
Aquaporins: Gatekeepers of Fluid Dynamics in Traumatic Brain Injury
 , , ,
, , ,
Abstract
:1. Introduction
2. Aquaporins in the CNS
3. Role of AQPs in TBI
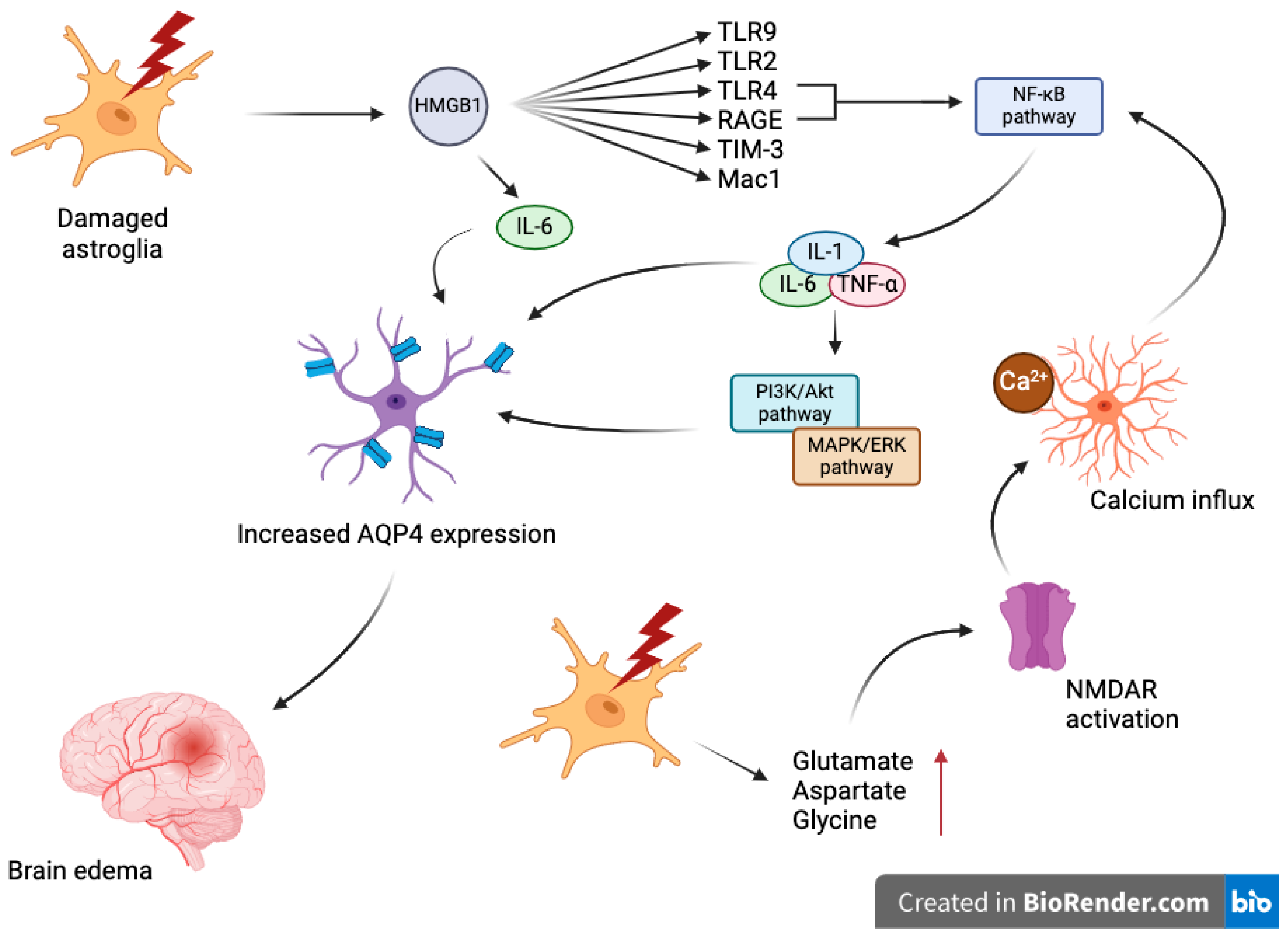
4. Mechanisms Underlying Aquaporin Dysregulation in TBI
5. Diagnostic and Prognostic Implications
6. Emerging Technologies and Future Directions
6.1. Vasopressin V1a Receptors
6.2. Antisense Oligonucleotides
6.3. Minocycline
6.4. Acetazolamide
6.5. miR-211-5p
6.6. Trifluoperazine
6.7. Adenine Dinucleotide Phosphate Oxidase 2
6.8. Hypertonic Saline
6.9. Monocyte Locomotion Inhibitor Factor
6.10. Lentivirus-Mediated AQP4 Gene Silencing
6.11. Omega-3 Polyunsaturated Fatty Acids
6.12. Angiotensin II Type 1 Receptor
6.13. Melatonin Receptors
6.14. Other Targets

{kind=link}
| Technology/Approach | Description | Reference |
|---|---|---|
| Gene deletion of AQPs | Reduces edema in preclinical studies but lacks direct AQP4 inhibitor | [141] |
| Bumetanide | Inhibits NKCC1, reduces astrocyte swelling after fluid percussion injury | [142] |
| AER-271 | Blocks acute brain edema, improves early outcomes in pediatric cardiac arrest models | [143] |
| Aquaporumab | Inhibits neuromyelitis optica progression in vivo, not tested in ischemic/traumatic models | [144] |
| Vasopressin V1a receptors | Regulates AQPs in the brain, potentially controlling cerebral edema through PKC activation and JNK pathways | [86] |
| Antisense oligonucleotides | Bind to mRINA, prevent AQP4 functional protein formation, reduce edema if administered early | [145] |
| Minocycline | Suppresses AQP4 expression post-TBI, protects BBB integrity, modulates astrocyte characteristics | [146] |
| Acetazolamide | Inhibits CA, prevents AQP4 redistribution post-TBI, mitigates cytotoxic edema | [147] |
| miR-211-5p | Regulates MMP9/AQP4 axis, therapeutic potential for TBI treatment by targeting this pathway | [148] |
| Trifluoperazine | Reduces AQP4 accumulation, mitigates brain edema, reduces neurological severity post-TBI | [149,150,151] |
| Adenine dinucleotide phosphate oxidase 2 | NOX2 inhibition reduces AQP4 levels, enhances cognitive function, reduces brain edema post-TBI | [152] |
| Hypertonic saline | Reduces AQP4, TNFa, IL-1B levels, and brain water contact, suppresses apoptosis post-TBI | [153] |
| Monocyte locomotion inhibitor factor | Reduces brain water content, suppresses AQP4 suppression, provides protection against TBI | [154,155] |
| Lentivirus-mediated AQP4 gene silencing | Reduces AQP4 expression, mitigates brain edema, reduces neurological deficits and neuronal apoptosis post-TBI | [156] |
| Omega-3 polyunsaturated fatty acids | Improve glymphatic clearance, reduce AB42 accumulation, protect BBB integrity post-TBI | [162] |
| Angiotensin II type 1 receptor | Deficiency preserves BBB integrity, reduces AQP4 polarization, improves glymphatic function, and AB clearance post-TBI | [158] |
| Melatonin receptors | Interaction with estrogen may reduce BBB permeability, influence AQP4 expression | [160] |
| Arginin and laminin | Regulate BBB integrity and AQP4 polarization, important for astrocyte migration and plasticity | [161] |
7. Conclusions
8. Final Thoughts
Funding
Conflicts of Interest
References
- Demlie, T.A.; Alemu, M.T.; Messelu, M.A.; Wagnew, F.; Mekonen, E.G. Incidence and predictors of mortality among traumatic brain injury patients admitted to Amhara region Comprehensive Specialized Hospitals, northwest Ethiopia, 2022. BMC Emerg. Med. 2023, 23, 55. [Google Scholar] [CrossRef]
- Haarbauer-Krupa, J.; Pugh, M.J.; Prager, E.M.; Harmon, N.; Wolfe, J.; Yaffe, K.C. Epidemiology of Chronic Effects of Traumatic Brain Injury. J. Neurotrauma 2021, 38, 3235–3247. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.L.; Wang, D.; Gottesman, R.F.; Selvin, E. Prevalence of Disability Associated with Head Injury with Loss of Consciousness in Adults in the United States: A Population-Based Study. Neurology 2021, 97, e124. [Google Scholar] [CrossRef] [PubMed]
- Keating, C.E.; Cullen, D.K. Mechanosensation in traumatic brain injury. Neurobiol. Dis. 2021, 148, 105210. [Google Scholar] [CrossRef] [PubMed]
- Andruszkow, H.; Deniz, E.; Urner, J.; Probst, C.; Grün, O.; Lohse, R.; Frink, M.; Krettek, C.; Zeckey, C.; Hildebrand, F. Physical and psychological long-term outcome after traumatic brain injury in children and adult patients. Health Qual. Life Outcomes 2014, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Howlett, J.R.; Nelson, L.D.; Stein, M.B. Mental health consequences of traumatic brain injury. Biol. Psychiatry 2022, 91, 413. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.S.; Arango-Lasprilla, J.C.; Andelic, N.; Nordenmark, T.H.; Soberg, H.L. Mental Health and Family Functioning in Patients and Their Family Members after Traumatic Brain Injury: A Cross-Sectional Study. Brain Sci. 2020, 10, 670. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Duan, H.; Hao, P.; Gao, Y.; Zhao, W.; Hao, F.; Li, X.; Yang, Z. Cellular regeneration treatments for traumatic brain injury. Med. Nov. Technol. Devices 2022, 16, 100182. [Google Scholar] [CrossRef]
- Inada, H.; Li, Q.; Bachani, A.; Hyder, A.A. Forecasting global road traffic injury mortality for 2030. Inj. Prev. 2020, 26, 339–343. [Google Scholar] [CrossRef]
- The Scope and Burden of Traumatic Brain Injury—Traumatic Brain Injury—NCBI Bookshelf. Available online: https://www.ncbi.nlm.nih.gov/books/NBK580076/ (accessed on 14 May 2024).
- Berwick, D.; Bowman, K.; Matney, C. Traumatic Brain Injury: A Roadmap for Accelerating Progress (2022); National Academies Press: Washington, DC, USA, 2022; pp. 1–228. [Google Scholar] [CrossRef]
- Datta, S.; Lin, F.; Jones, L.D.; Pingle, S.C.; Kesari, S.; Ashili, S. Traumatic brain injury and immunological outcomes: The double-edged killer. Future Sci. OA 2023, 9, FSO864. [Google Scholar] [CrossRef]
- Thapa, K.; Khan, H.; Singh, T.G.; Kaur, A. Traumatic Brain Injury: Mechanistic Insight on Pathophysiology and Potential Therapeutic Targets. J. Mol. Neurosci. 2021, 71, 1725–1742. [Google Scholar] [CrossRef] [PubMed]
- Jha, R.M.; Kochanek, P.M.; Simard, J.M. Pathophysiology and treatment of cerebral edema in traumatic brain injury. Neuropharmacology 2019, 145 Pt B, 230–246. [Google Scholar] [CrossRef]
- Busl, K.M. Management of Cerebral Edema, Brain Compression, and Intracranial Pressure. Continuum 2021, 27, 1172–1200. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.M.; Jones, G.M.; Hawryluk, G.W.J.; Mailloux, P.; McLaughlin, D.; Papangelou, A.; Samuel, S.; Tokumaru, S.; Venkatasubramanian, C.; Zacko, C.; et al. Guidelines for the Acute Treatment of Cerebral Edema in Neurocritical Care Patients. Neurocrit. Care 2020, 32, 647–666. [Google Scholar] [CrossRef] [PubMed]
- Zannetti, A.; Benga, G.; Brunetti, A.; Napolitano, F.; Avallone, L.; Pelagalli, A. Role of Aquaporins in the Physiological Functions of Mesenchymal Stem Cells. Cells 2020, 9, 2678. [Google Scholar] [CrossRef] [PubMed]
- Mader, S.; Brimberg, L. Aquaporin-4 Water Channel in the Brain and Its Implication for Health and Disease. Cells 2019, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.M.; Kitchen, P.; Halsey, A.; Wang, M.X.; Törnroth-Horsefield, S.; Conner, A.C.; Badaut, J.; Iliff, J.J.; Bill, R.M. Emerging roles for dynamic aquaporin-4 subcellular relocalization in CNS water homeostasis. Brain 2022, 145, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Shao, L.; Ma, L. Cerebral Edema Formation after Stroke: Emphasis on Blood–Brain Barrier and the Lymphatic Drainage System of the Brain. Front. Cell Neurosci. 2021, 15, 716825. [Google Scholar] [CrossRef] [PubMed]
- Dadgostar, E.; Rahimi, S.; Nikmanzar, S.; Nazemi, S.; Taheri, M.N.; Alibolandi, Z.; Aschner, M.; Mirzaei, H.; Tamtaji, O.R. Aquaporin 4 in Traumatic Brain Injury: From Molecular Pathways to Therapeutic Target. Neurochem. Res. 2022, 47, 860–871. [Google Scholar] [CrossRef]
- Zador, Z.; Bloch, O.; Yao, X.; Manley, G.T. Aquaporins: Role in cerebral edema and brain water balance. Prog. Brain Res. 2007, 161, 185–194. [Google Scholar] [CrossRef]
- Nakada, T.; Kwee, I.L. Fluid Dynamics Inside the Brain Barrier: Current Concept of Interstitial Flow, Glymphatic Flow, and Cerebrospinal Fluid Circulation in the Brain. Neuroscientist 2019, 25, 155. [Google Scholar] [CrossRef] [PubMed]
- Paul, L.; Madan, M.; Rammling, M.; Chigurupati, S.; Chan, S.L.; Pattisapu, J.V. Expression of aquaporin 1 and 4 in a congenital hydrocephalus rat model. Neurosurgery 2011, 68, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Chen, X.; Lei, Q.; Lu, W. AQP2 Promotes Astrocyte Activation by Modulating the TLR4/NFκB-p65 Pathway Following Intracerebral Hemorrhage. Front. Immunol. 2022, 13, 847360. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Gao, F.; Liu, H.; Yu, W.H.; Sun, S.Q. Temporal changes in expression of aquaporin-3, -4, -5 and -8 in rat brains after permanent focal cerebral ischemia. Brain Res. 2009, 1290, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Desai, B.; Hsu, Y.; Schneller, B.; Hobbs, J.G.; Mehta, A.I.; Linninger, A. Hydrocephalus: The role of cerebral aquaporin-4 channels and computational modeling considerations of cerebrospinal fluid. Neurosurg. Focus 2016, 41, E8. [Google Scholar] [CrossRef] [PubMed]
- Lambertz, N.; El Hindy, N.; Adler, C.; Rump, K.; Adamzik, M.; Keyvani, K.; Bankfalvi, A.; Siffert, W.; Sandalcioglu, I.E.; Bachmann, H.S. Expression of aquaporin 5 and the AQP5 polymorphism A(-1364)C in association with peritumoral brain edema in meningioma patients. J. Neurooncol. 2013, 112, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, R.; Schiera, G.; di Liegro, C.M.; Fricano, A.; Iacopino, D.G.; Di Liegro, I. Aquaporins and Brain Tumors. Int. J. Mol. Sci. 2016, 17, 1029. [Google Scholar] [CrossRef] [PubMed]
- Shin, I.; Kim, H.J.; Lee, J.E.; Gye, M.C. Aquaporin7 expression during perinatal development of mouse brain. Neurosci. Lett. 2006, 409, 106–111. [Google Scholar] [CrossRef]
- Zhu, S.J.; Wang, K.J.; Gan, S.W.; Xu, J.; Xu, S.Y.; Sun, S.Q. Expression of aquaporin8 in human astrocytomas: Correlation with pathologic grade. Biochem. Biophys. Res. Commun. 2013, 440, 168–172. [Google Scholar] [CrossRef]
- Badaut, J.; Regli, L. Distribution and possible roles of aquaporin 9 in the brain. Neuroscience 2004, 129, 969–979. [Google Scholar] [CrossRef]
- Koike, S.; Tanaka, Y.; Morishita, Y.; Ishibashi, K. Effects of osmolality on the expression of brain aquaporins in AQP11-null mice. Biochimie 2021, 188, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Koike, S.; Tanaka, Y.; Matsuzaki, T.; Morishita, Y.; Ishibashi, K. Aquaporin-11 (AQP11) Expression in the Mouse Brain. Int. J. Mol. Sci. 2016, 17, 861. [Google Scholar] [CrossRef] [PubMed]
- Amro, Z.; Ryan, M.; Collins-Praino, L.E.; Yool, A.J. Unexpected Classes of Aquaporin Channels Detected by Transcriptomic Analysis in Human Brain Are Associated with Both Patient Age and Alzheimer’s Disease Status. Biomedicines 2023, 11, 770. [Google Scholar] [CrossRef]
- Xu, M.; Xiao, M.; Li, S.; Yang, B. Aquaporins in Nervous System. Adv. Exp. Med. Biol. 2017, 969, 81–103. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.; Arnett, S.; Bukhari, W.; Khalilidehkordi, E.; Sanchez, S.J.; O’Gorman, C.; Sun, J.; Prain, K.M.; Woodhall, M.; Silvestrini, R.; et al. MRI Patterns Distinguish AQP4 Antibody Positive Neuromyelitis Optica Spectrum Disorder from Multiple Sclerosis. Front. Neurol. 2021, 12, 722237. [Google Scholar] [CrossRef] [PubMed]
- Szu, J.I.; Binder, D.K. Mechanisms Underlying Aquaporin-4 Subcellular Mislocalization in Epilepsy. Front. Cell Neurosci. 2022, 16, 900588. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin water channels in the nervous system. Nat. Rev. Neurosci. 2013, 14, 265–277. [Google Scholar] [CrossRef] [PubMed]
- MacAulay, N. Molecular mechanisms of brain water transport. Nat. Rev. Neurosci. 2021, 22, 326–344. [Google Scholar] [CrossRef]
- Goren, O.; Adorján, I.; Kálmán, M. Heterogeneous occurrence of aquaporin-4 in the ependyma and in the circumventricular organs in rat and chicken. Anat. Embryol. 2006, 211, 155–172. [Google Scholar] [CrossRef] [PubMed]
- Circumventricular Organs Dysregulation Syndrome (CODS) (1). Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9583022/ (accessed on 14 May 2024).
- Kiecker, C. The origins of the circumventricular organs. J. Anat. 2018, 232, 540–553. [Google Scholar] [CrossRef]
- Trillo-Contreras, J.L.; Toledo-Aral, J.J.; Echevarría, M.; Villadiego, J. AQP1 and AQP4 Contribution to Cerebrospinal Fluid Homeostasis. Cells 2019, 8, 197. [Google Scholar] [CrossRef] [PubMed]
- Shields, S.D.; Moore, K.D.; Phelps, P.E.; Basbaum, A.I. Olfactory ensheathing glia express aquaporin 1. J. Comp. Neurol. 2010, 518, 4329. [Google Scholar] [CrossRef] [PubMed]
- Potokar, M.; Jorgačevski, J.; Zorec, R. Astrocyte Aquaporin Dynamics in Health and Disease. Int. J. Mol. Sci. 2016, 17, 1121. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Aalling, N.N.; Förstera, B.; Ertürk, A.; Nedergaard, M.; Møllgård, K.; Xavier, A.L.R. Aquaporin 1 and the Na+/K+/2Cl− cotransporter 1 are present in the leptomeningeal vasculature of the adult rodent central nervous system. Fluids Barriers CNS 2020, 17, 15. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.Z.; Yao, J.; Meng, B.; Qin, Y.B.; Cao, S. Blood-nerve barrier enhances chronic postsurgical pain via the HIF-1α/ aquaporin-1 signaling axis. BMC Anesthesiol. 2023, 23, 381. [Google Scholar] [CrossRef]
- Volkart, S.; Kym, U.; Braissant, O.; Delgado-Eckert, E.; Al-Samir, S.; Angresius, R.; Huo, Z.; Holland-Cunz, S.; Gros, S.J. AQP1 in the Gastrointestinal Tract of Mice: Expression Pattern and Impact of AQP1 Knockout on Colonic Function. Int. J. Mol. Sci. 2023, 24, 3616. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Tan, M.; Gu, M.; Marshall, C.; Ding, J.; Hu, G.; Xiao, M. Cellular Localization of Aquaporin-1 in the Human and Mouse Trigeminal Systems. PLoS ONE 2012, 7, 46379. [Google Scholar] [CrossRef] [PubMed]
- Orešković, D.; Klarica, M. A new look at cerebrospinal fluid movement. Fluids Barriers CNS 2014, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, H.; Tsujita, M.; Kwee, I.L.; Nakada, T. Water influx into cerebrospinal fluid is primarily controlled by aquaporin-4, not by aquaporin-1: 17O JJVCPE MRI study in knockout mice. Neuroreport 2014, 25, 39–43. [Google Scholar] [CrossRef]
- Amiry-Moghaddam, M.; Lindland, H.; Zelenin, S.; Roberg, B.; Gundersen, B.B.; Petersen, P.; Rinvik, E.; Torgner, I.A.; Ottersen, O.P. Brain mitochondria contain aquaporin water channels: Evidence for the expression of a short AQP9 isoform in the inner mitochondrial membrane. FASEB J. 2005, 19, 1459–1467. [Google Scholar] [CrossRef]
- Badaut, J.; Lasbennes, F.; Magistretti, P.J.; Regli, L. Aquaporins in brain: Distribution, physiology, and pathophysiology. J. Cereb. Blood Flow Metab. 2002, 22, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Opdal, S.H.; Ferrante, L.; Rognum, T.O.; Stray-Pedersen, A. Aquaporin-1 and aquaporin-9 gene variations in sudden infant death syndrome. Int. J. Legal Med. 2021, 135, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Stahl, K.; Rahmani, S.; Prydz, A.; Skauli, N.; Macaulay, N.; Mylonakou, M.N.; Torp, R.; Skare, Ø.; Berg, T.; Leergaard, T.B.; et al. Targeted deletion of the aquaglyceroporin AQP9 is protective in a mouse model of Parkinson’s disease. PLoS ONE 2018, 13, e0194896. [Google Scholar] [CrossRef] [PubMed]
- Mobasheri, A.; Wray, S.; Marples, D. Distribution of AQP2 and AQP3 water channels in human tissue microarrays. J. Mol. Histol. 2005, 36, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.H.; Gao, H.W.; Fang, X.D.; Yang, H. Expression and function of aquaporins in peripheral nervous system. Acta Pharmacol. Sin. 2011, 32, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Bielewicz, J.; Kamieniak, M.; Szymoniuk, M.; Litak, J.; Czyżewski, W.; Kamieniak, P. Diagnosis and Management of Neuropathic Pain in Spine Diseases. J. Clin. Med. 2023, 12, 1380. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Yoneda, K.; Asai, K.; Sobue, K.; Tada, T.; Fujita, Y.; Katsuya, H.; Fujita, M.; Aihara, N.; Mase, M.; et al. Alterations in the expression of the AQP family in cultured rat astrocytes during hypoxia and reoxygenation. Mol. Brain Res. 2001, 90, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Marlar, S.; Jensen, H.H.; Login, F.H.; Nejsum, L.N. Aquaporin-3 in Cancer. Int. J. Mol. Sci. 2017, 18, 2106. [Google Scholar] [CrossRef] [PubMed]
- Maeda, N.; Hibuse, T.; Funahashi, T. Role of Aquaporin-7 and Aquaporin-9 in Glycerol Metabolism; Involvement in Obesity. Handb. Exp. Pharmacol. 2009, 190, 233–249. [Google Scholar] [CrossRef]
- Maeda, N. Implications of aquaglyceroporins 7 and 9 in glycerol metabolism and metabolic syndrome. Mol. Aspects Med. 2012, 33, 665–675. [Google Scholar] [CrossRef]
- Hao, Z.; Huajun, S.; Zhen, G.; Yu, X.; Qian, L.; Ziling, C.; Zihao, S.; Qingqian, X.; Shujuan, Z. AQP8 promotes glioma proliferation and growth, possibly through the ROS/PTEN/AKT signaling pathway. BMC Cancer 2023, 23, 516. [Google Scholar] [CrossRef] [PubMed]
- Gorelick, D.A.; Praetorius, J.; Tsunenari, T.; Nielsen, S.; Agre, P. Aquaporin-11: A channel protein lacking apparent transport function expressed in brain. BMC Biochem. 2006, 7, 14. [Google Scholar] [CrossRef]
- Szczygielski, J.; Kopańska, M.; Wysocka, A.; Oertel, J. Cerebral Microcirculation, Perivascular Unit, and Glymphatic System: Role of Aquaporin-4 as the Gatekeeper for Water Homeostasis. Front. Neurol. 2021, 12, 767470. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zhang, Y.; Liao, X.; Yang, R.; Lei, Y.; Luo, J. Potential Therapies for Cerebral Edema after Ischemic Stroke: A Mini Review. Front. Aging Neurosci. 2021, 12, 618819. [Google Scholar] [CrossRef] [PubMed]
- Keep, R.F.; Andjelkovic, A.V.; Xi, G. Cytotoxic and Vasogenic Brain Edema. In Primer on Cerebrovascular Diseases, 2nd ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 145–149. [Google Scholar] [CrossRef]
- Gu, Y.; Zhou, C.; Piao, Z.; Yuan, H.; Jiang, H.; Wei, H.; Zhou, Y.; Nan, G.; Ji, X. Cerebral edema after ischemic stroke: Pathophysiology and underlying mechanisms. Front. Neurosci. 2022, 16, 988283. [Google Scholar] [CrossRef] [PubMed]
- Management of Vasogenic Edema in Patients with Primary and Metastatic Brain Tumors—UpToDate. Available online: https://www.uptodate.com/contents/management-of-vasogenic-edema-in-patients-with-primary-and-metastatic-brain-tumors/print (accessed on 9 June 2024).
- Xiong, A.; Xiong, R.; Yu, J.; Liu, Y.; Liu, K.; Jin, G.; Xu, J.; Yan, J. Aquaporin-4 is a potential drug target for traumatic brain injury via aggravating the severity of brain edema. Burns Trauma 2021, 9, tkaa050. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Yang, G.Y. Aquaporin-4: A Potential Therapeutic Target for Cerebral Edema. Int. J. Mol. Sci. 2016, 17, 1413. [Google Scholar] [CrossRef] [PubMed]
- Solar, P.; Hendrych, M.; Barak, M.; Valekova, H.; Hermanova, M.; Jancalek, R. Blood-Brain Barrier Alterations and Edema Formation in Different Brain Mass Lesions. Front. Cell Neurosci. 2022, 16, 922181. [Google Scholar] [CrossRef] [PubMed]
- Patabendige, A.; Chen, R. Astrocytic aquaporin 4 subcellular translocation as a therapeutic target for cytotoxic edema in ischemic stroke. Neural Regen. Res. 2022, 17, 2666. [Google Scholar] [CrossRef]
- Sweeney, A.M.; Plá, V.; Du, T.; Liu, G.; Sun, Q.; Peng, S.; Plog, B.A.; Kress, B.T.; Wang, X.; Mestre, H.; et al. In Vivo Imaging of Cerebrospinal Fluid Transport through the Intact Mouse Skull using Fluorescence Macroscopy. J. Vis. Exp. 2019, 2019, e59774. [Google Scholar] [CrossRef]
- Braun, M.; Sevao, M.; Keil, S.A.; Gino, E.; Wang, M.X.; Lee, J.; Haveliwala, M.A.; Klein, E.; Agarwal, S.; Pedersen, T.; et al. Macroscopic changes in aquaporin-4 underlie blast traumatic brain injury-related impairment in glymphatic function. Brain 2024, 147, 2214–2229. [Google Scholar] [CrossRef] [PubMed]
- Cartagena, C.M.; Phillips, K.L.; Tortella, F.C.; Dave, J.R.; Schmid, K.E. Temporal alterations in aquaporin and transcription factor HIF1α expression following penetrating ballistic-like brain injury (PBBI). Mol. Cell Neurosci. 2014, 60, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Saadoun, S.; Verkman, A.S. Aquaporins in the Central Nervous System. In Handbook of Neurochemistry and Molecular Neurobiology: Neural Membranes and Transport; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2007; pp. 171–190. [Google Scholar] [CrossRef]
- Rao, K.V.R.; Verkman, A.S.; Curtis, K.M.; Norenberg, M.D. Aquaporin-4 deletion in mice reduces encephalopathy and brain edema in experimental acute liver failure. Neurobiol. Dis. 2014, 63, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.C.; Honey, C.R.; Berk, C.; Wong, N.L.M.; Tsui, J.K.C. Regulation of aquaporin-4 in a traumatic brain injury model in rats. J. Neurosurg. 2003, 98, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, S.-N.; Zhou, X.-Y.; Zhang, L.-X.; Chen, G.-X.; Wang, T.-H.; Xia, Q.-J.; Liang, N.; Zhang, X. The Dual Role of AQP4 in Cytotoxic and Vasogenic Edema Following Spinal Cord Contusion and Its Possible Association with Energy Metabolism via COX5A. Front. Neurosci. 2019, 13, 584. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Zhang, Y.; Wei, J.; Wu, R.; Cai, H. Overexpression of long noncoding RNA Malat1 ameliorates traumatic brain injury induced brain edema by inhibiting AQP4 and the NF-κB/IL-6 pathway. J. Cell Biochem. 2019, 120, 17584–17592. [Google Scholar] [CrossRef]
- Manley, G.T.; Binder, D.K.; Papadopoulos, M.C.; Verkman, A.S. New insights into water transport and edema in the central nervous system from phenotype analysis of aquaporin-4 null mice. Neuroscience 2004, 129, 981–989. [Google Scholar] [CrossRef]
- Jeon, H.; Kim, M.; Park, W.; Lim, J.S.; Lee, E.; Cha, H.; Ahn, J.S.; Kim, J.H.; Hong, S.H.; Park, J.E.; et al. Upregulation of AQP4 Improves Blood-Brain Barrier Integrity and Perihematomal Edema Following Intracerebral Hemorrhage. Neurotherapeutics 2021, 18, 2692–2706. [Google Scholar] [CrossRef]
- Farr, G.W.; Hall, C.H.; Farr, S.M.; Wade, R.; Detzel, J.M.; Adams, A.G.; Buch, J.M.; Beahm, D.L.; Flask, C.A.; Xu, K.; et al. Functionalized Phenylbenzamides Inhibit Aquaporin-4 Reducing Cerebral Edema and Improving Outcome in Two Models of CNS Injury. Neuroscience 2019, 404, 484. [Google Scholar] [CrossRef]
- Rauen, K.; Pop, V.; Trabold, R.; Badaut, J.; Plesnila, N. Vasopressin V1a Receptors Regulate Cerebral Aquaporin 1 after Traumatic Brain Injury. J. Neurotrauma 2020, 37, 665–674. [Google Scholar] [CrossRef]
- Tran, N.D.; Kim, S.; Vincent, H.K.; Rodriguez, A.; Hinton, D.R.; Bullock, M.R.; Young, H.F. Aquaporin-1–mediated cerebral edema following traumatic brain injury: Effects of acidosis and corticosteroid administration: Laboratory investigation. J. Neurosurg. 2010, 112, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Qiu, B.; Li, X.; Sun, X.; Wang, Y.; Jing, Z.; Zhang, X.; Wang, Y. Overexpression of aquaporin-1 aggravates hippocampal damage in mouse traumatic brain injury models. Mol. Med. Rep. 2014, 9, 916–922. [Google Scholar] [CrossRef]
- Hollborn, M.; Rehak, M.; Iandiev, I.; Pannicke, T.; Ulbricht, E.; Reichenbach, A.; Wiedemann, P.; Bringmann, A.; Kohen, L. Transcriptional regulation of aquaporins in the ischemic rat retina: Upregulation of aquaporin-9. Curr. Eye Res. 2012, 37, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Hirt, L.; Price, M.; Mastour, N.; Brunet, J.; Barrière, G.; Friscourt, F.; Badaut, J. Increase of aquaporin 9 expression in astrocytes participates in astrogliosis. J. Neurosci. Res. 2018, 96, 194–206. [Google Scholar] [CrossRef]
- D’Agostino, C.; Parisis, D.; Chivasso, C.; Hajiabbas, M.; Soyfoo, M.S.; Delporte, C. Aquaporin-5 Dynamic Regulation. Int. J. Mol. Sci. 2023, 24, 1889. [Google Scholar] [CrossRef]
- Xi, T.; Jin, F.; Zhu, Y.; Wang, J.; Tang, L.; Wang, Y.; Liebeskind, D.S.; Scalzo, F.; He, Z. miR-27a-3p protects against blood-brain barrier disruption and brain injury after intracerebral hemorrhage by targeting endothelial aquaporin-11. J. Biol. Chem. 2018, 293, 20041–20050. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Shaikh, M.F.; Chakraborti, A.; Kumari, Y.; Aledo-Serrano, Á.; Aleksovska, K.; Alvim, M.K.M.; Othman, I. HMGB1: A Common Biomarker and Potential Target for TBI, Neuroinflammation, Epilepsy, and Cognitive Dysfunction. Front. Neurosci. 2018, 12, 628. [Google Scholar] [CrossRef] [PubMed]
- Ravizza, T.; Terrone, G.; Salamone, A.; Frigerio, F.; Balosso, S.; Antoine, D.J.; Vezzani, A. High Mobility Group Box 1 is a novel pathogenic factor and a mechanistic biomarker for epilepsy. Brain Behav. Immun. 2018, 72, 14–21. [Google Scholar] [CrossRef]
- Edwards, K.A.; Pattinson, C.L.; Guedes, V.A.; Peyer, J.; Moore, C.; Davis, T.; Devoto, C.; Turtzo, L.C.; Latour, L.; Gill, J.M. Inflammatory Cytokines Associate with Neuroimaging after Acute Mild Traumatic Brain Injury. Front. Neurol. 2020, 11, 348. [Google Scholar] [CrossRef]
- da Silva, I.V.; Soveral, G. Aquaporins in Immune Cells and Inflammation: New Targets for Drug Development. Int. J. Mol. Sci. 2021, 22, 1845. [Google Scholar] [CrossRef]
- Richard, S.A.; Min, W.; Su, Z.; Xu, H. High Mobility Group Box 1 and Traumatic Brain Injury. J. Behav. Brain Sci. 2017, 7, 50–61. [Google Scholar] [CrossRef]
- Tang, Z.; Yang, G.; Liao, Z.; Chen, F.; Chen, S.; Wang, W.; Huo, G.; Sun, X.; Wang, X. Tanshinone IIA reduces AQP4 expression and astrocyte swelling after OGD/R by inhibiting the HMGB1/RAGE/NF-κB/IL-6 pro-inflammatory axis. Sci. Rep. 2022, 12, 14110. [Google Scholar] [CrossRef]
- Dixon, C.E.; Kline, A.E. Neurotransmitters and Electrophysiology in Traumatic Brain Injury. In Handbook of Neurochemistry and Molecular Neurobiology; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2009; pp. 179–202. [Google Scholar] [CrossRef]
- Skowrońska, K.; Obara-Michlewska, M.; Zielińska, M.; Albrecht, J. NMDA Receptors in Astrocytes: In Search for Roles in Neurotransmission and Astrocytic Homeostasis. Int. J. Mol. Sci. 2019, 20, 309. [Google Scholar] [CrossRef] [PubMed]
- Toro-Fernández, L.F.; Zuluaga-Monares, J.C.; Saldarriaga-Cartagena, A.M.; Cardona-Gómez, G.P.; Posada-Duque, R. Targeting CDK5 in Astrocytes Promotes Calcium Homeostasis Under Excitotoxic Conditions. Front. Cell Neurosci. 2021, 15, 643717. [Google Scholar] [CrossRef]
- Anilkumar, S.; Wright-Jin, E. NF-κB as an Inducible Regulator of Inflammation in the Central Nervous System. Cells 2024, 13, 485. [Google Scholar] [CrossRef]
- Birck, C.; Ginolhac, A.; Pavlou, M.A.S.; Michelucci, A.; Heuschling, P.; Grandbarbe, L. Nf-κb and tnf affect the astrocytic differentiation from neural stem cells. Cells 2021, 10, 840. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Ai, L.; Zhang, B. TNF-α induces AQP4 overexpression in astrocytes through the NF-κB pathway causing cellular edema and apoptosis. Biosci. Rep. 2022, 42, BSR20212224. [Google Scholar] [CrossRef]
- Li, N.; Ying, Y.; Yang, B. Aquaporins in Edema. Adv. Exp. Med. Biol. 2023, 1398, 281–287. [Google Scholar] [CrossRef]
- Nito, C.; Kamada, H.; Endo, H.; Narasimhan, P.; Lee, Y.S.; Chan, P.H. Involvement of Mitogen-Activated Protein Kinase Pathways in Expression of the Water Channel Protein Aquaporin-4 after Ischemia in Rat Cortical Astrocytes. J. Neurotrauma 2012, 29, 2404. [Google Scholar] [CrossRef]
- Hsu, Y.; Tran, M.; Linninger, A.A. Dynamic regulation of aquaporin-4 water channels in neurological disorders. Croat Med. J. 2015, 56, 401. [Google Scholar] [CrossRef]
- Li, B.; Liu, C.; Tang, K.; Dong, X.; Xue, L.; Su, G.; Zhang, W.; Jin, Y. Aquaporin-1 attenuates macrophage-mediated inflammatory responses by inhibiting p38 mitogen-activated protein kinase activation in lipopolysaccharide-induced acute kidney injury. Inflamm. Res. 2019, 68, 1035. [Google Scholar] [CrossRef] [PubMed]
- Szymoniuk, M.; Litak, J.; Sakwa, L.; Dryla, A.; Zezuliński, W.; Czyżewski, W.; Kamieniak, P.; Blicharski, T. Molecular Mechanisms and Clinical Application of Multipotent Stem Cells for Spinal Cord Injury. Cells 2022, 12, 120. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Sen, N. Traumatic brain injury: A risk factor for neurodegenerative diseases. Rev. Neurosci. 2016, 27, 93–100. [Google Scholar] [CrossRef]
- Audesse, A.J.; Karashchuk, G.; Gardell, Z.A.; Lakis, N.S.; Maybury-Lewis, S.Y.; Brown, A.K.; Leeman, D.S.; Teo, Y.V.; Neretti, N.; Anthony, D.C.; et al. FOXO3 regulates a common genomic program in aging and glioblastoma stem cells. Aging Cancer 2021, 2, 137. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Zhao, T.; Ni, H.; Li, Y.; Zhu, Y.; Gao, R.; Zhang, L.; Jia, Z.; Chen, G. USP11 exacerbates neuronal apoptosis after traumatic brain injury via PKM2-mediated PI3K/AKT signaling pathway. Brain Res. 2023, 1807, 148321. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, S.; Kim, S.M.; Farook, J.M.; Mir, S.; Saha, R.; Sen, N. Foxo3a Transcriptionally Upregulates AQP4 and Induces Cerebral Edema Following Traumatic Brain Injury. J. Neurosci. 2013, 33, 17398. [Google Scholar] [CrossRef] [PubMed]
- Fazzina, G.; Amorini, A.M.; Marmarou, C.R.; Fukui, S.; Okuno, K.; Dunbar, J.G.; Glisson, R.; Marmarou, A.; Kleindienst, A. The Protein Kinase C Activator Phorbol Myristate Acetate Decreases Brain Edema by Aquaporin 4 Downregulation after Middle Cerebral Artery Occlusion in the Rat. J. Neurotrauma 2010, 27, 453. [Google Scholar] [CrossRef] [PubMed]
- Amiry-Moghaddam, M.; Otsuka, T.; Hurn, P.D.; Traystman, R.J.; Haug, F.-M.; Froehner, S.C.; Adams, M.E.; Neely, J.D.; Agre, P.; Ottersen, O.P.; et al. An alpha-syntrophin-dependent pool of AQP4 in astroglial end-feet confers bidirectional water flow between blood and brain. Proc. Natl. Acad. Sci. USA 2003, 100, 2106–2111. [Google Scholar] [CrossRef] [PubMed]
- Marmarou, C.R.; Liang, X.; Abidi, N.H.; Parveen, S.; Taya, K.; Henderson, S.C.; Young, H.F.; Filippidis, A.S.; Baumgarten, C.M. Selective Vasopressin-1a receptor antagonist prevents brain edema, reduces astrocytic cell swelling and GFAP, V1aR and AQP4 expression after focal traumatic brain injury. Brain Res. 2014, 1581, 89. [Google Scholar] [CrossRef]
- Ikeshima-Kataoka, H. Neuroimmunological Implications of AQP4 in Astrocytes. Int. J. Mol. Sci. 2016, 17, 1306. [Google Scholar] [CrossRef]
- Czyżewski, W.; Mazurek, M.; Sakwa, L.; Szymoniuk, M.; Pham, J.; Pasierb, B.; Litak, J.; Czyżewska, E.; Turek, M.; Piotrowski, B.; et al. Astroglial Cells: Emerging Therapeutic Targets in the Management of Traumatic Brain Injury. Cells 2024, 13, 148. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.; Tham, D.K.L.; Perronnet, C.; Joshi, B.; Nabi, I.R.; Moukhles, H. The oxidative stress-induced increase in the membrane expression of the water-permeable channel aquaporin-4 in astrocytes is regulated by caveolin-1 phosphorylation. Front. Cell Neurosci. 2017, 11, 298524. [Google Scholar] [CrossRef]
- Mogoanta, L.; Ciurea, M.; Pirici, I.; Margaritescu, C.; Simionescu, C.; Ion, D.A.; Pirici, D. Different dynamics of aquaporin 4 and glutamate transporter-1 distribution in the perineuronal and perivascular compartments during ischemic stroke. Brain Pathol. 2014, 24, 475–493. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Wang, W.; Lutton, A.D.; Kiyoshi, C.M.; Ma, B.; Taylor, A.T.; Olesik, J.W.; McTigue, D.M.; Askwith, C.C.; Zhou, M. Dissipation of transmembrane potassium gradient is the main cause of cerebral ischemia-induced depolarization in astrocytes and neurons. Exp. Neurol. 2018, 303, 1. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Guan, S.; Wang, Z.; Ni, H.; Ding, D.; Xu, W.; Li, G. HIF-1α aggravated traumatic brain injury by NLRP3 inflammasome-mediated pyroptosis and activation of microglia. J. Chem. Neuroanat. 2021, 116, 101994. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.Y.; Kreipke, C.W.; Speirs, S.L.; Schafer, P.; Schafer, S.; Rafols, J.A. Hypoxia-inducible factor-1alpha signaling in aquaporin upregulation after traumatic brain injury. Neurosci. Lett. 2009, 453, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Xu, Z.; Lu, L.; Nie, H.; Ding, J.; Ying, W.; Tian, H. SIRT2 inhibition exacerbates neuroinflammation and blood-brain barrier disruption in experimental traumatic brain injury by enhancing NF-κB p65 acetylation and activation. J. Neurochem. 2016, 136, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Nesverova, V.; Törnroth-Horsefield, S. Phosphorylation-Dependent Regulation of Mammalian Aquaporins. Cells 2019, 8, 82. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, F.; Ge, Y.; Chen, S.; Xin, Y.; Umali, M.U.; De Gasperi, R.; Sosa, M.A.G.; Ahlers, S.T.; Elder, G.A. Neuronal DNA Methylation Profiling of Blast-Related Traumatic Brain Injury. J. Neurotrauma 2015, 32, 1200. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Ashiqueali, R.; Lin, C.I.; Walchale, A.; Clendaniel, V.; Matheson, R.; Fisher, M.; Lo, E.H.; Selim, M.; Shehadah, A. Histone Deacetylase 3 Inhibition Decreases Cerebral Edema and Protects the Blood–Brain Barrier After Stroke. Mol. Neurobiol. 2023, 60, 235. [Google Scholar] [CrossRef]
- Trillo-Contreras, J.L.; Ramírez-Lorca, R.; Villadiego, J.; Echevarría, M. Cellular Distribution of Brain Aquaporins and Their Contribution to Cerebrospinal Fluid Homeostasis and Hydrocephalus. Biomolecules 2022, 12, 530. [Google Scholar] [CrossRef]
- Neri, M.; Frati, A.; Turillazzi, E.; Cantatore, S.; Cipolloni, L.; Di Paolo, M.; Frati, P.; La Russa, R.; Maiese, A.; Scopetti, M.; et al. Immunohistochemical Evaluation of Aquaporin-4 and its Correlation with CD68, IBA-1, HIF-1α, GFAP, and CD15 Expressions in Fatal Traumatic Brain Injury. Int. J. Mol. Sci. 2018, 19, 3544. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, M.L.; Schiera, G.; Di Liegro, I.; Di Liegro, C.M.; Pál, J.; Czeiter, E.; Sulyok, E.; Dóczi, T. Aquaporin-4 distribution in control and stressed astrocytes in culture and in the cerebrospinal fluid of patients with traumatic brain injuries. Neurol. Sci. 2013, 34, 1309–1314. [Google Scholar] [CrossRef] [PubMed]
- Shenaq, M.; Kassem, H.; Peng, C.; Schafer, S.; Ding, J.Y.; Fredrickson, V.; Guthikonda, M.; Kreipke, C.W.; Rafols, J.A.; Ding, Y. Neuronal damage and functional deficits are ameliorated by inhibition of aquaporin and HIF1α after traumatic brain injury (TBI). J. Neurol. Sci. 2012, 323, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Tomura, S.; Nawashiro, H.; Otani, N.; Uozumi, Y.; Toyooka, T.; Ohsumi, A.; Shima, K. Effect of Decompressive Craniectomy on Aquaporin-4 Expression after Lateral Fluid Percussion Injury in Rats. J. Neurotrauma 2011, 28, 237–243. [Google Scholar] [CrossRef]
- Li, X.; Han, Y.; Xu, H.; Sun, Z.; Zhou, Z.; Long, X.; Yang, Y.; Zou, L. Aquaporin 4 expression and ultrastructure of the blood-brain barrier following cerebral contusion injury. Neural Regen. Res. 2013, 8, 338–345. [Google Scholar] [CrossRef]
- Liu, X.; Xie, Y.; Wan, X.; Wu, J.; Fan, Z.; Yang, L. Protective Effects of Aquaporin-4 Deficiency on Longer-term Neurological Outcomes in a Mouse Model. Neurochem. Res. 2021, 46, 1380–1389. [Google Scholar] [CrossRef]
- Oliva, A.; Kang, Y.; Truettner, J.; Sanchez-Molano, J.; Furones, C.; Yool, A.; Atkins, C. Fluid-percussion brain injury induces changes in aquaporin channel expression. Neuroscience 2011, 180, 272–279. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, J.; Lu, H. Expression of aquaporin-4 and pathological characteristics of brain injury in a rat model of traumatic brain injury. Mol. Med. Rep. 2015, 12, 7351. [Google Scholar] [CrossRef]
- Badaut, J.; Fukuda, A.M.; Jullienne, A.; Petry, K.G. Aquaporin and brain diseases. Biochim. Biophys. Acta 2014, 1840, 1554–1565. [Google Scholar] [CrossRef]
- Haj-Yasein, N.N.; Vindedal, G.F.; Eilert-Olsen, M.; Gundersen, G.A.; Skare, Ø.; Laake, P.; Klungland, A.; Thorén, A.E.; Burkhardt, J.M.; Ottersen, O.P.; et al. Glial-conditional deletion of aquaporin-4 (Aqp4) reduces blood-brain water uptake and confers barrier function on perivascular astrocyte endfeet. Proc. Natl. Acad. Sci. USA 2011, 108, 17815–17820. [Google Scholar] [CrossRef] [PubMed]
- Abir-Awan, M.; Kitchen, P.; Salman, M.M.; Conner, M.T.; Conner, A.C.; Bill, R.M. Inhibitors of Mammalian Aquaporin Water Channels. Int. J. Mol. Sci. 2019, 20, 1589. [Google Scholar] [CrossRef] [PubMed]
- Vella, J.; Zammit, C.; Di Giovanni, G.; Muscat, R.; Valentino, M. The central role of aquaporins in the pathophysiology of ischemic stroke. Front. Cell Neurosci. 2015, 9, 108. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Ying, X.; Qian, Y.; Liu, H.; Lan, Y.; Xie, A.; Zhu, X. Physiological and pathological impact of AQP1 knockout in mice. Biosci. Rep. 2019, 39, 20182303. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.V.R.; Reddy, P.V.B.; Curtis, K.M.; Norenberg, M.D. Aquaporin-4 expression in cultured astrocytes after fluid percussion injury. J. Neurotrauma 2011, 28, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Wallisch, J.S.; Janesko-Feldman, K.; Alexander, H.; Jha, R.M.; Farr, G.W.; McGuirk, P.R.; Kline, A.E.; Jackson, T.C.; Pelletier, M.F.; Clark, R.S.B.; et al. The aquaporin-4 inhibitor AER-271 blocks acute cerebral edema and improves early outcome in a pediatric model of asphyxial cardiac arrest. Pediatr. Res. 2019, 85, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.W.; Ovcjak, A.; Wong, R.; Yang, B.X.; Feng, Z.P.; Sun, H.S. Drug development in targeting ion channels for brain edema. Acta Pharmacol. Sin. 2020, 41, 1272–1288. [Google Scholar] [CrossRef] [PubMed]
- Hekimoglu, M.; Lule, S.; Ozer, H.; Cakir-Aktas, C.; Oguz, K.K.; Mut, M. Suppression of Aquaporin-4 By Antisense Oligonucleotides Reduces Brain Edema In Experimental Traumatic Brain Injury. Turk. Neurosurg. 2021, 32, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Xiong, J.; Yuan, Y.; Ruan, Z.; Zhang, Y.; Chai, B.; Li, L.; Cai, S.; Xiao, J.; Wu, Y.; et al. Minocycline improves the functional recovery after traumatic brain injury via inhibition of aquaporin-4. Int. J. Biol. Sci. 2022, 18, 441–458. [Google Scholar] [CrossRef]
- Glober, N.K.; Sprague, S.; Ahmad, S.; Mayfield, K.G.; Fletcher, L.M.; Digicaylioglu, M.H.; Sayre, N.L. Acetazolamide Treatment Prevents Redistribution of Astrocyte Aquaporin 4 after Murine Traumatic Brain Injury. Neurosci. J. 2019, 2019, 2831501. [Google Scholar] [CrossRef]
- Wang, M.; Yu, X.; Li, B.; Gao, C.; Chen, Y.; Zhang, X.; Li, W.; Yang, L.; Fan, Z. miR-211-5p targeting MMP9 regulates the expressions of AQP4 in traumatic brain injury. Acta Neurol. Belg. 2023, 123, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; Macdonald, J.A.; Ishida, H.; Vogel, H.J.; Almutiri, S.; Logan, A.; Kreida, S.; et al. Targeting Aquaporin-4 Subcellular Localization to Treat Central Nervous System Edema. Cell 2020, 181, 784–799.e19. [Google Scholar] [CrossRef]
- Xing, R.; Cheng, J.; Yu, J.; Li, S.; Ma, H.; Zhao, Y. Trifluoperazine reduces apoptosis and inflammatory responses in traumatic brain injury by preventing the accumulation of Aquaporin4 on the surface of brain cells. Int. J. Med. Sci. 2023, 20, 797–809. [Google Scholar] [CrossRef] [PubMed]
- Sylvain, N.J.; Salman, M.M.; Pushie, M.J.; Hou, H.; Meher, V.; Herlo, R.; Peeling, L.; Kelly, M.E. The effects of trifluoperazine on brain edema, aquaporin-4 expression and metabolic markers during the acute phase of stroke using photothrombotic mouse model. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183573. [Google Scholar] [CrossRef]
- He, R.; Zhang, X.; Pang, C.; Lin, L.; Li, S.; Jin, L.; Ding, L.; Wang, W. Inhibition of NADPH oxidase 2 improves cognitive abilities by modulating aquaporin-4 after traumatic brain injury in mice. Heliyon 2023, 9, e22035. [Google Scholar] [CrossRef]
- Yin, J.; Zhang, H.; Chen, H.; Lv, Q.; Jin, X. Hypertonic Saline Alleviates Brain Edema after Traumatic Brain Injury via Downregulation of Aquaporin 4 in Rats. Med. Sci. Monit. 2018, 24, 1863. [Google Scholar] [CrossRef]
- Wang, X.; Wang, C.; Yang, Y.; Ni, J. New monocyte locomotion inhibitory factor analogs protect against cerebral ischemia-reperfusion injury in rats. Bosn. J. Basic Med. Sci. 2017, 17, 221. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Zhu, Q.; Li, X.; Deng, S.; Guo, Y.; Mao, J.; Zhang, Y. Network pharmacology and molecular docking-based analysis of protective mechanism of MLIF in ischemic stroke. Front. Cardiovasc. Med. 2022, 9, 1071533. [Google Scholar] [CrossRef]
- Li, B.; Wei, M.; Wan, X.; Chen, Z.; Liu, M.; Fan, Z.; Yang, L. Neuroprotective effects of lentivirus-mediated aquaporin-4 gene silencing in rat model of traumatic brain injury. Neurol. Res. 2022, 44, 692–699. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, J.; Feng, X.; Jiao, W. Omega-3 polyunsaturated fatty acids alleviate early brain injury after traumatic brain injury by inhibiting neuroinflammation and necroptosis. Transl. Neurosci. 2023, 14, 20220277. [Google Scholar] [CrossRef]
- Yang, L.; Chen, Z.; Wan, X.; Liu, M.; Wu, J.; Chen, Y.; Zhang, G.; Fan, Z. Angiotensin II type 1 receptor deficiency protects against the impairment of blood-brain barrier in a mouse model of traumatic brain injury. Int. J. Neurosci. 2023, 133, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Shahrokhi, N.; Khaksari, M.; Asadikaram, G.; Soltani, Z.; Shahrokhi, N. Role of melatonin receptors in the effect of estrogen on brain edema, intracranial pressure and expression of aquaporin 4 after traumatic brain injury. Iran. J. Basic Med. Sci. 2018, 21, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.; da Silva, I.V.; Rodrigues, C.M.P.; Castro, R.E.; Soveral, G. The Emerging Role of microRNAs in Aquaporin Regulation. Front. Chem. 2018, 6, 238. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.; Unger, L.; Salman, M.M.; Kitchen, P.; Bill, R.M.; Yool, A.J. Signaling Mechanisms and Pharmacological Modulators Governing Diverse Aquaporin Functions in Human Health and Disease. Int. J. Mol. Sci. 2022, 23, 1388. [Google Scholar] [CrossRef]
- Zhang, E.; Wan, X.; Yang, L.; Wang, D.; Chen, Z.; Chen, Y.; Liu, M.; Zhang, G.; Wu, J.; Han, H.; et al. Omega-3 Polyunsaturated Fatty Acids Alleviate Traumatic Brain Injury by Regulating the Glymphatic Pathway in Mice. Front. Neurol. 2020, 11, 707. [Google Scholar] [CrossRef]
| Subject | Key Findings | AQP Type | Sample Type | Study Type | Year |
|---|---|---|---|---|---|
| “Cellular distribution of brain aquaporins and their contribution to cerebrospinal fluid homeostasis” [128] | Explored changes in aquaporin expression post-TBI in rats | AQP4 | Brain tissue, CSF | Experimental | 2022 |
| “Dynamic regulation of aquaporin-4 water channels in neurological disorders” [107] | Analyzed AQP4 expression changes in CSF of severe head injury patients | AQP4 | CSF | Clinical | 2015 |
| “Immunohistochemical evaluation of aquaporin-4 and its correlation” [129] | Examined AQP4 expression and localization changes in TBI patients | AQP4 | Brain tissue | Clinical | 2018 |
| “Aquaporin-4 distribution in control and stressed astrocytes” [130] | Found elevated AQP4 levels in CSF after TBI | AQP4 | CSF | Experimental | 2013 |
| “Neuronal damage and functional deficits are ameliorated by inhibition of aquaporin” [131] | Linked changes in AQP expression post-TBI to functional outcomes | AQP4 | Brain tissue | Experimental | 2012 |
| “Effect of decompressive craniectomy on aquaporin-4 expression” [132] | Investigated AQP4 expression changes following TBI | AQP4 | Brain tissue | Experimental | 2011 |
| “Aquaporin 4 expression and ultrastructure of the blood–brain barrier” [133] | Studied the dynamic change of AQP4 expression after cerebral contusion injury | AQP4 | Brain tissue | Experimental | 2013 |
| “Protective effects of aquaporin-4 deficiency on neurological outcomes” [134] | Studied the effects of AQP4 deficiency on TBI outcomes | AQP4 | Brain tissue | Experimental | 2021 |
| “Fluid-percussion brain injury induces changes in aquaporin channel expression” [135] | Examined the changes in AQP expression levels in a rat model of TBI | Multiple AQPs | Brain tissue | Experimental | 2011 |
| “Regulation of aquaporin-4 in a traumatic brain injury model in rats” [80] | Explored the regulation of AQP4 expression in TBI and its clinical implications | AQP4 | Brain tissue | Experimental | 2003 |
| “Emerging roles for dynamic aquaporin-4 subcellular relocalization” [19] | Discussed the dynamic relocalization of AQP4 in CNS water homeostasis post-TBI | AQP4 | Brain tissue, CSF | Review | 2022 |
| “Expression of aquaporin-4 and pathological characteristics of brain injury” [136] | Explored the relationship between AQP4 expression and brain injury pathology | AQP4 | Brain tissue, CSF | Experimental | 2015 |
| “Aquaporin and brain diseases” [137] | Examined the impact of traumatic brain injury on aquaporin expression levels | Multiple AQPs | Brain tissue, CSF | Review | 2014 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czyżewski, W.; Litak, J.; Sobstyl, J.; Mandat, T.; Torres, K.; Staśkiewicz, G. Aquaporins: Gatekeepers of Fluid Dynamics in Traumatic Brain Injury. Int. J. Mol. Sci. 2024, 25, 6553. https://doi.org/10.3390/ijms25126553
Czyżewski W, Litak J, Sobstyl J, Mandat T, Torres K, Staśkiewicz G. Aquaporins: Gatekeepers of Fluid Dynamics in Traumatic Brain Injury. International Journal of Molecular Sciences. 2024; 25(12):6553. https://doi.org/10.3390/ijms25126553
Chicago/Turabian StyleCzyżewski, Wojciech, Jakub Litak, Jan Sobstyl, Tomasz Mandat, Kamil Torres, and Grzegorz Staśkiewicz. 2024. "Aquaporins: Gatekeepers of Fluid Dynamics in Traumatic Brain Injury" International Journal of Molecular Sciences 25, no. 12: 6553. https://doi.org/10.3390/ijms25126553