, K.; Cooper, T.J.; Thomas, G.; Simmen, T. The Subcellular Distribution of Calnexin Is Mediated by PACS-2. Mol. Biol. Cell 2008, 19, 2777–2788. [Google Scholar] [CrossRef]
- Marchi, S.; Bittremieux, M.; Missiroli, S.; Morganti, C.; Patergnani, S.; Sbano, L.; Rimessi, A.; Kerkhofs, M.; Parys, J.B.; Bultynck, G.; et al. Endoplasmic Reticulum-Mitochondria Communication through Ca2+ Signaling: The Importance of Mitochondria-Associated Membranes (MAMs). In Organelle Contact Sites: From Molecular Mechanism to Disease; Tagaya, M., Simmen, T., Eds.; Springer: Singapore, 2017; pp. 49–67. ISBN 978-981-10-4567-7. [Google Scholar]
- Daverkausen-Fischer, L.; Pröls, F. Regulation of Calcium Homeostasis and Flux between the Endoplasmic Reticulum and the Cytosol. J. Biol. Chem. 2022, 298, 102061. [Google Scholar] [CrossRef] [PubMed]
- Simmen, T.; Aslan, J.E.; Blagoveshchenskaya, A.D.; Thomas, L.; Wan, L.; Xiang, Y.; Feliciangeli, S.F.; Hung, C.-H.; Crump, C.M.; Thomas, G. PACS-2 Controls Endoplasmic Reticulum-Mitochondria Communication and Bid-Mediated Apoptosis. EMBO J. 2005, 24, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Aslan, J.E.; You, H.; Williamson, D.M.; Endig, J.; Youker, R.T.; Thomas, L.; Shu, H.; Du, Y.; Milewski, R.L.; Brush, M.H.; et al. Akt and 14-3-3 Control a PACS-2 Homeostatic Switch That Integrates Membrane Traffic with TRAIL-Induced Apoptosis. Mol. Cell 2009, 34, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, L.; Yang, M.; Zeng, L.; Sun, L. PACS-2: A Key Regulator of Mitochondria-Associated Membranes (MAMs). Pharmacol. Res. 2020, 160, 105080. [Google Scholar] [CrossRef] [PubMed]
- Athauda, D.; Foltynie, T. Insulin Resistance and Parkinson’s Disease: A New Target for Disease Modification? Prog. Neurobiol. 2016, 145–146, 98–120. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Gerle, C.; Halestrap, A.P.; Jonas, E.A.; Karch, J.; Mnatsakanyan, N.; Pavlov, E.; Sheu, S.-S.; Soukas, A.A. Identity, Structure, and Function of the Mitochondrial Permeability Transition Pore: Controversies, Consensus, Recent Advances, and Future Directions. Cell Death Differ. 2023, 30, 1869–1885. [Google Scholar] [CrossRef] [PubMed]
- Abe, F.; Van Prooyen, N.; Ladasky, J.J.; Edidin, M. Interaction of Bap31 and MHC Class I Molecules and Their Traffic out of the Endoplasmic Reticulum. J. Immunol. 2009, 182, 4776–4783. [Google Scholar] [CrossRef] [PubMed]
- Niu, K.; Xu, J.; Cao, Y.; Hou, Y.; Shan, M.; Wang, Y.; Xu, Y.; Sun, M.; Wang, B. BAP31 Is Involved in T Cell Activation through TCR Signal Pathways. Sci. Rep. 2017, 7, 44809. [Google Scholar] [CrossRef] [PubMed]
- Zen, K.; Utech, M.; Liu, Y.; Soto, I.; Nusrat, A.; Parkos, C.A. Association of BAP31 with CD11b/CD18: Potential role in intracellular trafficking of CD11b/CD18 in neutrophils. J. Biol. Chem. 2004, 279, 44924–44930. [Google Scholar] [CrossRef]
- Wang, T.; Chen, J.; Hou, Y.; Yu, Y.; Wang, B. BAP31 Deficiency Contributes to the Formation of Amyloid-β Plaques in Alzheimer’s Disease by Reducing the Stability of RTN3. FASEB J. 2019, 33, 4936–4946. [Google Scholar] [CrossRef]
- Jia, C.; Li, G.; Jiang, R.; Liu, X.; Yuan, Q.; Le, W.; Hou, Y.; Wang, B. B-Cell Receptor-Associated Protein 31 Negatively Regulates the Expression of Monoamine Oxidase A Via R1. Front. Mol. Biosci. 2020, 7, 64. [Google Scholar] [CrossRef] [PubMed]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.-F.; Vizcay-Barrena, G.; Lin, W.-L.; Xu, Y.-F.; Lewis, J.; et al. ER-Mitochondria Associations Are Regulated by the VAPB-PTPIP51 Interaction and Are Disrupted by ALS/FTD-Associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef] [PubMed]
- Obara, C.J.; Nixon-Abell, J.; Moore, A.S.; Riccio, F.; Hoffman, D.P.; Shtengel, G.; Xu, C.S.; Schaefer, K.; Pasolli, H.A.; Masson, J.-B.; et al. Motion of VAPB Molecules Reveals ER-Mitochondria Contact Site Subdomains. Nature 2024, 626, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Drouet, V.; Majounie, E.; Deramecourt, V.; Jacoupy, M.; Nicolas, A.; Cormier-Dequaire, F.; Hassoun, S.M.; Pujol, C.; Ciura, S.; et al. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am. J. Hum. Genet. 2016, 98, 500–513. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Leonzino, M.; Hancock-Cerutti, W.; Horenkamp, F.A.; Li, P.; Lees, J.A.; Wheeler, H.; Reinisch, K.M.; De Camilli, P. VPS13A and VPS13C Are Lipid Transport Proteins Differentially Localized at ER Contact Sites. J. Cell Biol. 2018, 217, 3625–3639. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Mari, M.; Parashar, S.; Liu, D.; Cui, Y.; Reggiori, F.; Novick, P.J.; Ferro-Novick, S. Vps13 Is Required for the Packaging of the ER into Autophagosomes during ER-Phagy. Proc. Natl. Acad. Sci. USA 2020, 117, 18530–18539. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Wang, J.; Xiong, J.; Fang, N.; Ji, W.-K. VPS13D Interacts with VCP/P97 and Negatively Regulates Endoplasmic Reticulum–Mitochondria Interactions. MBoC 2021, 32, 1474–1486. [Google Scholar] [CrossRef] [PubMed]
- Guyard, V.; Monteiro-Cardoso, V.F.; Omrane, M.; Sauvanet, C.; Houcine, A.; Boulogne, C.; Ben Mbarek, K.; Vitale, N.; Faklaris, O.; El Khallouki, N.; et al. ORP5 and ORP8 Orchestrate Lipid Droplet Biogenesis and Maintenance at ER–Mitochondria Contact Sites. J. Cell Biol. 2022, 221, e202112107. [Google Scholar] [CrossRef] [PubMed]
- Yeo, H.K.; Park, T.H.; Kim, H.Y.; Jang, H.; Lee, J.; Hwang, G.-S.; Ryu, S.E.; Park, S.H.; Song, H.K.; Ban, H.S.; et al. Phospholipid Transfer Function of PTPIP51 at Mitochondria-associated ER Membranes. EMBO Rep. 2021, 22, e51323. [Google Scholar] [CrossRef]
- Gomez-Suaga, P.; Paillusson, S.; Stoica, R.; Noble, W.; Hanger, D.P.; Miller, C.C.J. The ER-Mitochondria Tethering Complex VAPB-PTPIP51 Regulates Autophagy. Curr. Biol. 2017, 27, 371–385. [Google Scholar] [CrossRef]
- Gómez-Suaga, P.; Pérez-Nievas, B.G.; Glennon, E.B.; Lau, D.H.W.; Paillusson, S.; Mórotz, G.M.; Calì, T.; Pizzo, P.; Noble, W.; Miller, C.C.J. The VAPB-PTPIP51 Endoplasmic Reticulum-Mitochondria Tethering Proteins Are Present in Neuronal Synapses and Regulate Synaptic Activity. Acta Neuropathol. Commun. 2019, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and Endoplasmic Reticulum Calcium Homeostasis and Cell Death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Basso, V.; Marchesan, E.; Ziviani, E. A Trio Has Turned into a Quartet: DJ-1 Interacts with the IP3R-Grp75-VDAC Complex to Control ER-Mitochondria Interaction. Cell Calcium 2020, 87, 102186. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Patel, C.; Shenkman, M.; Kessel, A.; Ben-Tal, N.; Lederkremer, G.Z. The Sigma-1 Receptor Is an ER-Localized Type II Membrane Protein. J. Biol. Chem. 2021, 297, 101299. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 Coordinately Regulate Mitochondrial Fusion and Are Essential for Embryonic Development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- de Brito, O.M.; Scorrano, L. Mitofusin 2 Tethers Endoplasmic Reticulum to Mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- McLelland, G.-L.; Goiran, T.; Yi, W.; Dorval, G.; Chen, C.X.; Lauinger, N.D.; Krahn, A.I.; Valimehr, S.; Rakovic, A.; Rouiller, I.; et al. Mfn2 Ubiquitination by PINK1/Parkin Gates the P97-Dependent Release of ER from Mitochondria to Drive Mitophagy. eLife 2018, 7, e32866. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Li, G.J.; Davis, J.; Zhu, D.; Wang, Y.; Pan, C.; Zhang, J. Identification of Novel Proteins Associated with Both α-Synuclein and DJ-1. Mol. Cell. Proteom. 2007, 6, 845–859. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Shin, J.Y.; Lee, J.E.; Kim, H.N.; Chung, S.J.; Yoo, H.S.; Kim, S.J.; Cho, H.J.; Lee, E.-J.; Nam, S.J.; et al. A Selective ER-Phagy Exerts Neuroprotective Effects via Modulation of α-Synuclein Clearance in Parkinsonian Models. Proc. Natl. Acad. Sci. USA 2023, 120, e2221929120. [Google Scholar] [CrossRef] [PubMed]
- Gorbatyuk, M.S.; Shabashvili, A.; Chen, W.; Meyers, C.; Sullivan, L.F.; Salganik, M.; Lin, J.H.; Lewin, A.S.; Muzyczka, N.; Gorbatyuk, O.S. Glucose Regulated Protein 78 Diminishes α-Synuclein Neurotoxicity in a Rat Model of Parkinson Disease. Mol. Ther. 2012, 20, 1327–1337. [Google Scholar] [CrossRef]
- De Almeida, S.F.; Fleming, J.V.; Azevedo, J.E.; Carmo-Fonseca, M.; De Sousa, M. Stimulation of an Unfolded Protein Response Impairs MHC Class I Expression. J. Immunol. 2007, 178, 3612–3619. [Google Scholar] [CrossRef]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. α-Synuclein Binds TOM20 and Inhibits Mitochondrial Protein Import in Parkinson’s Disease. Sci. Transl. Med. 2016, 8, 342ra78. [Google Scholar] [CrossRef] [PubMed]
- Burbulla, L.F.; Fitzgerald, J.C.; Stegen, K.; Westermeier, J.; Thost, A.K.; Kato, H.; Mokranjac, D.; Sauerwald, J.; Martins, L.M.; Woitalla, D.; et al. Mitochondrial Proteolytic Stress Induced by Loss of Mortalin Function Is Rescued by Parkin and PINK1. Cell Death Dis. 2014, 5, e1180. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A.; Area-Gomez, E. Mitochondria-Associated ER Membranes in Alzheimer Disease. Mol. Cell. Neurosci. 2013, 55, 26–36. [Google Scholar] [CrossRef]
- Holland, P.; Knævelsrud, H.; Søreng, K.; Mathai, B.J.; Lystad, A.H.; Pankiv, S.; Bjørndal, G.T.; Schultz, S.W.; Lobert, V.H.; Chan, R.B.; et al. HS1BP3 Negatively Regulates Autophagy by Modulation of Phosphatidic Acid Levels. Nat. Commun. 2016, 7, 13889. [Google Scholar] [CrossRef]
- Underwood, R.; Gannon, M.; Pathak, A.; Kapa, N.; Chandra, S.; Klop, A.; Yacoubian, T.A. 14-3-3 Mitigates Alpha-Synuclein Aggregation and Toxicity in the in Vivo Preformed Fibril Model. Acta Neuropathol. Commun. 2021, 9, 13. [Google Scholar] [CrossRef]
- Szargel, R.; Shani, V.; Abd Elghani, F.; Mekies, L.N.; Liani, E.; Rott, R.; Engelender, S. The PINK1, Synphilin-1 and SIAH-1 Complex Constitutes a Novel Mitophagy Pathway. Hum. Mol. Genet. 2016, 25, 3476–3490. [Google Scholar] [CrossRef]
- Halperin, L.; Jung, J.; Michalak, M. The Many Functions of the Endoplasmic Reticulum Chaperones and Folding Enzymes. IUBMB Life 2014, 66, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Berwin, B.; Rosser, M.F.N.; Brinker, K.G.; Nicchitta, C.V. Transfer of GRP94(Gp96)-Associated Peptides onto Endosomal MHC Class I Molecules. Traffic 2002, 3, 358–366. [Google Scholar] [CrossRef]
- Zhang, Y.; Inaba, K. Structural Basis of the Conformational and Functional Regulation of Human SERCA2b, the Ubiquitous Endoplasmic Reticulum Calcium Pump. BioEssays 2022, 44, 2200052. [Google Scholar] [CrossRef]
- Britzolaki, A.; Saurine, J.; Flaherty, E.; Thelen, C.; Pitychoutis, P.M. The SERCA2: A Gatekeeper of Neuronal Calcium Homeostasis in the Brain. Cell Mol. Neurobiol. 2018, 38, 981–994. [Google Scholar] [CrossRef]
- Betzer, C.; Lassen, L.B.; Olsen, A.; Kofoed, R.H.; Reimer, L.; Gregersen, E.; Zheng, J.; Calì, T.; Gai, W.; Chen, T.; et al. Alpha-synuclein Aggregates Activate Calcium Pump SERCA Leading to Calcium Dysregulation. EMBO Rep. 2018, 19, e44617. [Google Scholar] [CrossRef] [PubMed]
- Solana-Manrique, C.; Muñoz-Soriano, V.; Sanz, F.J.; Paricio, N. Oxidative Modification Impairs SERCA Activity in Drosophila and Human Cell Models of Parkinson’s Disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2021, 1867, 166152. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Han, J.; Kim, H.; Park, S.M.; Joe, E.; Jou, I. Parkinson’s Disease-Associated LRRK2-G2019S Mutant Acts through Regulation of SERCA Activity to Control ER Stress in Astrocytes. Acta Neuropathol. Commun. 2019, 7, 68. [Google Scholar] [CrossRef] [PubMed]
- Szabo, L.; Cummins, N.; Paganetti, P.; Odermatt, A.; Papassotiropoulos, A.; Karch, C.; Götz, J.; Eckert, A.; Grimm, A. ER-Mitochondria Contacts and Cholesterol Metabolism Are Disrupted by Disease-Associated Tau Protein. EMBO Rep. 2023, 24, e57499. [Google Scholar] [CrossRef] [PubMed]
- Lalier, L.; Mignard, V.; Joalland, M.-P.; Lanoé, D.; Cartron, P.-F.; Manon, S.; Vallette, F.M. TOM20-Mediated Transfer of Bcl2 from ER to MAM and Mitochondria upon Induction of Apoptosis. Cell Death Dis. 2021, 12, 182. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; López-Doménech, G.; Halff, E.F.; Covill-Cooke, C.; Ivankovic, D.; Melandri, D.; Arancibia-Cárcamo, I.L.; Burden, J.J.; Lowe, A.R.; Kittler, J.T. Miro Clusters Regulate ER-Mitochondria Contact Sites and Link Cristae Organization to the Mitochondrial Transport Machinery. Nat. Commun. 2019, 10, 4399. [Google Scholar] [CrossRef] [PubMed]
- Berenguer-Escuder, C.; Grossmann, D.; Antony, P.; Arena, G.; Wasner, K.; Massart, F.; Jarazo, J.; Walter, J.; Schwamborn, J.C.; Grünewald, A.; et al. Impaired Mitochondrial–Endoplasmic Reticulum Interaction and Mitophagy in Miro1-Mutant Neurons in Parkinson’s Disease. Hum. Mol. Genet. 2020, 29, 1353–1364. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, D.; Berenguer-Escuder, C.; Bellet, M.E.; Scheibner, D.; Bohler, J.; Massart, F.; Rapaport, D.; Skupin, A.; Fouquier d’Hérouël, A.; Sharma, M.; et al. Mutations in RHOT1 Disrupt Endoplasmic Reticulum–Mitochondria Contact Sites Interfering with Calcium Homeostasis and Mitochondrial Dynamics in Parkinson’s Disease. Antioxid. Redox Signal. 2019, 31, 1213–1234. [Google Scholar] [CrossRef]
- Priyanka; Seth, P. Insights Into the Role of Mortalin in Alzheimer’s Disease, Parkinson’s Disease, and HIV-1-Associated Neurocognitive Disorders. Front. Cell Dev. Biol. 2022, 10, 903031. [Google Scholar]
- Liu, Y.; Ma, X.; Fujioka, H.; Liu, J.; Chen, S.; Zhu, X. DJ-1 Regulates the Integrity and Function of ER-Mitochondria Association through Interaction with IP3R3-Grp75-VDAC1. Proc. Natl. Acad. Sci. USA 2019, 116, 25322–25328. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, T.; Isobe, T.; Okuyama, T.; Takahashi, N.; Araki, K.; Kuwano, R.; Takahashi, Y. Molecular Cloning of cDNA Coding for Brain-Specific 14-3-3 Protein, a Protein Kinase-Dependent Activator of Tyrosine and Tryptophan Hydroxylases. Proc. Natl. Acad. Sci. USA 1988, 85, 7084–7088. [Google Scholar] [CrossRef] [PubMed]
- Ostrerova, N.; Petrucelli, L.; Farrer, M.; Mehta, N.; Choi, P.; Hardy, J.; Wolozin, B. α-Synuclein Shares Physical and Functional Homology with 14-3-3 Proteins. J. Neurosci. 1999, 19, 5782–5791. [Google Scholar] [CrossRef] [PubMed]
- Dzamko, N.; Deak, M.; Hentati, F.; Reith, A.D.; Prescott, A.R.; Alessi, D.R.; Nichols, R.J. Inhibition of LRRK2 Kinase Activity Leads to Dephosphorylation of Ser910/Ser935, Disruption of 14-3-3 Binding and Altered Cytoplasmic Localization. Biochem. J. 2010, 430, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Underwood, R.; Lavalley, N.; Yacoubian, T.A. 14-3-3 Inhibition Promotes Dopaminergic Neuron Loss and 14-3-3θ Overexpression Promotes Recovery in the MPTP Mouse Model of Parkinson’s Disease. Neuroscience 2015, 307, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Pozuelo-Rubio, M. 14-3-3 Proteins Are Regulators of Autophagy. Cells 2012, 1, 754–773. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.J.; Lombardi, R.Q.; Pucilowska, J.; Jankovic, J.; Tan, E.K.; Rooney, J.P. A Variant in the HS1-BP3 Gene Is Associated with Familial Essential Tremor. Neurology 2005, 64, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, Y.; Akiguchi, I.; Nakamura, S.; Honjyo, Y.; Shibasaki, H.; Budka, H. 14-3-3 Proteins in Lewy Bodies in Parkinson Disease and Diffuse Lewy Body Disease Brains. J. Neuropathol. Exp. Neurol. 2002, 61, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.K.; Zhang, Y.; Lim, K.L.; Tanaka, Y.; Huang, H.; Gao, J.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkin Ubiquitinates the Alpha-Synuclein-Interacting Protein, Synphilin-1: Implications for Lewy-Body Formation in Parkinson Disease. Nat. Med. 2001, 7, 1144–1150. [Google Scholar] [CrossRef]
- Cao, X.; Wu, X.; Zhao, L.; Zheng, J.; Jin, X.; Hao, X.; Winderickx, J.; Liu, S.; Chen, L.; Liu, B. Maturation and Detoxification of Synphilin-1 Inclusion Bodies Regulated by Sphingolipids. eLife 2023, 12, 92180. [Google Scholar] [CrossRef]
- Liu, J.; Li, T.; Thomas, J.M.; Pei, Z.; Jiang, H.; Engelender, S.; Ross, C.A.; Smith, W.W. Synphilin-1 Attenuates Mutant LRRK2-Induced Neurodegeneration in Parkinson’s Disease Models. Hum. Mol. Genet. 2016, 25, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Liu, J.; Guo, G.; Ning, B.; Li, X.; Zhu, G.; Yang, D.; Moran, T.H.; Smith, W.W. Synphilin-1 Interacts with AMPK and Increases AMPK Phosphorylation. Int. J. Mol. Sci. 2020, 21, 4352. [Google Scholar] [CrossRef] [PubMed]
- Haloi, N.; Wen, P.-C.; Cheng, Q.; Yang, M.; Natarajan, G.; Camara, A.K.S.; Kwok, W.-M.; Tajkhorshid, E. Structural Basis of Complex Formation between Mitochondrial Anion Channel VDAC1 and Hexokinase-II. Commun. Biol. 2021, 4, 667. [Google Scholar] [CrossRef] [PubMed]
- Ciscato, F.; Filadi, R.; Masgras, I.; Pizzi, M.; Marin, O.; Damiano, N.; Pizzo, P.; Gori, A.; Frezzato, F.; Chiara, F.; et al. Hexokinase 2 Displacement from Mitochondria-associated Membranes Prompts Ca2+-dependent Death of Cancer Cells. EMBO Rep. 2020, 21, e49117. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.J.; Miyamoto, S. Hexokinase II Integrates Energy Metabolism and Cellular Protection: Akting on Mitochondria and TORCing to Autophagy. Cell Death Differ. 2015, 22, 248. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, M.; Queralt-Martín, M.; Gurnev, P.A.; Rosencrans, W.M.; Rovini, A.; Jacobs, D.; Abrantes, K.; Hoogerheide, D.P.; Bezrukov, S.M.; Rostovtseva, T.K. Restricting α-Synuclein Transport into Mitochondria by Inhibition of α-Synuclein–VDAC Complexation as a Potential Therapeutic Target for Parkinson’s Disease Treatment. Cell. Mol. Life Sci. 2022, 79, 368. [Google Scholar] [CrossRef]
- Salganik, M.; Sergeyev, V.G.; Shinde, V.; Meyers, C.A.; Gorbatyuk, M.S.; Lin, J.H.; Zolotukhin, S.; Gorbatyuk, O.S. The Loss of Glucose-Regulated Protein 78 (GRP78) during Normal Aging or from siRNA Knockdown Augments Human Alpha-Synuclein (α-Syn) Toxicity to Rat Nigral Neurons. Neurobiol. Aging 2015, 36, 2213–2223. [Google Scholar] [CrossRef] [PubMed]
- Enogieru, A.B.; Omoruyi, S.I.; Hiss, D.C.; Ekpo, O.E. GRP78/BIP/HSPA5 as a Therapeutic Target in Models of Parkinson’s Disease: A Mini Review. Adv. Pharmacol. Pharm. Sci. 2019, 2019, e2706783. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Paya, C.; Sanz-Hernandez, M.; De Simone, A. Plasticity of Membrane Binding by the Central Region of α-Synuclein. Front. Mol. Biosci. 2022, 9, 857217. [Google Scholar] [CrossRef]
- Betzer, C.; Movius, A.J.; Shi, M.; Gai, W.P.; Zhang, J.; Jensen, P.H. Identification of Synaptosomal Proteins Binding to Monomeric and Oligomeric α-Synuclein. PLoS ONE 2015, 10, e0116473. [Google Scholar] [CrossRef]
- Lassen, L.B.; Reimer, L.; Ferreira, N.; Betzer, C.; Jensen, P.H. Protein Partners of α-Synuclein in Health and Disease. Brain Pathol. 2016, 26, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, B.; Qin, Y.; Li, A.; Gao, M.; Liu, H.; Gong, G. Mitochondrial Fusion Protein Mfn2 and Its Role in Heart Failure. Front. Mol. Biosci. 2021, 8, 681237. [Google Scholar] [CrossRef] [PubMed]
- Utton, M.A.; Noble, W.J.; Hill, J.E.; Anderton, B.H.; Hanger, D.P. Molecular Motors Implicated in the Axonal Transport of Tau and α-Synuclein. J. Cell Sci. 2005, 118, 4645–4654. [Google Scholar] [CrossRef] [PubMed]
- Amadeo, A.; Pizzi, S.; Comincini, A.; Modena, D.; Calogero, A.M.; Madaschi, L.; Faustini, G.; Rolando, C.; Bellucci, A.; Pezzoli, G.; et al. The Association between α-Synuclein and α-Tubulin in Brain Synapses. Int. J. Mol. Sci. 2021, 22, 9153. [Google Scholar] [CrossRef]
- Cartelli, D.; Aliverti, A.; Barbiroli, A.; Santambrogio, C.; Ragg, E.M.; Casagrande, F.V.M.; Cantele, F.; Beltramone, S.; Marangon, J.; De Gregorio, C.; et al. α-Synuclein Is a Novel Microtubule Dynamase. Sci. Rep. 2016, 6, 33289. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Role(s) at the MAM | Location | Association with PD Pathology | References |
|---|---|---|---|
| Associates to Calnexin |
|
| [194,195] |
| Binds to GRP78/BiP |
|
| [196] |
| Binds to GRP94 |
|
| [65,100,194,197] |
| Binds to VAPB |
|
| [52,187] |
| Binds to TOM20 |
|
| [198] |
| Binds to VDAC |
|
| [55] |
| Associates with GRP75 (Mortalin) |
|
| [194,199] |
| Binds to MIRO1 |
|
| [80] |
| Binds to 14-3-3 |
|
| [200,201,202] |
| Binds to Synphilin-1 |
|
| [203] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbuti, P.A. A-Syn(ful) MAM: A Fresh Perspective on a Converging Domain in Parkinson’s Disease. Int. J. Mol. Sci. 2024, 25, 6525. https://doi.org/10.3390/ijms25126525
Barbuti PA. A-Syn(ful) MAM: A Fresh Perspective on a Converging Domain in Parkinson’s Disease. International Journal of Molecular Sciences. 2024; 25(12):6525. https://doi.org/10.3390/ijms25126525
Chicago/Turabian StyleBarbuti, Peter A. 2024. "A-Syn(ful) MAM: A Fresh Perspective on a Converging Domain in Parkinson’s Disease" International Journal of Molecular Sciences 25, no. 12: 6525. https://doi.org/10.3390/ijms25126525
APA StyleBarbuti, P. A. (2024). A-Syn(ful) MAM: A Fresh Perspective on a Converging Domain in Parkinson’s Disease. International Journal of Molecular Sciences, 25(12), 6525. https://doi.org/10.3390/ijms25126525