

Immunogenetic and Environmental Factors in Age-Related Macular Disease

 , , , , and
, , , , and 
Abstract

1. Introduction

- Presentation of the current state of knowledge about the impact of environmental and genetic factors on the development of AMD in relation to our research conducted on the Polish population in the Kuyavian–Pomeranian and Lubusz Regions (Central and SW Poland).
- Analyzing how the polluted environment of large agglomerations affects the development of AMD, because this disease has only recently been a research subject and its etiology is still not well known.
- Indication of new markers, because the review of the current literature from this field is very optimistic, which means that emphasis are changes in the protein and enzyme activity, metal concentration, gene polymorphism. This is the starting point for the analyses that we intend to perform within our studies. We investigated concentration of chemical elements Na, K, Ca, Mg, Be, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Al, Si, P, S, As, Se, Mo, Cd, activity of antioxidant enzymes (SOD, CAT, GPx, GR); intensity of lipoperoxidation (MDA), stress proteins (ceruloplasmin CP, haptoglobin HPT, ferritin FRT), non-enzymatic mechanisms (total antioxidant status TAS, uric acid, bilirubin).
- Analysis of all the above relationships, which can bring us closer to a multi-level response to the question of the causes and development of pathophysiological changes that accompany AMD.
2. Role of Toxic Heavy Metals in AMD Development
2.1. Element-Element Interactions in AMD
2.2. Accumulation of Heavy Metal Ions in Eye Tissues
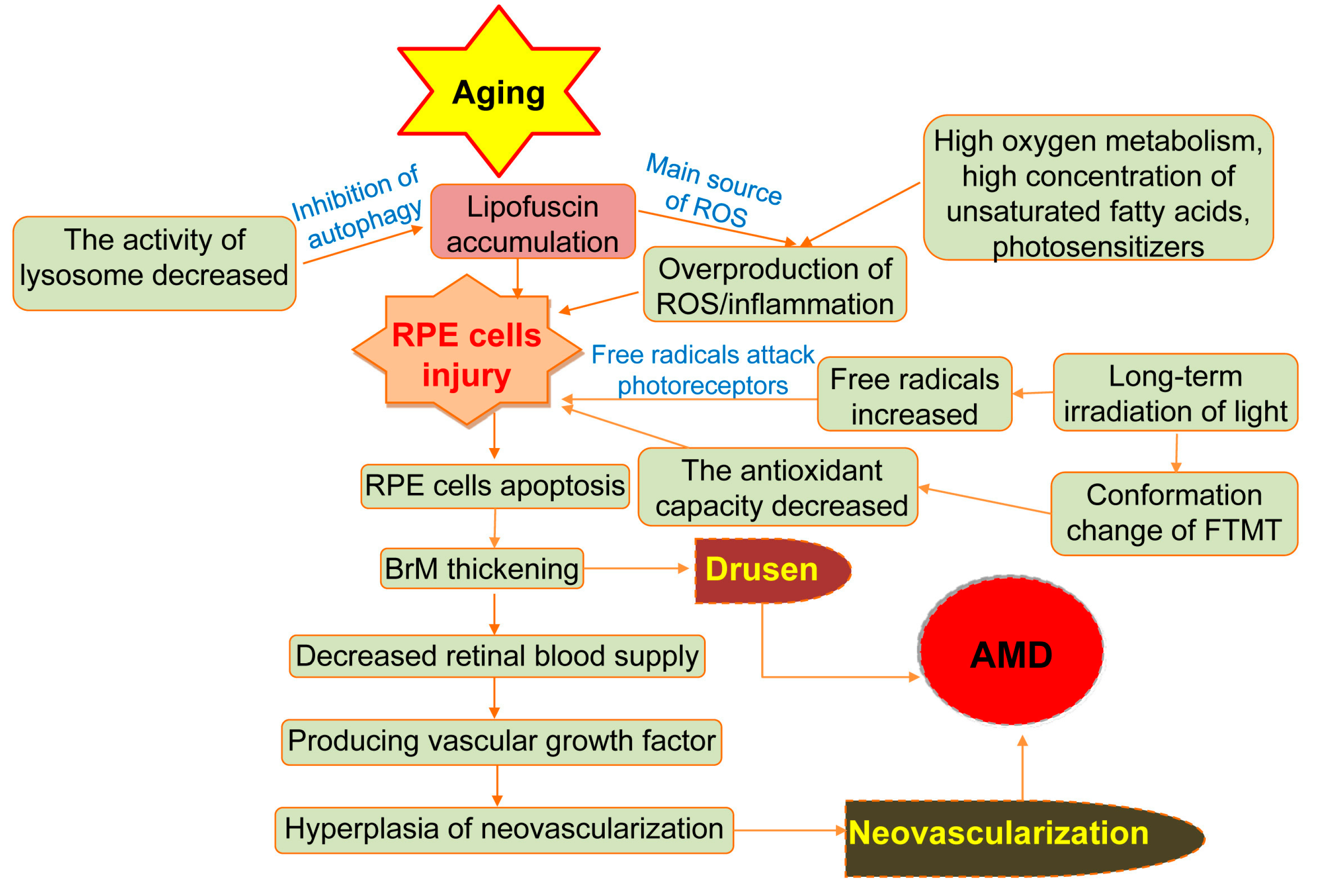
3. Role of Oxidative Stress
4. Role of Metallothioneins in AMD
5. Lifestyle and Environmental Risk Factors
5.1. Smoking
5.2. The Impact of Light
5.3. Diet and BMI
5.4. Other Risk Factors
6. The Influence of Genetic Factors on the Development of AMD
6.1. CFH
6.2. CFHR1 and CFHR3/CFHR1
6.3. ARMS2 and ARMS2/HTRA1
6.4. Complement Factor C3
6.5. Vascular Endothelial Growth Factor VEGFA
6.6. CFB
6.7. SKIV2L
6.8. LIPC
6.9. CETP
6.10. CFI
6.11. IL-8
6.12. GST Polymorphisms
6.13. Other Polymorphisms Promoting the Development of AMD
7. Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Congdon, N.; O’Colmain, B.; Klaver, C.C.; Klein, R.; Muñoz, B.; Friedman, D.S.; Kempen, J.; Taylor, H.R.; Mitchell, P. Eye Diseases Prevalence Research Group. Causes and prevalence of visual impairment among adults in the United States. Arch. Ophthalmol. 2004, 122, 477–485. [Google Scholar]
- Tuo, J.; Bojanowski, C.M.; Chan, C.C. Genetic factors of age-related macular degeneration. Prog. Retin. Eye Res. 2004, 23, 229–249. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Rodrigues, G.A. Progress and perspectives on the role of RPE cell inflammatory responses in the development of age-related macular degeneration. J. Inflam. Res. 2008, 1, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Handa, J.T. How Does the Macula Protect Itself from Oxidative Stress? Mol. Aspects Med. 2012, 33, 418–435. [Google Scholar] [CrossRef]
- Zeng, S.; Hernández, J.; Mullins, R.F. Effects of Antioxidant Components of AREDS Vitamins and Zinc Ions on Endothelial Cell Activation: Implications for Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2012, 53, 1041–1047. [Google Scholar] [CrossRef]
- Tokarz, P.; Kaarniranta, K.; Błasiak, J. Role of antioxidant enzymes and small molecular weight antioxidants in the pathogenesis of age-related macular degeneration (AMD). Biogerontology 2013, 14, 461–482. [Google Scholar] [CrossRef]
- Błasiak, J.; Petrovski, G.; Veréb, Z.; Facskó, A.; Kaarniranta, K. Oxidative Stress, Hypoxia, and Autophagy in the Neovascular Processes of Age-Related Macular Degeneration. Biomed. Res. Int. 2014, 2014, 768026. [Google Scholar] [CrossRef]
- Plestina-Borjan, I.; Katusic, D.; Medvidovic-Grubisic, M.; Supe-Domic, D.; Bucan, K.; Tandara, L.; Rogosic, V. Association of Age-Related Macular Degeneration with Erythrocyte Antioxidant Enzymes Activity and Serum Total Antioxidant Status. Oxid. Med. Cell. Longev. 2015, 2015, 804054. [Google Scholar] [CrossRef] [PubMed]
- Pugazhendhi, A.; Hubbell, M.; Jairam, P.; Ambati, B. Neovascular Macular Degeneration: A Review of Etiology, Risk Factors, and Recent Advances in Research and Therapy. Int. J. Mol. Sci. 2021, 22, 1170. [Google Scholar] [CrossRef]
- Heesterbeek, T.J.; Lores-Motta, L.; Hoyng, C.B.; Lechanteur, Y.T.E.; den Hollander, A.I. Risk factors for progression of age-related macular degeneration. Ophthalmic. Physiol. Opt. 2020, 40, 140–170. [Google Scholar] [CrossRef]
- Shughoury, A.; Sevgi, D.D.; Ciulla, T.A. Molecular Genetic Mechanisms in Age-Related Macular Degeneration. Genes 2022, 13, 1233. [Google Scholar] [CrossRef] [PubMed]
- Kushwah, N.; Bora, K.; Maurya, M.; Pavlovich, M.C.; Chen, J. Oxidative Stress and Antioxidants in Age-Related Macular Degeneration. Antioxidants 2023, 12, 1379. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.R.; Zielińska, A.; Sanchez-Lopez, E.; dos Santos, T.; Garcia, M.L.; Silva, A.M.; Karczewski, J.; Souto, E.B. Exudative versus Nonexudative Age-Related Macular Degeneration: Physiopathology and Treatment Options. Int. J. Mol. Sci. 2022, 23, 2592. [Google Scholar] [CrossRef]
- Deng, Y.; Qiao, L.; Du, M.; Qu Ch Wan, L.; Li, J.; Huang, L. Age-related macular degeneration: Epidemiology, genetics, pathophysiology, diagnosis, and targeted therapy. Genes Dis. 2022, 9, 6279. [Google Scholar] [CrossRef] [PubMed]
- Hammadi, S.; Tzoumas, N.; Ferrara, M.; Porpino Meschede, I.; Lo, K.; Harris, C.; Lako, M.; Steel, D.H. Bruch’s Membrane: A Key Consideration with Complement-Based Therapies for Age-Related Macular Degeneration. J. Clin. Med. 2023, 12, 2870. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, L.; Fu, Y.; Zhang, M.; Yang, Q.; Peng, J. Rethinking the potential and necessity of drug delivery systems in neovascular age-related macular degeneration therapy. Front. Bioeng. Biotechnol. 2023, 11, 1199922. [Google Scholar] [CrossRef] [PubMed]
- Arslan, S.; Kadayifcilar, S.; Samur, G. The Potential Role of Dietary Antioxidant Capacity in Preventing Age-Related Macular Degeneration. J. Am. Coll. Nutr. 2018, 38, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Zarbin, M.A. Current concepts in the pathogenesis of age-related macular degeneration. Arch. Ophthalmol. 2004, 122, 598–614. [Google Scholar] [CrossRef] [PubMed]
- Ambati, J.; Ambati, B.K.; Yoo, S.H.; Ianchulev, S.; Adamis, A.P. Age-related macular degeneration: Etiology, pathogenesis, and therapeutic strategies. Surv. Ophthalmol. 2003, 48, 257–293. [Google Scholar] [CrossRef]
- Nachman-Clewner, M.; Giblin, F.J.; Dorey, C.K.; Blanks, R.H.I.; Dang, L.; Dougherty, C.J.; Blanks, J.C. Selective Degeneration of Central Photoreceptors after Hyperbaric Oxygen in Normal and Metallothionein-Knockout Mice. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3207–3215. [Google Scholar] [CrossRef]
- Bartosz, G. Druga Twarz Tlenu. Wolne Rodniki w Przyrodzie; PWN-Polish Scientific Publishers: Warszawa, Poland, 2019; 447p. [Google Scholar]
- Alvarez, L.; Gonzalez-Iglesias, H.; Garcia, M.; Ghosh, S.; Sanz-Medel, A.; Coca-Prados, M. The Stoichiometric Transition from Zn6Cu1-Metallothionein to Zn7-Metallothionein Underlies the Up-regulation of Metallothionein (MT) Expression—Quantitative analysis of MT-metal load in eye cells. J. Biol. Chem. 2012, 287, 28456–28469. [Google Scholar] [CrossRef]
- Honda, S.; Farboud, B.; Hjelmeland, L.M.; Handa, J.T. Induction of an aging mRNA retinal pigment epithelial cell phenotype by matrix-containing advanced glycation end products in vitro. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2419–2425. [Google Scholar]
- Cano, M.; Thimmalappula, R.; Fujihara, M.; Nagai, N.; Sporn, M.; Wang, A.L.; Neufeld, A.H.; Biswal, S.; Handa, J.T. Cigarette smoking, oxidative stress, the anti-oxidant response through Nrf2 signaling, and Age-related Macular Degeneration. Vis. Res. 2010, 50, 652–664. [Google Scholar] [CrossRef]
- Nguyen, T.; Sherratt, P.J.; Pickett, C.B. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Ann. Rev. Pharmacol. Toxicol. 2003, 43, 233–260. [Google Scholar] [CrossRef]
- Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke induced emphysema in mice. J. Clin. Investig. 2004, 114, 1248–1259. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar]
- Yates, J.R.; Sepp, T.; Matharu, B.K.; Khan, J.C.; Thurlby, D.A.; Shahid, H.; Clayton, D.G.; Hayward, C.; Morgan, J.; Wright, A.F.; et al. Complement C3 variant and the risk of age-related macular degeneration. N. Eng. J. Med. 2007, 357, 553–561. [Google Scholar] [CrossRef]
- Biswas, S.K.; Rahman, I. Environmental toxicity, redox signaling and lung inflammation: The role of glutathione. Mol. Asp. Med. 2009, 30, 60–76. [Google Scholar] [CrossRef]
- Walsh, A.C.; Michaud, S.G.; Malossi, J.A.; Lawrence, D.A. Glutathione depletion in human T lymphocytes: Analysis of activation-associated gene expression and the stress response. Toxicol. Appl. Pharmacol. 1995, 133, 249–261. [Google Scholar] [CrossRef]
- Will, O.; Mahler, H.C.; Arrigo, A.P.; Epe, B. Influence of glutathione levels and heat-shock on the steady-state levels of oxidative DNA base modifications in mammalian cells. Carcinogenesis 1999, 20, 333–337. [Google Scholar] [CrossRef]
- Handa, J.T.; Verzijl, N.; Matsunaga, H.; Aotaki-Keen, A.; Lutty, G.A.; Te Koppele, J.M.; Miyata, T.; Hjelmeland, L.M. Increase in the advanced glycation end product pentosidine in Bruch’s membrane with age. Investig. Ophthalmol. Vis. Sci. 1999, 40, 775–779. [Google Scholar]
- Nowak, J.Z. Age-related macular degeneration (AMD): Pathogenesis and therapy. Pharmacol. Rep. 2006, 58, 353–363. [Google Scholar]
- Fisher, R.F. The influence of age on some ocular basement membranes. Eye 1987, 1, 184–189. [Google Scholar] [CrossRef]
- Bok, D. Retinal photoreceptor-pigment epithelium interactions. Friedenwald lecture. Investig. Ophthalmol. Vis. Sci. 1985, 26, 1659–1694. [Google Scholar]
- Kell, D.B.; Pretorius, E. Serum ferritin is an important inflammatory disease marker, as it is mainly a leakage product from damaged cells. Metallomics 2014, 6, 748–773. [Google Scholar] [CrossRef]
- Klein, R.J.; Zeiss, C.; Chew, E.Y.; Tsai, J.Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; San Giovanni, J.P.; Mane, S.M.; Mayne, S.T.; et al. Complement Factor H Polymorphism in Age-Related Macular Degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef]
- Wang, Y.; Kinzie, E.; Berger, F.G.; Lim, S.K.; Baumann, H. Haptoglobin, an inflammation-inducible plasma protein. Redox Rep. 2001, 6, 379–385. [Google Scholar] [CrossRef]
- Fenkci, V.; Fenkci, S.; Yilmazer, M.; Serteser, M. Decreased total antioxidant status and increased oxidative stress in women with polycystic ovary syndrome may contribute to the risk of cardiovascular disease. Fertil. Steril. 2003, 80, 123–127. [Google Scholar] [CrossRef]
- Piotrowski, J. Podstawy Toksykologii; WNT: Warszawa, Poland, 2008; 467p. [Google Scholar]
- Haddad, H.H. The Effect of Heavy Metals Cadmium, Chromium and Iron Accumulation in Human Eyes. Am. J. Analyt. Chem. 2012, 3, 710–713. [Google Scholar] [CrossRef]
- Lutz, W.; Pałczyński, C. Immunotoksykologia; Publishing House, Institute of Occupational Medicine named after Professor George Nofer: Łódź, Poland, 2005; 395p. [Google Scholar]
- Kędryna, T. Chemia Ogólna z Elementami Biochemii; ZamKor: Kraków, Poland, 2006; 530p. [Google Scholar]
- Tanito, M.; Masutani, H.; Kim, Y.C.; Nishikawa, M.; Ohira, A.; Yodoi, J. Sulforaphane induces thioredoxin through the antioxidant-responsive element and attenuates retinal light damage in mice. Investig. Ophthalmol. Vis. Sci. 2005, 46, 979–987. [Google Scholar] [CrossRef]
- Gao, X.; Talalay, P. Induction of phase 2 genes by sulforaphane protects retinal pigment epithelial cells against photooxidative damage. Proc. Nat. Acad. Sci. USA 2004, 101, 10446–10451. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.C.; Armstrong, J.S.; Moriarty, S.; Cai, J.; Wu, M.W.; Sternberg, P. Jr, Jones D.P. Protection of retinal pigment epithelial cells from oxidative damage by oltipraz, a cancer chemo-preventive agent. Investig. Ophthalm. Vis. Sci. 2002, 43, 3550–3554. [Google Scholar]
- Ha, K.N.; Chen, Y.; Cai, J.; Sternberg, P., Jr. Increased glutathione synthesis through an ARE-Nrf2-dependent pathway by zinc in the RPE: Implication for protection against oxidative stress. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2709–2715. [Google Scholar] [CrossRef]
- Chlebda, E.; Antonowicz-Juchniewicz, E.; Andrzejak, R. Wpływ ekspozycji zawodowej na ołów i arsen na stężenie karotenoidów w surowicy u pracowników huty miedzi. Med. Pr. 2004, 55, 389–401. [Google Scholar]
- Ociepa-Kubicka, A.; Ociepa, E. Toksyczne oddziaływanie metali ciężkich na rośliny, zwierzęta i ludzi. Inż. Ochr. Środ. 2012, 15, 169–180. [Google Scholar]
- Wojciechowska, M.; Kołodziejczyk, J.; Gocki, J.; Bartuzi, Z. Nadwrażliwość na nikiel. Alerg. Astma Immunol. 2008, 13, 136–140. [Google Scholar]
- Brzóska, M.; Jurczuk, M.; Moniuszko-Jakoniuk, J. Interakcje kadmu z wybranymi biopierwiastkami. Terapia 1997, 7, 28–30. [Google Scholar]
- Bridges, C.C.; Zalups, R.K. Molecular and ionic mimicry and the transport of toxic metals. Toxicol. Appl. Pharmacol. 2005, 204, 274–308. [Google Scholar] [CrossRef]
- Klassen, C.D.; Liu, J.; Choudhuri, S. Metallothionein: An intracelluar protein to protect against cadmium toxicity. Ann. Rev. Pahrmacol. Toxicol. 1999, 39, 267–294. [Google Scholar] [CrossRef]
- Swindell, W.R. Metallothionein and the Biology of Aging. Ageing Res. Rev. 2011, 10, 132–145. [Google Scholar] [CrossRef]
- Krzywy, I.; Krzywy, E.; Pastuszak-Gabinowska, M.; Brodkiewicz, A. Ołów—Czy jest się czego obawiać? Ann. Acad. Med. Stet. 2010, 56, 118–128. [Google Scholar]
- Erie, J.C.; Butz, J.A.; Good, J.A.; Erie, E.A.; Burritt, M.F.; Cameron, J.D. Heavy Metal Concentrations in Human Eyes. Am. J. Ophthalmol. 2005, 139, 888–893. [Google Scholar] [CrossRef] [PubMed]
- Wills, N.K.; Ramanujam, V.M.; Kalariya, N.; Lewis, J.R.; van Kuijk, F.J. Copper and zinc distribution in the human retina: Relationship to cadmium accumulation, age, and gender. Exp. Eye Res. 2008, 87, 80–88. [Google Scholar] [CrossRef]
- Brodzka, S. Środowiskowe i Immunogenetyczne Uwarunkowania Zmian Patofizjologicznych w Zwyrodnieniu Plamki Żółtej. Environmental and Immunogenetic Determinants of Pathophysiological Changes in Macular Degeneration. Ph.D. Thesis, University of Zielona Góra, Zielona Góra, Poland, 2021; 388p. [Google Scholar]
- Park, S.J.; Lee, J.H.; Woo, S.J.; Kang, S.W.; Park, K.H. Five Heavy Metallic Elements and Age-Related Macular Degeneration: Korean National Health and Nutrition Examination Survey, 2008–2011. Ophthalmology 2015, 122, 129–137. [Google Scholar] [CrossRef]
- Bhattacharyya, M.H.; Wilson, A.K.; Rajan, S.S.; Jonah, M. Biochemical pathways in cadmium toxicity. In Molecular Biology and Toxicology of Metals; Zalups, R.K., Koropatnick, J., Eds.; Taylor and Francis: London, UK, 2000; pp. 34–74. [Google Scholar]
- Potts, A.M.; Au, P.C. The affinity of melanin for inorganic ions. Exp. Eye Res. 1976, 22, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Ulshafer, R.J.; Allen, C.B.; Rubin, M.L. Distributions of elements in the human retinal pigment epithelium. Arch. Ophthalmol. 1990, 108, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Pamphlett, R.; Cherepanoff, S.; Khoon Too, L.; Kum Jew, S.; Doble, P.A.; Bishop, D.P. The distribution of toxic metals in the human retina and optic nerve head: Implications for age-related macular degeneration. PLoS ONE 2020, 15, e0241054. [Google Scholar] [CrossRef] [PubMed]
- Filipoiu, D.C.; Bungau, S.G.; Endres, L.; Negru, P.A.; Bungau, A.F.; Pasca, B.; Radu, A.-F.; Tarce, A.G.; Bogdan, M.A.; Behl, T.; et al. Characterization of the Toxicological Impact of Heavy Metals on Human Health in Conjunction with Modern Analytical Methods. Toxics 2022, 10, 716. [Google Scholar] [CrossRef]
- Eichenbaum, J.W.; Zheng, W. Distribution of lead and transthyretin in human eyes. Clin. Toxicol. 2000, 38, 377–381. [Google Scholar] [CrossRef]
- Valko, M.; Morris, H.; Cronin, M.T. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed]
- Dantzig, P.I. Age related macular degeneration and cutaneous signs of mercury. Toxic Cutan. Ocular Toxicol. 2005, 24, 3–9. [Google Scholar] [CrossRef]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts mediate NALP-3 inflammasome activation via phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Erie, J.C.; Good, J.A.; Butz, J.A.; Pulido, J.S. Reduced zinc and copper in the retinal pigment epithelium and choroid in age-related macular degeneration. Am. J. Ophthalmol. 2009, 147, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Awh, C.C.; Lane, A.M.; Hawken, S.; Zanke, B.; Kim, I.K. CFH and ARMS2 genetic polymorphisms predict response to antioxidants and zinc in patients with age-related macular degeneration. Ophthalmology 2013, 120, 2317–2323. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.D.; Dizdaroglu, M.; Cooke, M.S. Oxidative DNA damage and disease: Induction, repair and significance. Mutation Res. 2004, 567, 1–61. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.K.; Dong, A.; Hackett, S.F.; Bell, W.R.; Green, W.R.; Campochiaro, P.A. Oxidative damage in age-related macular degeneration. Histol. Histopathol. 2007, 22, 1301–1308. [Google Scholar]
- Beatty, S.; Koh, H.; Phil, M.; Henson, D.; Boulton, M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 2000, 45, 115–134. [Google Scholar] [CrossRef] [PubMed]
- Hollyfield, J.G.; Bonilha, V.L.; Rayborn, M.E.; Yang, X.; Shadrach, K.G.; Lu, L.; Ufret, R.L.; Salomon, R.G.; Perez, V.L. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat. Med. 2008, 14, 194–198. [Google Scholar] [CrossRef]
- Thurman, J.M.; Renner, B.; Kunchithapautham, K.; Ferreira, V.P.; Pangburn, M.K.; Ablonczy, Z.; Tomlinson, S.; Holers, V.M.; Rohrer, B. Oxidative stress renders retinal pigment epithelial cells susceptible to complement-mediated injury. J. Biol. Chem. 2009, 284, 16939–16947. [Google Scholar] [CrossRef]
- Wu, Z.; Lauer, T.W.; Sick, A.; Hackett, S.F.; Campochiaro, P.A. Oxidative stress modulates complement factor H expression in retinal pigmented epithelial cells by acetylation of FOXO3. J. Biol. Chem. 2007, 282, 22414–22425. [Google Scholar] [CrossRef] [PubMed]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W.; Ding, J.; et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef] [PubMed]
- Rose, R.C.; Richer, S.P.; Bode, A.M. Ocular oxidants and antioxidant protection. Proc. Soc. Exp. Biol. Med. 1998, 217, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Winkler, B.S.; Boulton, M.E.; Gottsch, J.D.; Sternberg, P. Oxidative damage and age-related macular degeneration. Mol. Vis. 1999, 5, 32. [Google Scholar]
- Schneeweis, D.M.; Schnapf, J.L. Photovoltage of rods and cones in the macaque retina. Science 1995, 268, 1053–1056. [Google Scholar] [CrossRef] [PubMed]
- Rogers, B.S.; Symons, R.C.; Komeima, K.; Shen, J.; Xiao, W.; Swaim, M.E.; Gong, Y.Y.; Kachi, S.; Campochiaro, P.A. Differential sensitivity of cones to iron-mediated oxidative damage. Investig. Ophthalmol. Vis. Sci. 2007, 48, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Roth, F.; Bindewald, A.; Holz, F.G. Key pathophysiologic pathways in age-related macular disease. Graefe’s Arch. Clin. Exp. Ophthalmol. 2004, 242, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Tate Jr, D.J.; Miceli, M.V.; Newsome, D.A. Phagocytosis and H2O2 induce catalase and metallothionein gene expression in human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 1995, 36, 1271–1279. [Google Scholar]
- Kennedy, C.J.; Rakoczy, P.E.; Constable, I.J. Lipofuscin of the retinal pigment epithelium: A review. Eye 1995, 9, 763–771. [Google Scholar] [CrossRef]
- Murdaugh, L.S.; Avalle, L.B.; Mandal, S.; Dill, A.E.; Dillon, J.; Simon, J.D.; Gaillard, E.R. Compositional studies of human RPE lipofuscin. J. Mass Spectrom. 2010, 45, 1139–1147. [Google Scholar] [CrossRef]
- Iliescu, D.A.; Ghita, A.C.; Ilie, L.A.; Voiculescu, S.E.; Geamanu, A.; Ghita, A.M. Non-Neovascular Age-Related Macular Degeneration Assessment: Focus on Optical Coherence Tomography Biomarkers. Diagnostics 2024, 14, 764. [Google Scholar] [CrossRef] [PubMed]
- Jamrozik, D.; Dutczak, R.; Machowicz, J.; Wojtyniak, A.; Smędowski, A.; Pietrucha-Dutczak, M. Metallothioneins, a Part of the Retinal Endogenous Protective System in Various Ocular Diseases. Antioxidants 2023, 12, 1251. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Shimazawa, M.; Hara, H. Physiological Roles of Metallothioneins in Central Nervous System Diseases. Biol. Pharm. Bull. 2018, 41, 1006–1013. [Google Scholar] [CrossRef] [PubMed]
- Velilla, S.; García-Medina, J.J.; García-Layana, A.; Dolz-Marco, R.; Pons-Vázquez, S.; Pinazo-Durán, M.D.; Gómez-Ulla, F.; Arévalo, J.F.; Díaz-Llopis, M.; Gallego-Pinazo, R. Smoking and Age-Related Macular Degeneration: Review and Update. J. Ophthalmol. Hindawi. 2013, 2013, 895147. [Google Scholar] [CrossRef] [PubMed]
- Thornton, J.; Edwards, R.; Mitchell, P.; Harrison, R.A.; Buchan, I.; Kelly, S.P. Smoking and age-related macular degeneration: A review of association. Eye 2005, 19, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; Ajani, U.A.; Mitchell, B.D. Familial aggregation of age-related maculopathy. Am. J. Ophthalmol. 1997, 123, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Vingerling, J.R.; Hofman, A.; Grobbee, D.E.; de Jong, P.T. Age-related macular degeneration and smoking. The Rotterdam Study. Arch. Ophthalmol. 1996, 114, 1193–1196. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, M.; Hassan, Y.; Bakka Vemana, P.P.S.; Bellary Pattanashetty, M.S.; Abdin, Z.U.; Siddiqui, H.F. Age-Related Macular Degeneration: An Exponentially Emerging Imminent Threat of Visual Impairment and Irreversible Blindness. Cureus 2023, 15, e39624. [Google Scholar] [CrossRef]
- Seddon, J.M.; Gensler, G.; Milton, R.C.; Klein, M.L.; Rifai, N. Association between C-reactive protein and age-related macular degeneration. J. Am. Med. Assoc. 2004, 291, 704–710. [Google Scholar] [CrossRef]
- Molins, B.; Romero-Vázquez, S.; Fuentes-Prior, P.; Adan, A.; Dick, A.D. C-Reactive Protein as a Therapeutic Target in Age-Related Macular Degeneration. Front Immunol. 2018, 9, 808. [Google Scholar] [CrossRef]
- Khan, J.C.; Thurlby, D.A.; Shahid, H.; Clayton, D.G.; Yates, J.R.; Bradley, M.; Moore, A.T.; Bird, A.C.; Genetic Factors in AMD Study. Smoking and age related macular degeneration: The number of pack years of cigarette smoking is a major determinant of risk for both geographic atrophy and choroidal neovascularisation. Br. J. Ophthalmol. 2006, 90, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Sowa, P.; Rutkowska-Talipska, J.; Rutkowski, K.; Kosztyła-Hojna, B.; Rutkowski, R. Optical radiation in modern medicine. Post. Dermatol. Alergol. 2013, 30, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Youssef, P.N.; Sheibani, N.; Albert, D.M. Retinal light toxicity. Eye 2011, 25, 1–14. [Google Scholar] [CrossRef]
- Neelam, K.; Zhou, S.W.; Au Eong, K.G. The role of blue light in the pathogenesis of Age-related Macular Degeneration. Points De Vue 2014, 71, 77. [Google Scholar]
- Wu, M.; Guo, Y.; Ma, Y.; Zheng, Z.; Wang, Q.; Zhou, X. Association of Two Polymorphisms, rs1061170 and rs1410996, in Complement Factor H with Age-Related Macular Degeneration in an Asian Population: A Meta-Analysis. Ophthalm. Res. 2016, 55, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, A.E.; Bentham, G.C.; Agnew, M.; Young, I.S.; Augood, C.; Chakravarthy, U.; de Jong, P.T.; Rahu, M.; Seland, J.; Soubrane, G.; et al. Sunlight exposure, antioxidants, and age-related macular degeneration. Arch. Ophthalmol. 2008, 126, 1396–1403. [Google Scholar] [CrossRef]
- Brodzka, S.; Baszyński, J.; Rektor, K.; Hołderna-Bona, K.; Stanek, E.; Kurhaluk, N.; Tkaczenko, H.; Malukiewicz, G.; Woźniak, A.; Kamiński, P. The Role of Glutathione in Age-Related Macular Degeneration (AMD). Int. J. Mol. Sci. 2024, 25, 4158. [Google Scholar] [CrossRef] [PubMed]
- Rapp, L.M.; Smith, S.C. Morphologic comparisons between rhodopsin-mediated and short wavelength classes of retinal light damage. Investig. Ophthalmol. Vis. Sci. 1992, 33, 3367–3377. [Google Scholar]
- Braunstein, R.E.; Sparrow, J.R. A blue-blocking intraocular lens should be used in cataract surgery. Arch. Ophthalmol. 2005, 123, 547–549. [Google Scholar] [CrossRef]
- Remé, C.E.; Grimm, C.; Hafezi, F.; Wenzel, A.; Williams, T.P. Apoptosis in the Retina: The Silent Death of Vision. News Physiol. Sci. 2000, 15, 120–124. [Google Scholar] [CrossRef]
- Connell, P.P.; Keane AO’Neill, C.; Altaie, W.R.; Loane, E.; Neelam, K.; Nolan, M.J.; Beatty, S. Risk Factors for Age-Related Maculopathy. J. Ophthalmol. 2009, 2009, 360764. [Google Scholar] [CrossRef] [PubMed]
- Sui, G.Y.; Liu, G.C.; Liu, G.Y.; Gao, Y.Y.; Deng, Y.; Wang, W.Y.; Tong, S.H.; Wang, L. Is sunlight exposure a risk factor for age-related macular degeneration? A systematic review and meta-analysis. Br. J. Ophthalmol. 2013, 97, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Mainster, M.A. Violet and blue light blocking intraocular lenses: Photoprotection versus photoreception. Br. J. Ophthalmol. 2006, 90, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Taylor, H.R.; Muñoz, B.; West, S.; Bressler, N.M.; Bressler, S.B.; Rosenthal, F.S. Visible light and risk of age-related macular degeneration. Trans. Am. Ophthalmol. Soc. 1990, 88, 163–178. [Google Scholar] [PubMed]
- Tomany, S.C.; Cruickshanks, K.J.; Klein, R.; Klein, B.E.; Knudtson, M.D. Sunlight and the 10-year incidence of age-related maculopathy: The Beaver Dam Eye Study. Arch. Ophthalmol. 2004, 122, 750–757. [Google Scholar] [CrossRef]
- Delcourt, C.; Carrière, I.; Ponton-Sanchez, A.; Fourrey, S.; Lacroux, A.; Papoz, L. POLA Study Group. Light exposure and the risk of age-related macular degeneration: The Pathologies Oculaires Liées à l’Age (POLA) study. Arch. Ophthalmol. 2001, 119, 1463–1468. [Google Scholar] [CrossRef] [PubMed]
- Maurya, M.; Bora, K.; Blomfield, A.K.; Pavlovich, M.C.; Huang, S.; Liu, C.h.-H.; Chen, J. Oxidative stress in retinal pigment epithelium degeneration: From pathogenesis to therapeutic targets in dry age-related macular degeneration. Neural Regen. Res. 2023, 18, 2173–2181. [Google Scholar] [PubMed]
- Seddon, J.M.; Sobrin, L. Epidemiology of age-related macular degeneration. In Retina, 5th ed.; Ryan, S.J., Ed.; Saunders: London, UK; Elsevier: Amsterdam, The Netherlands, 2013; pp. 1134–1144. [Google Scholar]
- WHO. WHO World Report on Vision; WHO: Geneva, Switzerland, 2019; 180p.
- Chew, E.Y. Age-related Macular Degeneration: Nutrition, Genes and Deep Learning—The LXXVI Edward Jackson Memorial Lecture. Am. J. Ophthalmol. 2020, 217, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Merle, B.M.J.; Colijn, J.M.; Cougnard-Gregoire, A.; de Koning-Backus, A.P.; Delyfer, M.N.; Kiefte-de Jong, J.C.; Meester-Smoor, M.; Féart, C.; Verzijden, T.; Samieri, C.; et al. Mediterranean diet and incidence of advanced age-related macular degeneration: The EYE-RISK Consortium. Ophthalmology 2019, 126, 381–390. [Google Scholar] [CrossRef]
- McGuinness, M.B.; Karahalios, A.; Simpson, J.A.; Guymer, R.H.; Robman, L.D.; Hodge, A.M.; Cerin, E.; Giles, G.G.; Finger, R.P. Past physical activity and age-related macular degeneration: The Melbourne Collaborative Cohort Study. Br. J. Ophthalmol. 2016, 100, 1353–1358. [Google Scholar] [CrossRef]
- Bringmann, A.; Hollborn, M.; Kohen, L.; Wiedemann, P. Intake of dietary salt and drinking water: Implications for the development of age-related macular degeneration. Mol. Vis. 2016, 22, 1437–1454. [Google Scholar]
- Jaisankar, D.; Swaminathan, G.; Roy, R.; Kulothungan, V.; Sharma, T.; Raman, R. Association of obesity and age-related macular degeneration in Indian population. Indian J. Ophthalmol. 2018, 66, 976–983. [Google Scholar] [PubMed]
- Nielsen, F.H. The emergence of boron as nutritionally important throughout the life cycle. Nutrition 2000, 16, 512–514. [Google Scholar] [CrossRef]
- Kim, M.H.; Zhao, D.; Cho, J.; Guallar, E. Cadmium exposure and age-related macular degeneration. J. Exp. Sci. Environ. Epidemiol. 2016, 26, 214–218. [Google Scholar] [CrossRef]
- Zhang, M.; Jiang, N.; Chu, Y.; Postnikova, O.; Varghese, R.; Horvath, A.; Cheema, A.K.; Golestaneh, N. Dysregulated metabolic pathways in age-related macular degeneration. Sci. Rep. 2020, 10, 2464. [Google Scholar] [CrossRef] [PubMed]
- Buch, H.; Vinding, T.; la Cour, M.; Jensen, G.B.; Prause, J.U.; Nielsen, N.V. Risk factors for age-related maculopathy in a 14-year follow-up study: The Copenhagen City Eye Study. Acta Ophthalmol. Scand. 2005, 83, 409–418. [Google Scholar] [CrossRef]
- Choudhury, F.; Varma, R.; McKean-Cowdin, R.; Klein, R.; Azen, S.P. Los Angeles Latino Eye Study G. Risk factors for four-year incidence and progression of age-related macular degeneration: The los angeles latino eye study. Am. J. Ophthalmol. 2011, 152, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Erke, M.G.; Bertelsen, G.; Peto, T.; Sjolie, A.K.; Lindekleiv, H.; Njolstad, I. Prevalence of age-related macular degeneration in elderly Caucasians: The Tromso Eye Study. Ophthalmology 2012, 119, 1737–1743. [Google Scholar] [CrossRef]
- Klein, R.; Klein, B.E.; Knudtson, M.D.; Meuer, S.M.; Swift, M.; Gangnon, R.E. Fifteen-year cumulative incidence of age-related macular degeneration: The Beaver Dam Eye Study. Ophthalmology 2007, 114, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.J.; Ahn, J.; Morrison, M.A.; Ahn, S.Y.; Lee, J.; Kim, K.W.; DeAngelis, M.M.; Park, K.H. Analysis of Genetic and Environmental Risk Factors and Their Interactions in Korean Patients with Age-Related Macular Degeneration. PLoS ONE 2015, 10, e0132771. [Google Scholar] [CrossRef]
- Labarrere, C.A.; Kassab, G.S. Glutathione: A Samsonian life-sustaining small molecule that protects against oxidative stress, aging and damaging inflammation. Front. Nutr. 2022, 9, 1007816. [Google Scholar] [CrossRef]
- Lee, K.S.; Lin, S.; Copland, D.A.; Dick, A.D.; Liu, J. Cellular senescence in the aging retina and developments of genotherapies for age-related macular degeneration. J. Neuroinflam. 2021, 18, 32. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.I. Age-Related Macular Degeneration, 2nd ed.; Informa Healthcare: New York, NY, USA, 2008; 386p. [Google Scholar]
- Zetterberg, M. Age-related eye disease and gender. Maturitas 2016, 83, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Lou, L.; Miao, Q.; Wang, Y.; Jin, K.; Shan, P.; Xu, Y. The pattern and gender disparity in global burden of age-related macular degeneration. Eur. J. Ophthalmol. 2020, 31, 1120672120927256. [Google Scholar] [CrossRef] [PubMed]
- Jankowska-Lech, I.; Grabska-Liberek, I.; Krzyżewska-Niedziałek, A.; Pietruszyńska, M. Zwyrodnienie plamki związane z wiekiem (AMD)—choroba starzejących się społeczeństw. Post. Nauk Med. 2013, 26, 868–873. [Google Scholar]
- Pennington, K.L.; DeAngelis, M.M. Epidemiology of age-related macular degeneration (AMD): Associations with cardiovascular disease phenotypes and lipid factors. Eye Vis. 2016, 3, 34. [Google Scholar] [CrossRef] [PubMed]
- Rim, T.H.; Kim, H.K.; Kim, J.W.; Lee, J.S.; Kim, D.W.; Kim, S.S. A Nationwide Cohort Study on the Association Between Past Physical Activity and Neovascular Age-Related Macular Degeneration in an East Asian Population. JAMA Ophthalmol. 2018, 136, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Knudtson, M.D.; Lee, K.E.; Gangnon, R.E.; Klein, B.E. Age-period-cohort effect on the incidence of age-related macular degeneration: The Beaver Dam Eye Study. Ophthalmology 2008, 115, 1460–1467. [Google Scholar] [CrossRef] [PubMed]
- Merle, B.M.J.; Silver, R.E.; Rosner, B.; Seddon, J.M. Associations between vitamin D intake and progression to incident advanced age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2017, 58, 4569–4578. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Knudtson, M.D.; Cruickshanks, K.J.; Klein, B.E. Further observations on the association between smoking and the long-term incidence and progression of age-related macular degeneration: The Beaver Dam Eye Study. Arch. Ophthalmol. 2008, 126, 115–121. [Google Scholar] [CrossRef]
- Klein, R.; Klein, B.E.; Knudtson, M.D.; Wong, T.Y.; Cotch, M.F.; Liu, K.; Burke, G.; Saad, M.F.; Jacobs, D.R., Jr. Prevalence of age-related macular degeneration in 4 racial/ethnic groups in the multi-ethnic study of atherosclerosis. Ophthalmology 2006, 113, 373–380. [Google Scholar] [CrossRef]
- Age-Related Eye Disease Study Research Group. Risk factors associated with age-related macular degeneration. A case-control study in the age-related eye disease study: Age-Related Eye Disease Study Report Number 3. Ophthalmology 2000, 107, 2224–2232. [Google Scholar]
- Pang, C.P.; Baum, L.; Chan, W.M.; Lau, T.C.; Poon, P.M.; Lam, D.S. The apolipoprotein E epsilon4 allele is unlikely to be a major risk factor of age-related macular degeneration in Chinese. Ophthalmologica 2000, 214, 289–291. [Google Scholar] [CrossRef]
- Krishnaiah, S.; Das, T.; Nirmalan, P.K.; Nutheti, R.; Shamanna, B.R.; Rao, G.N.; Thomas, R. Risk factors for age-related macular degeneration: Findings from the Andhra Pradesh eye disease study in South India. Investig. Ophthalmol Vis Sci. 2005, 46, 4442–4449. [Google Scholar] [CrossRef]
- Cugati SMitchell, P.; Rochtchina, E.; Tan, A.G.; Smith, W.; Wang, J.J. Cataract surgery and the 10-year incidence of age-related maculopathy: The Blue Mountains Eye Study. Ophthalmology 2006, 113, 2020–2025. [Google Scholar] [CrossRef]
- Ho, L.; Boekhoorn, S.S.; Liana van Duijn, C.M.; Uitterlinden, A.G.; Hofman, A.; de Jong, P.T.; Stijnen, T.; Vingerling, J.R. Cataract surgery and the risk of aging macula disorder: The Rotterdam study. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4795–4800. [Google Scholar] [CrossRef]
- Rosen, E.S. Age-related macular degeneration and cataract surgery. J. Cataract Refract. Surg. 2014, 40, 173–174. [Google Scholar] [CrossRef]
- Casparis, H.; Lindsley, K.; Kuo, I.C.; Sikder, S.; Bressler, N.M. Surgery for cataracts in people with age-related macular degeneration. Cochrane Database Syst. Rev. 2017, 2, CD006757. [Google Scholar] [CrossRef]
- Shim, S.H.; Kim, S.G.; Bae, J.H.; Yu, H.G.; Song, S.J. Risk factors for progression of early age-related macular degeneration in Koreans. Ophthalmic Epidemiol. 2016, 23, 80–87. [Google Scholar] [CrossRef]
- Wang, I.K.; Lin, H.J.; Wan, L.; Lin, C.L.; Yen, T.H.; Sung, F.C. Risk of age-related macular degeneration in end-stage renal disease patients receiving long-term dialysis. Retina 2016, 36, 1866–1873. [Google Scholar] [CrossRef]
- Klein, R.; Klein, B.E.; Knudtson, M.D.; Cotch, M.F.; Wong, T.Y.; Liu, K.; Burke, G.L.; Saad, M.F.; Jacobs, D.R., Jr.; Sharrett, A.R. Subclinical atherosclerotic cardiovascular disease and early age-related macular degeneration in a multiracial cohort: The Multiethnic Study of Atherosclerosis. Arch. Ophthalmol. 2007, 125, 534–543. [Google Scholar] [CrossRef]
- Yip, J.L.; Khawaja, A.P.; Chan, M.P.; Broadway, D.C.; Peto, T.; Tufail, A.; Luben, R.; Hayat, S.; Bhaniani, A.; Wareham, N.J.; et al. Cross sectional and longitudinal associations between cardiovascular risk factors and age related macular degeneration in the EPIC-Norfolk Eye Study. PLoS ONE 2015, 10, e0132565. [Google Scholar] [CrossRef]
- Saunier, V.; Merle, B.M.J.; Delyfer, M.N.; Cougnard-Grégoire, A.; Rougier, M.B.; Amouyel, P.; Lambert, J.C.; Dartigues, J.F.; Korobelnik, J.F.; Delcourt, C. Incidence of and Risk Factors Associated with Age-Related Macular Degeneration: Four-Year Follow-up From the ALIENOR Study. JAMA Ophthalmol. 2018, 136, 473–481. [Google Scholar] [CrossRef]
- Chen, Y.J.; Yeung, L.; Sun, C.C.; Huang, C.C.; Chen, K.S.; Lu, Y.H. Age-related macular degeneration in chronic kidney disease: A meta-analysis of observational studies. Am. J. Nephrol. 2018, 48, 278–291. [Google Scholar] [CrossRef]
- Gopinath, B.; Liew, G.; Kifley, A.; Mitchell, P. Thyroid dysfunction and ten-year incidence of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5273–5277. [Google Scholar] [CrossRef]
- Chaker, L.; Buitendijk, G.H.; Dehghan, A.; Medici, M.; Hofman, A.; Vingerling, J.R.; Franco, O.H.; Klaver, C.C.; Peeters, R.P. Thyroid function and age-related macular degeneration: A prospective population-based cohort study–The Rotterdam Study. BMC Med. 2015, 13, 94. [Google Scholar] [CrossRef]
- Tsai, C.C.; Kao, S.C.; Cheng, C.Y.; Kau, H.C.; Hsu, W.M.; Lee, C.F.; Wei, Y.H. Oxidative stress change by systemic corticosteroid treatment among patients having active graves ophthalmopathy. Arch. Ophthalmol. 2007, 125, 1652–1656. [Google Scholar] [CrossRef]
- Duncan, K.G.; Bailey, K.R.; Baxter, J.D.; Schwartz, D.M. The human fetal retinal pigment epithelium: A target tissue for thyroid hormones. Ophthalmic Res. 1999, 31, 399–406. [Google Scholar] [CrossRef]
- Ma, H.W.; Thapa, A.; Morris, L.; Redmond, T.M.; Baehr, W.; Ding, X.Q. Suppressing thyroid hormone signaling preserves cone photoreceptors in mouse models of retinal degeneration. Proc. Nat. Acad. Sci. USA 2014, 111, 3602–3607. [Google Scholar] [CrossRef]
- Jonasson, F.; Fisher, D.E.; Eiriksdottir, G.; Sigurdsson, S.; Klein, R.; Launer, L.J.; Harris, T.; Gudnason, V.; Cotch, M.F. Five-year incidence, progression, and risk factors for age-related macular degeneration: The age, gene/environment susceptibility study. Ophthalmology 2014, 121, 1766–1772. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.; Al-Shabrawey, M.; Caldwell, R.W.; Caldwell, R.B. Inflammation and diabetic retinal microvascular complications. J. Cardiovasc. Dis. Res. 2011, 2, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Ratnayaka, J.A.; Serpell, L.C.; Lotery, A.J. Dementia of the eye: The role of amyloid beta in retinal degeneration. Eye 2015, 29, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Bojanowski, C.M.; Shen, D.; Chew, E.Y.; Ning, B.; Csaky, K.G.; Green, W.R.; Chan, C.C.; Tuo, J. An apolipoprotein E variant may protect against age-related macular degeneration through cytokine regulation. Environ. Mol. Mutagen. 2006, 47, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Keenan, T.D.; Goldacre, R.; Goldacre, M.J. Associations between age-related macular degeneration, Alzheimer disease, and dementia: Record linkage study of hospital admissions. JAMA Ophthalmol. 2014, 132, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Brilliant, M.H.; Vaziri, K.; Connor, T.B., Jr.; Schwartz, S.G.; Carroll, J.J.; McCarty, C.A.; Schrodi, S.J.; Hebbring, S.J.; Kishor, K.S.; Flynn, H.W., Jr.; et al. Mining Retrospective Data for Virtual Prospective Drug Repurposing: L-DOPA and Age-related Macular Degeneration. Am. J. Med. 2016, 129, 292–298. [Google Scholar] [CrossRef]
- Locke, C.J.; Congrove, N.R.; Stamer, W.D.; Bowen, T.J.; Stamer, W.D.; McKay, B.S. Controlled exosome release from the retinal pigment epithelium in situ. Exp. Eye Res. 2014, 129, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Cascella, R.; Ragazzo, M.; Strafella, C.; Missiroli, F.; Borgiani, P.; Angelucci, F.; Marsella, L.T.; Cusumano, A.; Novelli, G.; Ricci, F.; et al. Age-related macular degeneration: Insights into inflammatory genes. J. Ophthalmol. 2014, 2014, 582842. [Google Scholar] [CrossRef] [PubMed]
- Meyers, S.M.; Zachary, A.A. Monozygotic twins with age-related macular degeneration. Arch. Ophthalmol. 1988, 106, 651–653. [Google Scholar] [CrossRef] [PubMed]
- Meyers, S.M. A twin study on age-related macular degeneration. Trans Am. Ophthalmol. Soc. 1994, 92, 775–843. [Google Scholar] [CrossRef]
- Gottfredsdottir, M.S.; Sverrisson, T.; Musch, E.; Stefansson, D.C. Age related macular degeneration in monozygotic twins and theirspouses in Iceland. Acta Ophthalmol. Scand. 1999, 77, 422–425. [Google Scholar] [CrossRef]
- Klein, B.E.K.; Klein, R.; Lee, K.E.; Moore, E.L.; Danforth, L. Risk of incident age-related eye diseases in people with an affected sibling: The Beaver Dam Eye Study. Am. J. Epidemiol. 2001, 154, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Haines, J.H.; Hauser, M.A.; Schmidt, S. Complement factor H variant increases the risk of age-related macular degeneration. Science 2005, 308, 419–421. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, U.; McKay, G.J.; de Jong, P.T.; Rahu, M.; Seland, J.; Soubrane, G.; Tomazzoli, L.; Topouzis, F.; Vingerling, J.R.; Vioque, J.; et al. ARMS2 increases the risk of early and late age-related macular degeneration in the European Eye Study. Ophthalmology 2013, 120, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Kenney, M.C.; Hertzog, D.; Chak, G.; Atilano, S.R.; Khatibi, N.; Soe, K.; Nobe, A.; Yang, E.; Chwa, M.; Zhu, F.; et al. Mitochondrial DNA haplogroups confer differences in risk for age-related macular degeneration: A case control study. BMC Med. Genet. 2013, 14, 4. [Google Scholar] [CrossRef] [PubMed]
- Nazari Khanamiri, H.; Ghasemi Falavarjani, K.; Sanati, M.H.; Aryan, H.; Irani, A.; Hashemi, M.; Modarres, M.; Parvaresh, M.M.; Nikeghbali, A. Complement Factor H Y402H and LOC387715 A69S Polymorphisms in Association with Age-Related Macular Degeneration in Iran. J. Ophthalm. Vis. Res. 2014, 9, 181–187. [Google Scholar]
- Francis, P.J.; Schultz, D.W.; Hamon, S.; Ott, J.; Weleber, R.G.; Klein, M.L. Haplotypes in the complement factor H (CFH) gene: Associations with drusen and advanced age-related macular degeneration. PLoS ONE 2007, 2, e1197. [Google Scholar] [CrossRef] [PubMed]
- Thakkinstian, A.; Han, P.; McEvoy, M.; Smith, W.; Hoh, J.; Magnusson, K.; Zhang, K.; Attia, J. Systematic review and meta-analysis of the association between complement factor H Y402H polymorphisms and age-related macular degeneration. Human Mol. Genet. 2006, 15, 2784–2790. [Google Scholar] [CrossRef] [PubMed]
- Seitsonen, S.P.; Onkamo, P.; Peng, G.; Xiong, M.; Tommila, P.V.; Ranta, P.H.; Holopainen, J.M.; Moilanen, J.A.; Palosaari, T.; Kaarniranta, K.; et al. Multifactor Effects and Evidence of Potential Interaction between Complement Factor H Y402H and LOC387715 A69S in Age-Related Macular Degeneration. PLoS ONE 2008, 3, e3833. [Google Scholar] [CrossRef]
- Cao, S.; Ko, A.; Partanen, M.; Pakzad-Vaezi, K.; Merkur, A.B.; Albiani, D.A.; Kirker, A.W.; Wang, A.; Cui, J.Z.; Forooghian, F.; et al. Relationship between systemic cytokines and complement factor H Y402H polymorphism in patients with dry age-related macular degeneration. Am. J. Ophthalmol. 2013, 156, 1176–1183. [Google Scholar] [CrossRef]
- Wang, J.C.; Cao, S.; Wang, A.; To, E.; Law, G.; Gao, J.; Zhang, D.; Cui, J.Z.; Matsubara, J.A. CFH Y402H polymorphism is associated with elevated vitreal GM-CSF and choroidal macrophages in the postmortem human eye. Mol. Vis. 2015, 21, 264–272. [Google Scholar]
- Fang, K.; Gao, P.; Tian, J.; Qin, X.; Yu, W.; Li, J.; Chen, Q.; Huang, L.; Chen, D.; Hu, Y.; et al. Joint Effect of CFH and ARMS2/HTRA1 Polymorphisms on Neovascular Age-Related Macular Degeneration in Chinese Population. J. Ophthalmol. 2015, 2015, 821918. [Google Scholar] [CrossRef]
- Meyers, K.J.; Liu, Z.; Millen, A.E.; Iyengar, S.K.; Blodi, B.A.; Johnson, E.; Snodderly, D.M.; Klein, M.L.; Gehrs, K.M.; Tinker, L.; et al. Joint Associations of Diet, Lifestyle, and Genes with Age-Related Macular Degeneration. Ophthalmology 2015, 122, 2286–2294. [Google Scholar] [CrossRef]
- Hughes, A.E.; Orr, N.; Esfandiary, H.; Diaz-Torres, M.; Goodship, T.; Chakravarthy, U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat. Genet. 2006, 38, 1173–1177. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.; McKeigue, P.M.; Skerka, C.; Hayward, C.; Rudan, I.; Vitart, V.; Polasek, O.; Armbrecht, A.M.; Yates, J.R.; Vatavuk, Z.; et al. Genetic influences on plasma CFH and CFHR1 concentrations and their role in susceptibility to age-related macular degeneration. Human Mol. Genet. 2013, 22, 4857–4869. [Google Scholar] [CrossRef]
- Emilsson, V.; Gudmundsson, E.F.; Jonmundsson, T.; Jonsson, B.G.; Twarog, M.; Gudmundsdottir, V.; Li, Z.; Finkel, N.; Poor, S.; Liu, X.; et al. A proteogenomic signature of age-related macular degeneration in blood. Nat. Commun. 2022, 13, 3401. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Lauer, N.; Hartmann, A.; Stippa, S.; Keilhauer, C.N.; Oppermann, M.; Pandey, M.K.; Köhl, J.; Zipfel, P.F.; Weber, B.H.; et al. An imbalance of human complement regulatory proteins CFHR1, CFHR3 and factor H influences risk for age-related macular degeneration (AMD). Human Mol. Genet. 2010, 19, 4694–4704. [Google Scholar] [CrossRef] [PubMed]
- Hageman, G.S.; Hancox, L.S.; Taiber, A.J.; Gehrs, K.M.; Anderson, D.H.; Johnson, L.V.; Radeke, M.J.; Kavanagh, D.; Richards, A.; Atkinson, J.; et al. AMD Clinical Study Group. Extended haplotypes in the complement factor H (CFH) and CFH-related (CFHR) family of genes protect against age-related macular degeneration: Characterization, ethnic distribution and evolutionary implications. Ann. Med. 2006, 38, 592–604. [Google Scholar] [CrossRef]
- Sawitzke, J.; Im, K.M.; Kostiha, B.; Dean, M.; Gold, B. Association Assessment of Copy Number Polymorphism and Risk of Age-Related Macular Degeneration. Ophthalmology 2011, 118, 2442–2446. [Google Scholar] [CrossRef]
- Kato, Y.; Oguchi, Y.; Omori, T.; Kasai, A.; Ogasawara, M.; Sugano, Y.; Itagaki, K.; Ojima, A.; Ishida, Y.; Machida, T.; et al. Age-Related Maculopathy Susceptibility 2 and Complement Factor H Polymorphism and Intraocular Complement Activation in Neovascular Age-Related Macular Degeneration. Ophthalmol. Sci. 2022, 2, 100167. [Google Scholar] [CrossRef]
- He, F.; Li, X.; Cai, S.; Lu, L.; Zhang, T.; Yang, M.; Fan, N.; Wang, X.; Liu, X. Polymorphism rs11200638 enhanced HtrA1 responsiveness and expression are associated with age-related macular degeneration. Eye 2022, 36, 1631–1638. [Google Scholar] [CrossRef]
- Anand, A.; Sharma, K.; Sharma, S.K.; Singh, R.; Sharma, N.K.; Prasad, K. AMD genetics in India: The missing links. Front. Aging Neurosci. 2016, 8, 115. [Google Scholar] [CrossRef] [PubMed]
- Peter, I.; Huggins, G.S.; Ordovas, J.M.; Haan, M.; Seddon, J.M. Evaluation of new and established age-related macular degeneration susceptibility genes in the Women’s Health Initiative Sight Exam (WHI-SE) Study. Am. J. Ophthalmol. 2011, 152, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Ristau, T.; Paun, C.; Ersoy, L.; Hahn, M.; Lechanteur, Y.; Hoyng, C.; de Jong, E.K.; Daha, M.R.; Kirchhof, B.; den Hollander, A.I.; et al. Impact of the common genetic associations of age-related macular degeneration upon systemic complement component C3d levels. PLoS ONE 2014, 9, e93459. [Google Scholar] [CrossRef] [PubMed]
- Zerbib, J.; Richard, F.; Puche, N.; Leveziel, N.; Cohen, S.Y.; Korobelnik, J.F.; Sahel, J.; Munnich, A.; Kaplan, J.; Rozet, J.M.; et al. R102G polymorphism of the C3 gene associated with exudative age-related macular degeneration in a French population. Mol. Vis. 2010, 16, 1324–1330. [Google Scholar] [PubMed]
- Despriet, D.D.; van Duijn, C.M.; Oostra, B.A. Complement component C3 and risk of age-related macular degeneration. Ophthalmology 2009, 116, 474–480.e2. [Google Scholar] [CrossRef] [PubMed]
- Neto, J.M.; Viturino, M.G.; Ananina, G.; Bajano, F.F.; Costa, S.M.D.S.; Roque, A.B.; Borges, G.F.; Franchi, R.; Rim, P.H.; Medina, F.M.; et al. Association of genetic variants rs641153 (CFB), rs2230199 (C3), and rs1410996 (CFH) with age-related macular degeneration in a Brazilian population. Exp. Biol. Med. 2021, 246, 2290–2296. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Arima, K.; Noguchi, Y.; Yamanashi, H.; Kawashiri, S.Y.; Nobusue, K.; Nonaka, F.; Aoyagi, K.; Nagata, Y.; Maeda, T. Vascular endothelial growth factor (VEGF) polymorphism rs3025039 and atherosclerosis among older with hypertension. Sci. Rep. 2022, 12, 5564. [Google Scholar] [CrossRef] [PubMed]
- Villani, L.; Carolei, A.; Rosti, V.; Massa, M.; Campanelli, R.; Catarsi, P.; Abbà, C.; Gale, R.P.; Barosi, G. Clinical Relevance of VEGFA (rs3025039) +936 C>T Polymorphism in Primary Myelofibrosis: Susceptibility, Clinical Co-Variates, and Outcomes. Genes 2021, 12, 1271. [Google Scholar] [CrossRef]
- Lin, J.M.; Wan, L.; Tsai, Y.Y.; Lin, H.J.; Tsai, Y.; Lee, C.C.; Tsai, C.H.; Tseng, S.H.; Tsai, F.J. Vascular endothelial growth factor gene polymorphisms in age-related macular degeneration. Am. J. Ophthalmol. 2008, 145, 1045–1051. [Google Scholar] [CrossRef]
- Wang, D.; Zhou, J.; Hou, X.; Nguyen, D.H.; Cao, G.; Li, G.; Qiu, G.; Zhang, K.; Zhang, M.; Su, Z. CETP Gene may be Associated with Advanced Age-Related Macular Degeneration in the Chinese Population. Ophthalmic Genet. 2015, 36, 303–308. [Google Scholar] [CrossRef]
- Hughes, A.E.; Mullan, G.M.; Bradley, D.T. Complement factor B polymorphism 32W protects against age-related macular degeneration. Mol. Vis. 2011, 20, 983–988. [Google Scholar]
- Kopplin, L.J.; Igo, R.P., Jr.; Wang, Y.; Sivakumaran, T.A.; Hagstrom, S.A.; Peachey, N.S.; Francis, P.J.; Klein, M.L.; SanGiovanni, J.P.; Chew, E.Y.; et al. Genome-wide association identifies SKIV2L and MYRIP as protective factors for age-related macular degeneration. Genes Immunol. 2010, 11, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Shi, Y.; Qu, C.; Zhao, P.; Liu, X.; Gong, B.; Ma, S.; Zhou, Y.; Zhang, Q.; Fei, P.; et al. A genetic variant in the SKIV2L gene is significantly associated with age-related macular degeneration in a Han Chinese population. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2911–2917. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, R.; Rosner, B.; Seddon, J.M. Serum Lipid Biomarkers and Hepatic Lipase Gene Associations with Age-Related Macular Degeneration. Ophthalmology 2010, 117, 1989–1995. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Reynolds, R.; Fagerness, J.; Rosner, B.; Daly, M.J.; Seddon, J.M. Association of variants in the LIPC and ABCA1 genes with intermediate and large drusen and advanced age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4663–4670. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; Reynolds, R.; Rosne, B. Associations of smoking, body mass index, dietary lutein, and the LIPC gene variant rs10468017 with advanced age-related macular degeneration. Mol. Vis. 2010, 16, 2412–2424. [Google Scholar] [PubMed]
- Neale, B.M.; Fagerness, J.; Reynolds, R.; Sobrin, L.; Parker, M.; Raychaudhuri, S.; Tan, P.L.; Oh, E.C.; Merriam, J.E.; Souied, E.; et al. Genome-wide association study of advanced age-related macular degeneration identifies a role of the hepatic lipase gene (LIPC). Proc. Nat. Acad. Sci. USA 2010, 107, 7395–7400. [Google Scholar] [CrossRef] [PubMed]
- Merle, B.M.J.; Maubaret, C.; Korobelnik, J.F.; Delyfer, M.N.; Rougier, M.B. Association of HDL-Related Loci with Age-Related Macular Degeneration and Plasma Lutein and Zeaxanthin: The Alienor Study. PLoS ONE 2013, 8, e79848. [Google Scholar] [CrossRef] [PubMed]
- Restrepo, N.A.; Spencer, K.L.; Goodloe, R.; Garrett, T.A.; Heiss, G.; Bůžková, P.; Jorgensen, N.; Jensen, R.A.; Matise, T.C.; Hindorff, L.A.; et al. Genetic Determinants of Age-Related Macular Degeneration in Diverse Populations from the PAGE Study. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6839–6850. [Google Scholar] [CrossRef]
- Tian, J.; Yu, W.; Qin, X.; Fang, K.; Chen, Q.; Hou, J.; Li, J.; Chen, D.; Hu, Y.; Li, X. Association of genetic polymorphisms and age-related macular degeneration in Chinese population. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4262–4269. [Google Scholar] [CrossRef]
- Chen, W.; Stambolian, D.; Edwards, A.O.; Branham, K.E.; Othman, M.; Jakobsdottir, J.; Tosakulwong, N.; Pericak-Vance, M.A.; Campochiaro, P.A.; Klein, M.L.; et al. Genetic variants near TIMP3 and high-density lipoprotein-associated loci influence susceptibility to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 7401–7406. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, V.; Leung, H.T.; Plagnol, V.; Bunce, C.; Khan, J.C.; Shahid, H.; Moore, A.T.; Harding, S.P.; Bishop, P.N.; Hayward, C.; et al. French AMD Investigators; Cree A.J., Gibson J., Ennis S., Lotery A.J., Wright A.F., Clayton D.G., Yates J.R. Genome-wide association study of age-related macular degeneration identifies associated variants in the TNXB-FKBPL-NOTCH4 region of chromosome 6p21.3. Human Mol. Genet. 2012, 21, 4138–4150. [Google Scholar] [CrossRef] [PubMed]
- Ghorbani, R.; Rasouli, M.; Sefat, F.; Heidari Keshel, S. Pathogenesis of Common Ocular Diseases: Emerging Trends in Extracellular Matrix Remodeling. Semin. Ophthalmol. 2023, 39, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Fagerness, J.A.; Maller, J.B.; Neale, B.M.; Reynolds, R.C.; Daly, M.J.; Seddon, J.M. Variation near complement factor I is associated with risk of advanced AMD. Eur. J. Human Genet. 2009, 17, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhao, H.S.; Li, L. Association between complement factor I gene polymorphisms and the risk of age-related macular degeneration: A Meta-analysis of literature. Int. J. Ophthalmol. 2016, 9, 298–305. [Google Scholar] [PubMed]
- Goverdhan, S.V.; Ennis, S.; Hannan, S.R.; Madhusudhana, K.C.; Cree, A.J.; Luff, A.J.; Lotery, A.J. Interleukin-8 promoter polymorphism -251A/T is a risk factor for age-related macular degeneration. Br. J. Ophthalmol. 2008, 92, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.Y.; Lin, J.M.; Wan, L.; Lin, H.J.; Tsai, Y.; Lee, C.C.; Tsai, C.H.; Tsai, F.J.; Tseng, S.H. Interleukin gene polymorphisms in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2008, 49, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Ricci, F.; Staurenghi, G.; Lepre, T.; Missiroli, F.; Zampatti, S.; Cascella, R.; Borgiani, P.; Marsella, L.T.; Eandi, C.M.; Cusumano, A.; et al. Haplotypes in IL-8 gene are associated to age-related macular degeneration: A case-control study. PLoS ONE 2013, 8, e66978. [Google Scholar] [CrossRef]
- Oz, O.; Aras Ates, N.; Tamer, L.; Yildirim, O.; Adigüzel, U. Glutathione S-transferase M1, T1, and P1 gene polymorphism in exudative age-related macular degeneration: A preliminary report. Eur. J. Ophthalmol. 2006, 16, 105–110. [Google Scholar] [CrossRef]
- Haines, J.L.; Schnetz-Boutaud, N.; Schmidt, S.; Scott, W.K.; Agarwal, A.; Postel, E.A.; Olson, L.; Kenealy, S.J.; Hauser, M.; Gilbert, J.R.; et al. Functional Candidate Genes in Age-Related Macular Degeneration: Significant Association with VEGF, VLDLR, and LRP6, IOVS. Investig. Ophthalmol. Vis. Sci. 2006, 47, 329–335. [Google Scholar] [CrossRef]
- Güven, M.; Görgün, E.; Ünal, M.; Yenerel, M.; Batar, B.; Küçümen, B.; Dinç, U.A.; Güven, G.S.; Ulus, T.; Yüksel, A. Glutathione S-transferase M1, GSTT1 and GSTP1 genetic polymorphisms and the risk of age-related macular degeneration. Ophthalm. Res. 2011, 46, 31–37. [Google Scholar] [CrossRef]
- Othman, H.; Gholampour, A.R.; Saadat, I.; Farvardin-Jahromoi, M.; Saadat, M. Age-related macular degeneration and genetic polymorphisms of glutathione S-transferases M1 (GSTM1) and T1 (GSTT1). Mol. Biol. Rep. 2012, 39, 3299–3303. [Google Scholar] [CrossRef]
- Chen, X.; Luo, Y. Association of GSTM1, GSTT1, and GSTP1 Ile105Val polymorphisms with risk of age-related macular degeneration: A meta-analysis. Ophthalmic Genet. 2022, 43, 615–621. [Google Scholar] [CrossRef]
- Gu, H.; Sun, E.; Cui, L.; Yang, X.; Lim, A.; Xu, J.; Snellingen, T.; Liu, X.; Wang, N.; Liu, N. Association of glutathione S-transferase pi isoform single-nucleotide polymorphisms with exudative age-related macular degeneration in a Chinese population. Retina 2012, 32, 1967–1972. [Google Scholar] [CrossRef]
- Zhan, X.; Larson, D.E.; Wang, C.; Koboldt, D.C.; Sergeev, Y.V.; Fulton, R.S.; Fulton, L.L.; Fronick, C.C.; Branham, K.E.; Bragg-Gresham, J.; et al. Identification of a rare coding variant in complement 3 associated with age-related macular degeneration. Nat. Genet. 2013, 45, 1375–1379. [Google Scholar] [CrossRef]
- Raychaudhuri, S.; Iartchouk, O.; Chin, K.; Tan, P.L.; Tai, A.K.; Ripke, S.; Gowrisankar, S.; Vemuri, S.; Montgomery, K.; Yu, Y.; et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat. Genet. 2011, 43, 1232–1236. [Google Scholar] [CrossRef]
- Told, R.; Palkovits, S.; Boltz, A.; Schmidl, D.; Napora, K.J.; Werkmeister, R.M.; Haslacher, H.; Frantal, S.; Popa-Cherecheanu, A.; Schmetterer, L.; et al. Flicker-induced retinal vasodilatation is not dependent on complement factor H polymorphism in healthy young subjects. Acta Ophthalmol. 2014, 92, e540–e545. [Google Scholar] [CrossRef]
- Ersoy, L.; Schick, T.; de Graft, D.; Felsch, M.; Hoyng, C.B.; den Hollander, A.I.; Kirchhof, B.; Fauser, S.; Liakopoulos, S. Extramacular drusen are highly associated with age-related macular degeneration, but not with CFH and ARMS2 genotypes. Br. J. Ophthalmol. 2015, 100, 1047–1051. [Google Scholar] [CrossRef]
- Cruz-González, F.; Cieza-Borrella, C.; López Valverde, G.; Lorenzo-Pérez, R.; Hernández-Galilea, E.; González-Sarmiento, R. CFH (rs1410996), HTRA1 (rs112000638) and ARMS2 (rs10490923) gene polymorphisms are associated with AMD risk in Spanish patients. Ophthalm. Genet. 2014, 35, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Brión, M.; Sanchez-Salorio, M.; Cortón, M.; de la Fuente, M.; Pazos, B.; Othman, M.; Swaroop, A.; Abecasis, G.; Sobrino, B.; Carracedo, A. Genetic association study of age-related macular degeneration in the Spanish population. Acta Ophthalmol. 2011, 89, e12–e22. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Tao, Q.; Chen, W.; Wang, Z.; Song, Y.; Sheng, S.; Li, P.; Zhou, J. Association between Polymorphisms of Complement Pathway Genes and Age-Related Macular Degeneration in a Chinese Population. Investig. Ophthalmol Vis. Sci. 2013, 54, 170–174. [Google Scholar] [CrossRef]
- Babanejad, M.; Moein, H.; Akbari, M.R. Investigating the CFH Gene Polymorphisms as a Risk Factor for Age-related Macular Degeneration in an Iranian Population. Ophthalmol. Genet. 2015, 21, 1–6. [Google Scholar] [CrossRef]
- García, M.; Álvarez, L.; Nogacka, A.M.; González-Iglesias, H.; Escribano, J.; Fernández-Vega, B.; Fernández-Vega, Á.; Fernández-Vega, L.; Coca-Prados, M. CFH polymorphisms in a Northern Spanish population with neovascular and dry forms of age-related macular degeneration. Acta Ophthalmol. 2015, 93, e658–e666. [Google Scholar] [CrossRef]
- Hyttinen, J.M.T.; Błasiak, J.; Kaarniranta, K. Non-Coding RNAs Regulating Mitochondrial Functions and the Oxidative Stress Response as Putative Targets against Age-Related Macular Degeneration (AMD). Int. J. Mol. Sci. 2023, 24, 2636. [Google Scholar] [CrossRef]
- Seddon, J.M.; Francis, P.J.; George, S.; Schultz, D.W.; Rosner, B.; Klein, M.L. Association of CFH Y402H and LOC387715 A69S with progression of age-related macular degeneration. JAMA 2007, 297, 1793–1800. [Google Scholar] [CrossRef]
- Tuo, J.; Ross, R.J.; Reed, G.F.; Yan, Q.; Wang, J.J.; Bojanowski, C.M.; Chew, E.Y.; Feng, X.; Olsen, T.W.; Ferris, F.L., 3rd; et al. The HtrA1 promotor polymorphism, smoking, and age-related macular degeneration in multiple case-control samples. Ophthalmology 2008, 115, 1891–1898. [Google Scholar] [CrossRef]
- Contreras, A.V.; Zenteno, J.C.; Fernández-López, J.C.; Rodríguez-Corona, U.; Falfán-Valencia, R.; Sebastian, L.; Morales, F.; Ochoa-Contreras, D.; Carnevale, A.; Silva-Zolezzi, I. CFH haplotypes and ARMS2, C2, C3, and CFB alleles show association with susceptibility to age-related macular degeneration in Mexicans. Mol. Vis. 2014, 20, 105–116. [Google Scholar]
- Botto, M.; Fong, K.Y.; So, A.K.; Koch, C.; Walport, M.J. Molecular basis of polymorphisms of human complement component C3. J. Exp. Med. 1990, 172, 1011–1017. [Google Scholar] [CrossRef]
- Thakkinstian, A.; McKay, G.J.; McEvoy, M.; Chakravarthy, U.; Chakrabarti, S.; Silvestri, G.; Kaur, I.; Li, X.; Attia, J. Systematic review and meta-analysis of the association between complement component 3 and age-related macular degeneration: A HuGE review and meta-analysis. Am. J. Epidemiol. 2011, 173, 1365–1379. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, S.; Kondo, N.; Miki, A.; Matsumiya, W.; Kusuhara, S.; Tsukahara, Y.; Honda, S.; Negi, A. A Common Complement C3 Variant Is Associated with Protection against Wet Age-Related Macular Degeneration in a Japanese Population. PLoS ONE 2011, 6, e28847. [Google Scholar] [CrossRef] [PubMed]
- Penn, J.S.; Madanet, A.; Caldwell, R.B.; Bartoli, M.; Caldwell, R.W.; Hartnett, M.E. Vascular endothelial growth factor in eye disease. Progr. Retin. Eye Res. 2008, 27, 331–371. [Google Scholar] [CrossRef] [PubMed]
- Janik-Papis, K.; Zaras, M.; Krzyżanowska, A.; Woźniak, K.; Błasiak, J.; Szaflik, J.; Szaflik, J.P. Association between vascular endothelial growth factor gene polymorphisms and age-related macular degeneration in a Polish population. Exp. Mol. Pathol. 2009, 87, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Bhangale, T.R.; Fagerness, J.; Ripke, S.; Thorleifsson, G.; Tan, P.L.; Souied, E.H.; Richardson, A.J.; Merriam, J.E.; Buitendijk, G.H.; et al. Common variants near FRK/COL10A1 and VEGFA are associated with advanced age-related macular degeneration. Human Mol. Genet. 2011, 20, 3699–3709. [Google Scholar] [CrossRef]
- Ambreen, F.; Ismail, M.; Qureshi, I.Z. Association of gene polymorphism with serum levels of inflammatory and angiogenic factors in Pakistani patients with age-related macular degeneration. Mol. Vis. 2015, 21, 985–999. [Google Scholar]
- Kaur, I.; Katta, S.; Reddy, R.K.; Narayanan, R.; Mathai, A.; Majji, A.B.; Chakrabarti, S. The involvement of complement factor B and complement component C2 in an Indian cohort with age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2010, 51, 59–63. [Google Scholar] [CrossRef]
- Mantel, I.; Ambresin, A.; Moetteli, L.; Droz, I.; Roduit, R.; Munier, F.L.; Schorderet, D.F. Complement factor B polymorphism and the phenotype of early age-related macular degeneration. Ophthalm. Genet. 2014, 35, 12–17. [Google Scholar] [CrossRef]
- Nasr, A.S.; Sami, R.M.; Ibrahim, N.Y.; Darwish, D.O. Glutathione S transferase (GSTP 1, GSTM 1, and GSTT 1) gene polymorphisms in Egyptian patients with acute myeloid leukemia. Indian J. Cancer. 2015, 52, 490–495. [Google Scholar] [CrossRef]
- Grassmann, F.; Friedrich, U.; Fauser, S.; Schick, T.; Milenkovic, A.; Schulz, H.L.; von Strachwitz, C.N.; Bettecken, T.; Lichtner, P.; Meitinger, T.; et al. A Candidate Gene Association Study Identifies DAPL1 as a Female-Specific Susceptibility Locus for Age-Related Macular Degeneration (AMD). Neuromol. Med. 2015, 17, 111–120. [Google Scholar] [CrossRef]
- Grizzard, S.W.; Arnett, D.; Haag, S.L. Twin study of age-related macular degeneration. Ophthalm. Epidemiol. 2003, 10, 315–322. [Google Scholar] [CrossRef]
- Arakawa, S.; Takahashi, A.; Ashikawa, K.; Hosono, N.; Aoi, T.; Yasuda, M.; Oshima, Y.; Yoshida, S.; Enaida, H.; Tsuchihashi, T.; et al. Genome-wide association study identifies two susceptibility loci for exudative age-related macular degeneration in the Japanese population. Nat. Genet. 2011, 43, 1001–1004. [Google Scholar] [CrossRef] [PubMed]
- Zerbib, J.; Seddon, J.M.; Richard, F.; Reynolds, R.; Leveziel, N.; Benlian, P.; Borel, P.; Feingold, J.; Munnich, A.; Soubrane, G.; et al. rs5888 variant of SCARB1 gene is a possible susceptibility factor for age-related macular degeneration. PLoS ONE 2009, 4, e7341. [Google Scholar] [CrossRef]
- Sheu, S.J.; Ger, L.P.; Kuo, N.W.; Liu, N.C.; Wu, T.T.; Lin, M.C. Association of IL-4 gene polymorphism and age-related macular degeneration in Taiwanese adults. Taiwan J. Ophthalmol. 2012, 2, 51–55. [Google Scholar] [CrossRef]
- Terluk, M.R.; Kapphahn, R.J.; Soukup, L.M.; Gong, H.; Gallardo, C.; Montezuma, S.R.; Ferrington, D.A. Investigating mitochondria as a target for treating age-related macular degeneration. J. Neurosci. 2015, 35, 7304–7311. [Google Scholar] [CrossRef] [PubMed]
- Restrepo, N.A.; Mitchell, S.L.; Goodloe, R.J.; Murdock, D.G.; Haines, J.L.; Crawford, D.C. Mitochondrial variation and the risk of age-related macular degeneration across diverse populations. Pac. Symp. Biocomput. 2015, 2015, 243–254. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Risk Factor | Phenotypic Risk Factors |
| Drusen | Predictive potential in context of AMD |
| Deposits of extracellular debris are located between Bruch’s membrane and retinal pigment epithelium (RPE). Different types of drusen and its location, volume, measured area, or total number of drusen may serve as predictive factors for AMD progression. Small drusen may be connected with a low probability (0.4%) of developing late AMD within five years in patients older than 55 years. Medium or large drusen with or without accompanying pigmentary abnormalities may inform about early or intermediate AMD stage. Individuals with medium drusen are burdened with probability between 2% and 20% of developing late AMD within five years, while large drusen increases such probability to between 13% and 47%. | |
| Different types of drusen and possible types of AMD | |
| Individuals with calcified drusen have a probability of 26% of developing geographic atrophy (GA, subtype of late AMD) within 5 years. The presence of reticular pseudo-drusen is highly prevalent in fellow-eyes of individuals with unilateral neovascular AMD. Progression rate in cases of neovascular AMD is estimated at the level of 31% in a period of two years. Finally, cuticular drusen may be connected with variants of CFH gene, and may also be helpful in the prediction of the development of nAMD and GA (progression probabilities of 8.7–12.5% and 25–28% in a period of 5 years). | |
| Alternations of the retinal pigment epithelium | Pigmentary changes and hyperreflective foci in context of AMD risk |
| The presence of pigmentary changes in addition to drusen is connected with dramatic increase of risk of developing late AMD (probability of 47% within 5 years in cases of large drusen and accompanying pigmentary changes). Hyperreflective foci may be connected with progression rates of both nAMD (47% of eyes with hyperreflective foci develop this form of AMD after two years) and GA (approximately 50% of eyes with hyperreflective foci develop GA within a period of 28 months). | |
| Types of pigment epithelial detachment and AMD | |
| Fibrovascular and serious types are both connected with nAMD. Drusenoid type may be engaged in the progression of different kinds of AMD, as well as being connected with the development of pigmentary alternations and calcified drusen. In cases of GA, features like lesion size, number, location (increased rates of GA growth in eyes with multifocal and extrafoveal lesions compared to unifocal and foveal lesions) and shape (lesions with irregular shape are connected with a faster growth rate compared to lesions with a circular shape) are important. | |
| Vascular and other features | Choroid vascular structures alternations and flow anomalies in the context of AMD |
| There is a possibility that choroid vascular structures deplete in eyes with early AMD, nAMD, and GA, as well as that irregular choroidal vessels may be a predictive factor for the development of nAMD and GA. Similarly, flow abnormalities in the choriocapillaris surrounding the atrophic lesions may be connected with the escalation in GA growth rate. Moreover, in cases of eyes with a quiescent choroidal neovascularization, there is considerably higher risk of becoming exudative (15–18) compared to eyes lacking a precursor quiescent choroidal neovascularization. | |
| Outer retinal tubulations and dynamics of AMD | |
| Outer retinal tubulations can be present in areas of fibrosis after the development of exudative nAMD or at the border of GA. In cases of individuals with GA, the localization of outer retinal tubulations at the border of GA lesion promote slower rate of GA growth compared to individuals without outer retinal tubulations. | |
| Risk factor | Demographic and Environmental Risk Factors |
| Age | The prevalence of early AMD increases from 3.5% in individuals aged 55–59 years up to 17.6% in individuals 85 years and older. Furthermore, the prevalence rates increase from 0.1% to 9.8% respectively, as well as for late AMD. |
| Sex | There is a tendency of a higher progression rate to early AMD and late AMD (mainly nAMD) in cases of female sex. |
| Smoking | Toxic compounds contained in cigarette smoke may trigger the formation of retinal oxidative stress, vascular changes in the choroidal vessels as well as the inflammation in RPE cells. Thus, smoking is connected with a two- to four-fold increased risk for any form of AMD. Smoking may also be associated with escalation in GA growth. Quitting smoking generally mitigates the risk of developing AMD; however, risk probabilities appear to be comparable to that of non-smoking individuals after as long as 20 years cessation of smoking. |
| Body composition | Higher body mass index may be connected with greater probability of AMD developing. Especially obese individuals (body mass index BMI > 30) are in danger of developing late AMD compared with individuals with normal weight (BMI 20–25). |
| Diet | The number of vegetables and fruits in a diet is considered to be a protective factor in context of AMD progression, because these components of diet are rich in such antioxidants as carotenoids and vitamins. Similarly, fish consumption (rich in protective fatty acids) tends to mitigate the risk of AMD progression. |
| Physical activity | Physical activity tends to be connected with lower odds of both late and early AMD, because regular exercise is considered to be positively associated with activity of antioxidative enzymes. |
| Education | Higher education level generally acts as alleviating factor for early AMD and late AMD development (inverse relation between education level and development of AMD). |
| Sunlight exposure | Sunlight exposure escalates oxidative stress in the retina and triggers the development of AMD. This factor may be unreliable (difficulties in quantification of the total amount of sunlight exposure). |
| Ethnicity | There are evident differences between ethnic groups in context of prevalence rates of any form of AMD (5.4% Caucasians, 4.6% Chinese, 4.2% Hispanics, 2.4% Africans). |
| Comorbidity | Certain accompanying diseases may exert influence on AMD development and progression, e.g., cataract surgery may increase the incidence of AMD and intensify its progression because of augmented inflammatory processes during surgery and increased exposure to ultraviolet light afterward. |
| Causative Factor | Immune Dysregulation and the Complement System Factors |
|---|---|
| Innate immune system | Immune complex deposition is engaged in the formation and biomolecular makeup of drusen. AMD pathogenesis may be mediated by localized inflammation and microglial cell recruitment. |
| Complement components | Terminal fragment C5b is engaged in the uprising of a membrane attack complex (MAC) along with such complement factors as C6, C7, C8, C9. Finally, MAC is responsible for disruption of the lipid bilayer that ultimately leads to cell lysis. In this context, complement factor H (CFH gene) on chromosome 1 is recognized as a major susceptibility locus for AMD development. In vitro knockdown of this gene may be connected with escalated MAC deposition in choroidal endothelial cells. Common loss-of-function CFH variants rs1061170 (Y402H) and rs1410996 constitute important genetic risk factors of AMD. Another CFH rs121913059 (p.Arg1210Cys) may confer approximately a 20-fold increment in AMD threat. Complement Factor I (CFI) gene on chromosome 4 and such variants as p.Gly119Arg and p.Leu131Arg may be implicated in a reduction of CFI concentration and activity subsequently leading to the development of AMD. The variant in the complement C3 gene of chromosome 19, rs2230199 (p.Arg102Gly), is connected with disrupted CFH binding, conferring resistance to CFH-mediated inactivation. This variant, as well as the associated polymorphism rs1047286 (p.Pro292Leu), exist commonly in Caucasian populations and considerably augment the risk of AMD. Another candidate factor potentially connected with AMD may be complement component 5 because of its presence in drusen, as well as in the context of elevated serum C5a levels in course of disease. In turn, variants in the gene coding complement component 9 may be engaged in progression to more advanced stages of AMD. Especially rs34882957 (p.Pro167Ser) is connected with elevated serum concentration of C9 and increased polymerization rates, which results in increased MAC formation and advances in course of AMD. Contrariwise, certain rare variants in the complement component 2 and complement factor B genes on chromosome 6 may act as protective factors against the development of AMD. Variants mitigating the function of coded enzymes subsequently influence on the activity of certain complement cascades and exert this beneficial effect. |
| Extracellular Matrix/Metabolism of Lipids/Angiogenesis/Oxidative Stress/Multiple Pathways | |
| Extracellular Matrix Remodeling | The structure of Bruch’s membrane depends on the balance between certain matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs) (especially MMP-1, MMP-2, MMP-3, MMP-9, TIMP-1, TIMP-2, TIMP-3). TIMP-3 excess leads to impaired extracellular matrix turnover as well as pathologic thickening of Bruch’s membrane. Decreased TIMP-3 activity may lead to escalated angiogenesis. Nine rare variants in TIMP-3 are considered to be cumulatively connected with over 30-fold increased risk of AMD (rs5754227, rs713685, rs743751, rs5749482 TIMP-3 intron variants constitute particularly important risk factors). In the matter of MMPs -gene MMP-2 appear to be significant. There are admissions that T allele (TT and CT genotypes) of rs243865 polymorphism may influences protectively against AMD, whereas homozygous CC genotype is possibly connected with hard drusen development in the course of disease. Variants of MMP-9 (rs142450006, rs3918241, rs3918241, rs3918242, rs4810482, rs17576, rs17577) appear to be connected with threat of AMD and possible progression to the form of macular neovascularization. Also MMP-9 CA (13-27) microsatellite expansion variant may be related with threat of the progression to macular neovascularization. Among other genes coding for extracellular matrix components, gene variant rs140647181, near the Collagen Type 8 α1 (COL8A1) gene is considered to be a risk factor for AMD. Presumptions about connections with AMD also apply to certain rare, protein-altering variants in the COL8A1 gene itself. |
| Lipid metabolism | Lipids constitute important components of drusen making up over 40% of its volume. Pathologic accumulation of lipids results in RPE and photoreceptor loss in AMD while. Oxidation of lipoproteins participate in the progression of AMD. Therefore, variants in several genes coding for proteins engaged in lipid metabolism and cholesterol transport may be considered to be risk factors of AMD. Genes involved in functionality of high density lipoprotein (HDL) appear particularly important due to its fundamental transport and anti-inflammatory significance. The apolipoprotein E (APOE) gene is recognized as possibly connected with AMD in various contexts. E.g., allelic variant ApoE4 may exert protective effect against the disease and confers up to 40% reduction in the threat of AMD developing. Oppositely, ApoE2 is connected with a slightly escalated risk of AMD or pathogenesis of macular neovascularization. Important modulator is hepatic lipase (LIPC) involved in intra-retinal lipid transport and regulation of plasma HDL levels. Effects are diversified from protective (against AMD development) connected with such LIPC variants as rs493258, rs10468017, rs9621532 or rs11755724 to escalating the risk of disease associated with such LIPC variants as rs13095226 and rs3748391. Moreover, cholesteryl ester transfer protein (CETP) may be engaged in AMD due to its role in the transport of cholesterol from peripheral tissue to the liver through transfer of cholesterol esters from low-density lipoproteins to HDLs. Mainly CETP rs3764261 is connected with escalated risk of AMD, while two intronic variants of CETP (rs17231506 and rs5817082) are associated respectively with slightly increased and reduced risk of AMD. Another factor is ATP-binding cassette transporter A1 (ABCA1) protein because its function in the elimination of excessive tissue cholesterol by triggering the formation of HDL. The rs1883025 ABCA1 protein seems to exert the opposite influence due to its two alleles (C allele connected with increment of plasma HDL levels and escalated threat of AMD and T allele related to decreased HDL and lower risk of AMD). |
| Angiogenesis | Angiogenic processes may be engaged in the development of macular neovascularization. Vascular endothelial growth factor (VEGF) is considered to be a driving force of neovascularization. Among its isoforms elevated levels of VEGF-A are connected with ocular neovascular diseases. The T allele of VEGF-A rs3025000 variant augments possibility of clinical response to anti-VEGF therapy in macular neovascularization. The VEGF-A rs3025033 variant and haplotype of rs1570360A-rs699947A-rs3025033G-rs2146323A may be connected with decreased threat of neovascular AMD. Finally, fibulin 5 (FBLN5) dysfunction may pose important factor in AMD pathogenesis because this extracellular matrix protein is engaged in modulation of angiogenesis and antagonizing VEGF. |
| Oxidative stress | Certain genetic mutations heighten susceptibility to oxidative damages that leads to photoreceptor dysfunction and AMD pathogenesis. Rare mutations in RAD51B gene are connected with an augmented threat of disease and individuals with AMD may characterize themselves with abnormally decreased serum concentrations of RAD51B. Additionally, the tumor necrosis factor receptor superfamily member 10A (TNFRSF10A) gene mutations may increase the risk of AMD, mainly in Asian populations. Animal models confirmed that decreased expression of oxidative stress is connected with reduced RPE cell viability and escalated apoptosis. Mutations in excision repair cross complex 6 (ERCC6) gene may exert certain influence on the augmentation of AMD risk because individuals with AMD may demonstrate decreased retinal ERCC6 expression. Subsequently, in such patients ERCC6 functionality in context of transcription-coupled excision repair of DNA mutations undergoes perturbation. |
| Genes implicated in multiple pathways | The region of chromosome 10q26 spanning high-temperature requirement factor A serine peptidase 1 (HTRA1) gene promoter and the age-related maculopathy susceptibility 2 (ARMS2) gene coding region constitutes loci of particular significance in context of AMD susceptibility. In cases of Caucasians and East Asians, ARMS2-HTRA1 region together with CFH, is responsible for over half of the genetic threat related to AMD. ARMS2-HTRA1 variants are connected with more rapid course of AMD. Certain local ARMS2 dysfunction may be associated with oxidative damages of the retina. In turn, HTRA1 shows implication in extracellular matrix remodeling, TGF-β cytokine signaling as well as acts as regulator of local subretinal inflammatory processes and angiogenesis. Both gene variants HTRA1 rs11200638 and ARMS2 rs10490924 are connected with an escalated AMD threat and younger onset of the disease. ARMS2 rs10490924 may implicate inflammatory response due to correlation with elevated C-reactive protein levels, while G allele is connected with augmented threat of AMD progression to advanced stages. However, this variant pose as more significant risk factor of the disease in Asian populations compared to Europeans (risk allele frequencies of 40% versus 20%). This fact also gains importance in the context of a slight connection between ARMS2 dysfunction and progression to neovascular disease. Other ARMS-HTRA1 variant rs2284665 seems to be associated with development of macular neovascularization. Finally, HTRA1 variants rs1049331, rs2293870, and rs2284665 are reported as connected with AMD progression, possibly through disrupted HTRA1-mediated inhibition of cellular apoptosis and insulin-like growth factor 1. |
| Chemical Element | Harmful Compounds | Mechanism of Action/Health Consequences of Exposition |
|---|---|---|
| Arsenic [As] | As2S3, AsS, AsO6, FeAsS | Absorption |
| The most common arsenic compounds are mainly absorbed by respiratory tract and accumulated in keratin-rich structures (hair, nails, skin), and in the placenta [48]. | ||
| Health effect and biochemical impact | ||
| Arsenic and its compounds are carcinogenic for respiratory system, skin and other organs triggering tumors, and impaired metabolic processes of hepatocytes and kidneys. Arsenic blocks sulfhydryl groups of proteins and inhibits enzymes. It can react directly with thiols, especially glutathione, cysteine, and dithiides [48]. | ||
| Chromium [Cr] | Valuable III active compounds | Consequences of reactivity |
| Compounds react with substances that allow the diffusion of ions (pyrophosphates, methionine, glycine, leucine, lysine, proline). They form stable protein complexes and generate the mechanisms of harmful influence of chromium [49]. | ||
| Health effect | ||
| Cr compounds damage the digestive system, cause skin lesions, mutagenic, embryotoxic, and teratogenic effects [49]. | ||
| Nickel [Ni] | Ni2+ compounds and ions | Physiological significance |
| Nickel in low concentrations is needed for normal functioning, whereas deficiency causes a reduction of oxygen consumption by the liver and an increase of fat accumulation. Physiologically, Ni activates certain enzymes, increases the activity of humoral response, stabilizes the nucleic acids, and plays a role in lipid metabolism [50]. | ||
| Absorption and health effect | ||
| Nickel is a major causative agent of contact allergy [50]. A total of 75–80% of inhaled nickel remains in the lungs, when administered orally 90% is excreted. Nickel allergy is most often manifested as allergic contact dermatitis (Ni-ACD). Hand eczema is also observed, resulting from prolonged exposure to Ni contained in detergents or nickel-plated items. There are known cases of vesicular eczema following the consumption of foods containing Ni [50]. | ||
| Cadmium [Cd] | CdO, CdCl2, CdSO4 | Absorption |
| When absorbed from food, cadmium effects undergo modulation in the range of digestive tract thanks to protein agents, Zn, Cu, Ca, and Fe contained in the diet. Their low content in the food cause increases in the absorption of Cd from the gastrointestinal tract and influence its accumulation in the body [51]. The greatest amount of Cd is absorbed in the duodenum. During absorption in the enterocytes, the transport of Cd occurs (non-specific divalent metal transporter (DMT1) bivalent conveyor and metal tolerance protein (MTP1) conveyor). Absorption can also occur through calcium channels of transporters responsible for Zn transport or can be absorbed from gastrointestinal tract in the combination with cysteine or glutathione-thiol groups [52]. | ||
| Accumulation and health effect | ||
| Cd accumulates in the liver and kidneys, which are the target organ of toxicity. In the liver, Cd induces the synthesis of low molecular weight metallothionein (MT) proteins that bind Cd (II) ions to Cd-MT complexes. In the kidneys, Cd-MT complexes are easily filtered in glomeruli and resorbed in proximal tubules, where they degrade Cd ions. These structures are particularly exposed to metal toxicity and causing subjected to damages them and resulting that may result in resorption disorders [20,21,22,43,53,54]. | ||
| AMD risk assessment | ||
| The results suggest that people with higher levels of Cd exposure are at increased risk for AMD. Concentration of Cd in the blood is a good reflection of recent exposure, especially in exposed workers. Additionally, nicotine is one of the main risk factors for AMD development and the main source of human exposure to Cd [53]. | ||
| Lead [Pb] | All Pb compounds and metallic Pb | Absorption and health effect |
| Lead is mainly absorbed through respiratory and digestive tract [43,49,55]. In children exposed to Pb in the pre- or postnatal period, a number of deviations in mental development were demonstrated. The most commonly observed aggression seizures, emotional lability, memory disorders, and learning and reading difficulties [43,55]. The most common disease is saturnism. It is chronic poisoning with lead and its salts. It occurs in employees of printers, battery factories and lead paint factories [43,49]. Pb can cause high blood pressure. Through nephrotoxic activity lead may cause renal dysfunction and develop the renal hypertension. Pb can generate oxidative stress, which in turn results in dysfunction of sympathetic system. It also reduces baroreceptor sensitivity and vagus nerve tension. Pb significantly influences the conduction of stimuli in the parasympathetic system. This can result in ventricular arrhythmias or myocardial infarction [21,43,55]. Pb and Cd induce changes in vascular endothelium and thus contribute the development of atherosclerotic lesions. Lead affects the course of early and late inflammatory reactions, T and B lymphocytes, macrophages and cytotoxic cells. Bone marrow damage is a common complication of Pb poisoning. Hematopoietic disorders manifest as thrombocytopaenia, leukopenia or anemia. In advanced bone marrow failure, all marrow cell lines undergo perturbation. The higher levels of immunoglobulins, especially IgA, IgE and IgG, were also observed in patients with higher Pb. Neurological disorders after long exposure to Pb usually take the form of Pb neuropathy. It usually involves the motor neurons of the upper limbs, and in particular the radial nerves. Exposure of men to Pb results in an increased risk of spontaneous abortion in their partners. In men, gonadal dysfunction occurs in the form of decreased sperm quality [43,55]. | ||
| AMD risk assessment | ||
| The present levels of Pb exposure in adult Americans are lower than in the past, and recent levels of exposure determined by blood lead measurements are not high enough to increase the risk of AMD. Such a hypothesis is probable, however, Pb accumulates in RPE may participate in ocular cancer as well as promote chronic disease by increasing chronic oxidative stress. As the level of antioxidant enzymes decreases with age, the retina is susceptible to stress and free radicals, which can lead to AMD development [56,57]. | ||
| Mercury [Hg] | Inorganic compounds HgCl2, HgCn2, HgS | Absorption and consequence of exposure |
| In people exposed to professional exposure to mercury, it penetrates through respiratory tract. A total of 80% is retained in the body. Sulfhydryl ligands (RSH) form complexes with Hg (reduced glutathione) and amino acids with cysteine and glycine. Directly high mercury poisoning occurs in cases of metallic form, from which 70% is removed to the outside of lungs. It partly penetrates into the brain [43,49]. | ||
| Accumulation and mechanism of toxicity | ||
| Mercury remains in the blood and penetrates the blood–brain barrier and placental barrier. Consequently, it accumulates in the brain and fetal tissues. Hg2+ ions have the ability to form complexes with proteins and with compounds containing SH-groups. Fetal hemoglobin has a high affinity for Hg, and fetuses have a higher concentration of Hg than mothers. Due to the fact that mercury easily diffuses into lipids it is also present in breast milk of mothers. It is deposited in the hair, its concentration there is proportional to the concentration in the blood. The first mercury-invaded cell element is the cell membrane. This is due to the presence of SH-groups. Most proteins and enzymes in their composition contain amino acids of these groups, therefore Hg poisoning can interfere with enzymatic reactions [43,49]. Irrespective of the form of Hg, it is deposited in the form of a complex with metallothioneins (MT), because of the induction of MT production. Synthesis of these proteins play a key role in the detoxification process [20,21,22,43,49,54]. |
| Isoform | Location of Expression |
|---|---|
| MT-1 | brain (mainly astrocytes), liver, kidney, eye |
| MT-2 | brain (mainly astrocytes), liver, kidney, eye |
| MT-3 | brain (astrocytes, neurons, cortex, hippocampus), heart, kidney, eye |
| MT-4 | squamous epithelia (upper gastrointestinal tract and skin), maternal deciduum |
| Gene | Polymorphism | Mechanism | Role in the AMD | Reference |
|---|---|---|---|---|
| CFH | rs1061170 (Y402H) | Deregulation of proinflammatory cytokines, changes in alternative complement pathway | Progression of AMD | [166,171,172,173,174,175,176,177,178,179,180,181] |
| rs6677604 | Regulation of complement system | Protection against AMD | [182,183,184] | |
| CFHR1 | rs6677604 | Decrease level of CFHR1 in serum | Protection against AMD | [183] |
| CFHR3/CFHR1 | delCFHR1/CFHR3 (CNP147) | Enhanced complement inhibitory activity | Protection against AMD | [185,186,187] |
| ARMS2 | rs10490924 | Activation of intraocular complement | Progression of AMD | [166,188] |
| HTRA1 | rs11200638 | Increased the expression of HtrA1 | Progression of AMD | [189,190] |
| C3 | rs2230199 (R102G) | Changes in the expression level and binding affinity of complement factors | Progression of AMD | [28,191,192,193,194,195] |
| VEGFA | rs3025039 | Lower concentration of VEGFA | Progression of AMD | [196,197,198] |
| rs3025020 | Elevated level of VEGFA | Progression of AMD | [196] | |
| CFB | rs4151667, rs641153 | Suppressing the inflammation process | Protection against AMD | [166,190,199,200] |
| SKIV2L | rs429608 | Unknown | Protection against AMD | [201,202] |
| LIPC | rs493258, rs10468017 | Elevated HDL levels | Protection against AMD | [190,191,203,204,205,206,207] |
| CETP | rs2230199, rs3764261 | Changes in the concentration of cholesteryl esters in HDL | Progression of AMD | [44,179,199,204,206,208,209,210,211,212] |
| CFI | rs10033900 | Changes in CFI production | Progression of AMD | [213,214] |
| Il-8 | IL8 −251AA, IL-8 +781C/T, rs2227306 | Increase in inflammation and angiogenesis | Progression of AMD | [166,215,216,217] |
| GSTM1 | rs1183423000 | Weaker antioxidant protection | [218,219,220,221,222] | |
| GSTT1 | rs1601993659 | Weaker antioxidant protection | [218,222] | |
| GSTP1 | rs1695, rs1138272 | Weaker antioxidant protection | [218,222,223] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brodzka, S.; Baszyński, J.; Rektor, K.; Hołderna-Bona, K.; Stanek, E.; Kurhaluk, N.; Tkaczenko, H.; Malukiewicz, G.; Woźniak, A.; Kamiński, P. Immunogenetic and Environmental Factors in Age-Related Macular Disease. Int. J. Mol. Sci. 2024, 25, 6567. https://doi.org/10.3390/ijms25126567
Brodzka S, Baszyński J, Rektor K, Hołderna-Bona K, Stanek E, Kurhaluk N, Tkaczenko H, Malukiewicz G, Woźniak A, Kamiński P. Immunogenetic and Environmental Factors in Age-Related Macular Disease. International Journal of Molecular Sciences. 2024; 25(12):6567. https://doi.org/10.3390/ijms25126567
Chicago/Turabian StyleBrodzka, Sylwia, Jędrzej Baszyński, Katarzyna Rektor, Karolina Hołderna-Bona, Emilia Stanek, Natalia Kurhaluk, Halina Tkaczenko, Grażyna Malukiewicz, Alina Woźniak, and Piotr Kamiński. 2024. "Immunogenetic and Environmental Factors in Age-Related Macular Disease" International Journal of Molecular Sciences 25, no. 12: 6567. https://doi.org/10.3390/ijms25126567
APA StyleBrodzka, S., Baszyński, J., Rektor, K., Hołderna-Bona, K., Stanek, E., Kurhaluk, N., Tkaczenko, H., Malukiewicz, G., Woźniak, A., & Kamiński, P. (2024). Immunogenetic and Environmental Factors in Age-Related Macular Disease. International Journal of Molecular Sciences, 25(12), 6567. https://doi.org/10.3390/ijms25126567