
Structural Evaluation of a Nitroreductase Engineered for Improved Activation of the 5-Nitroimidazole PET Probe SN33623

 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
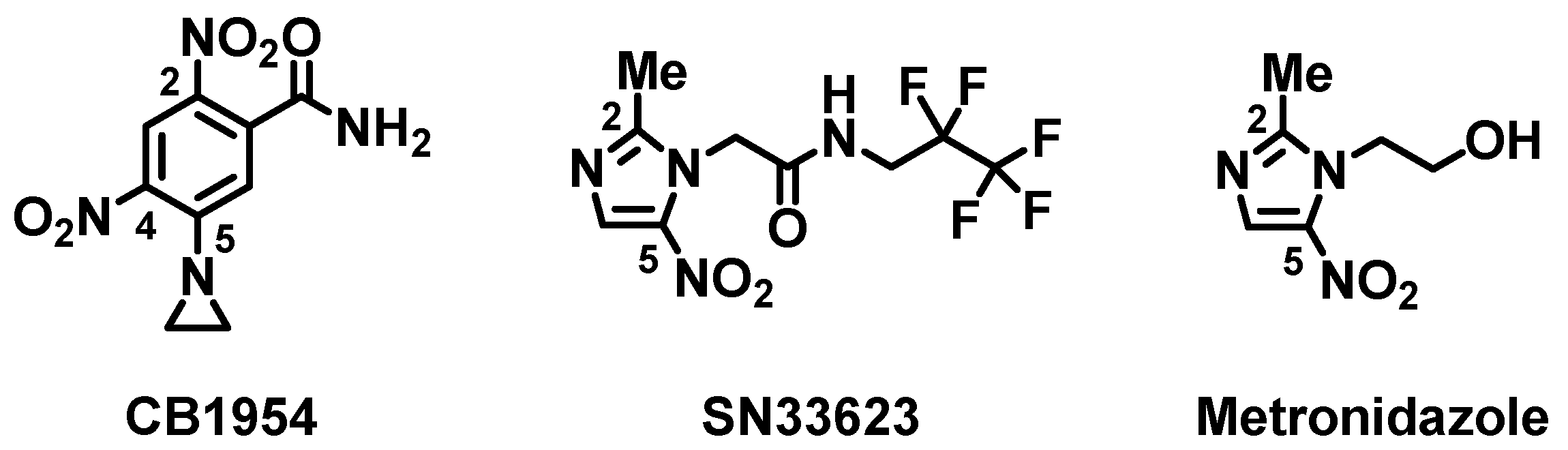
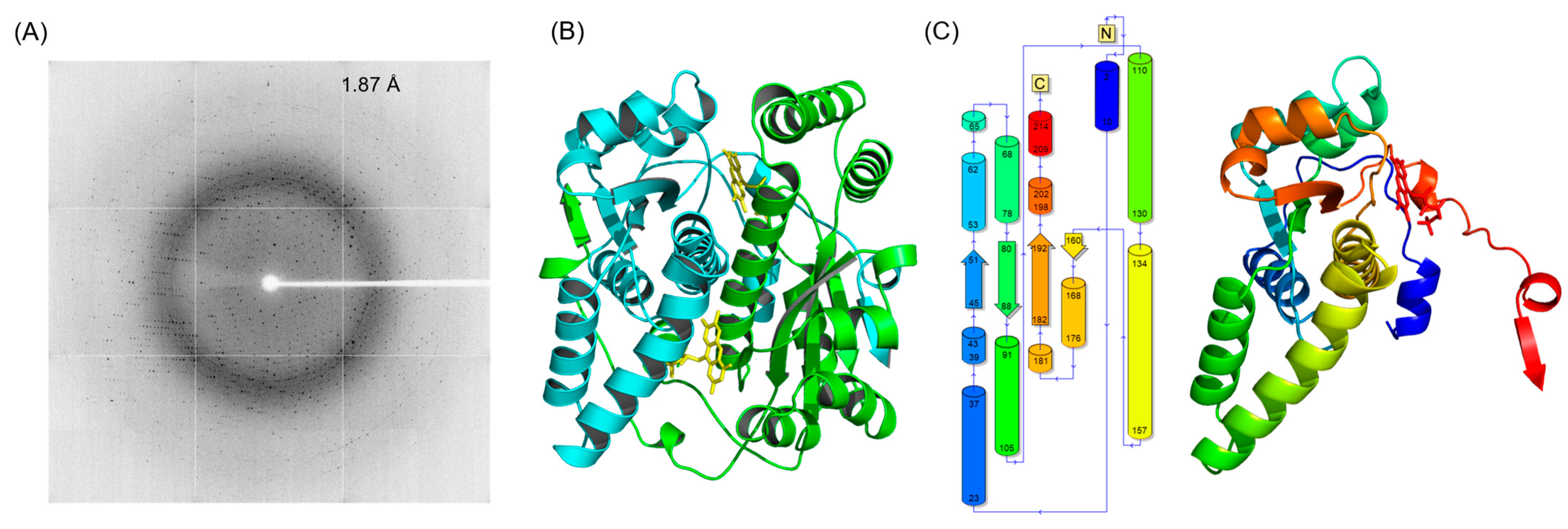
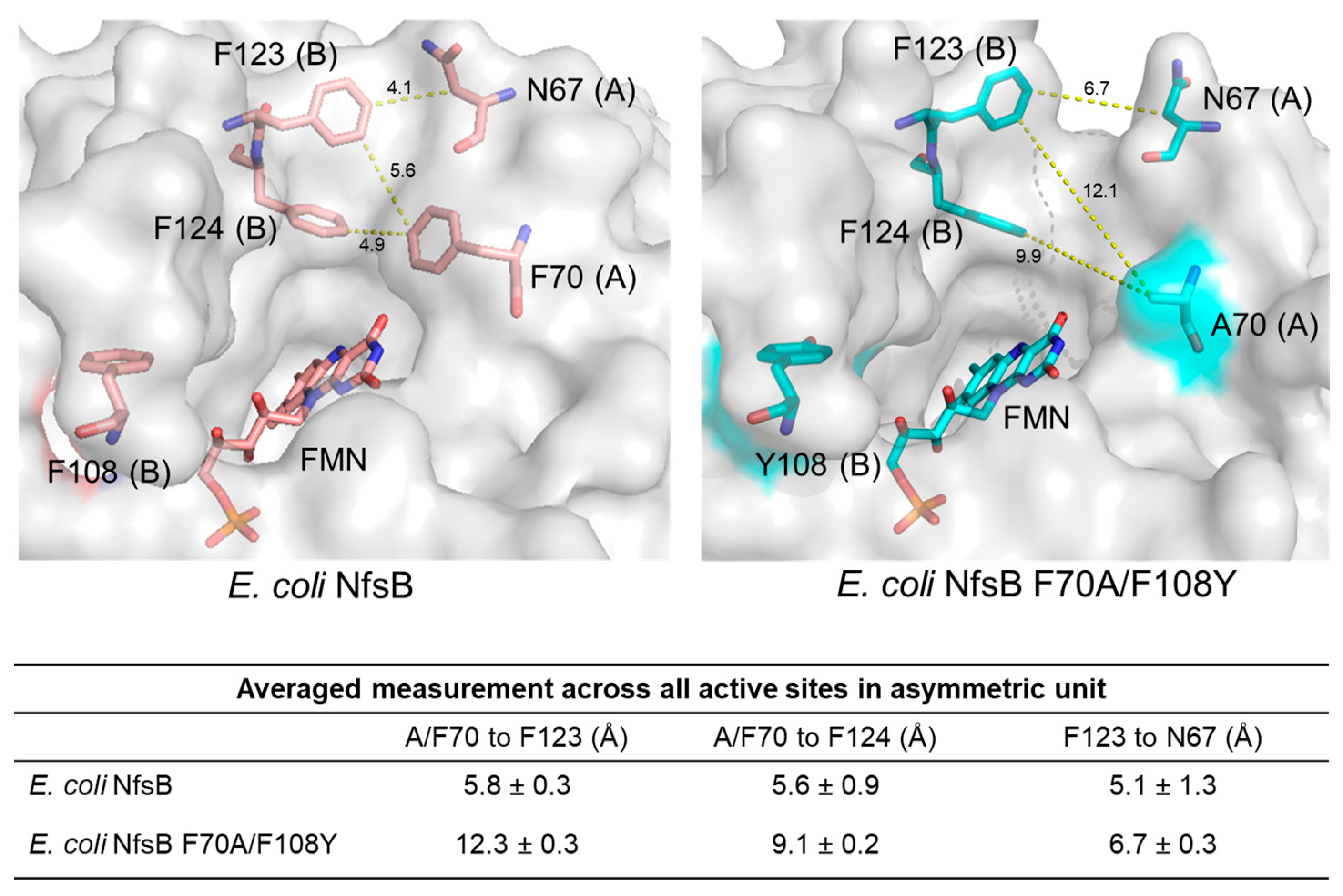
2.1. Crystal Structure of the Double-Mutant F70A/F108Y
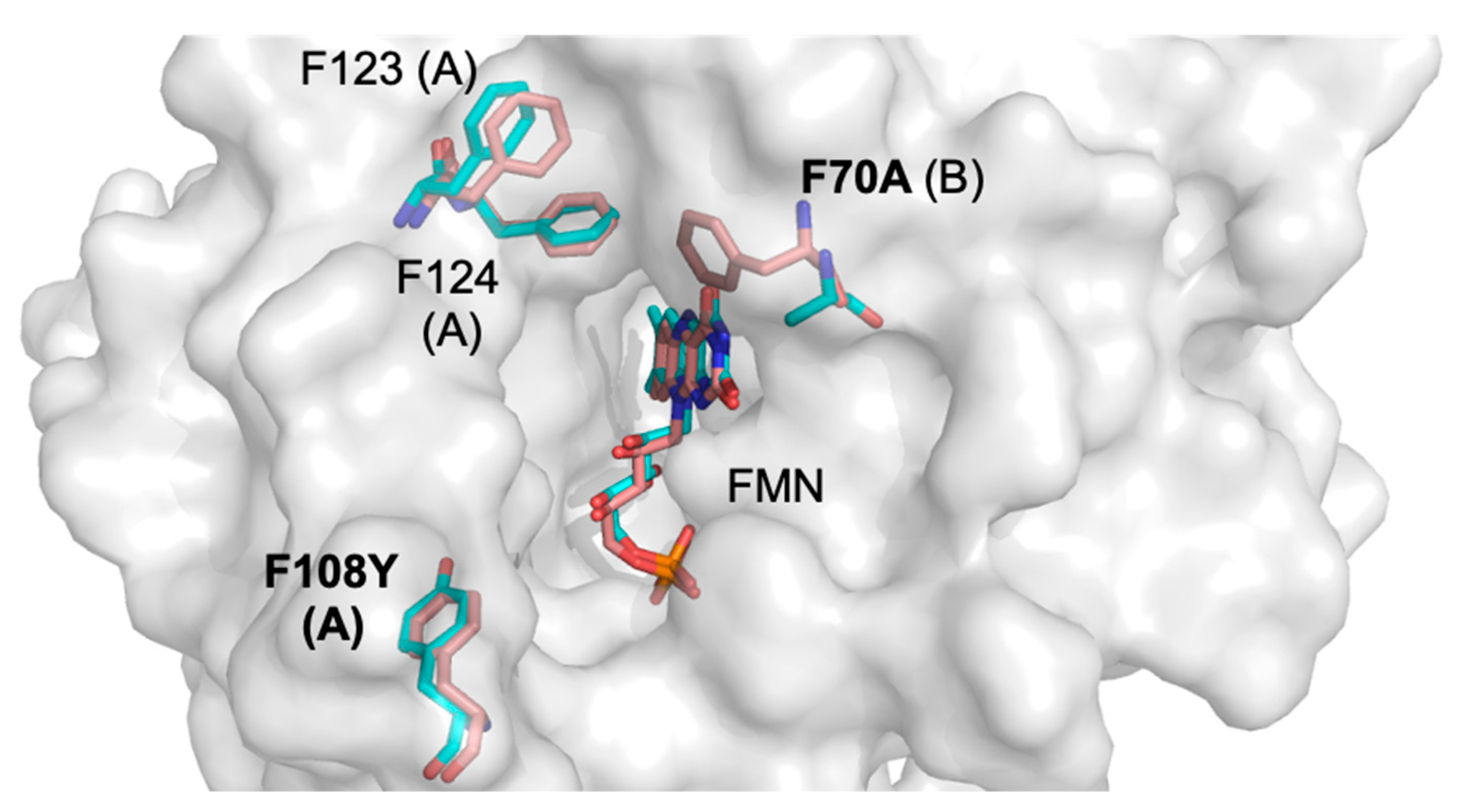
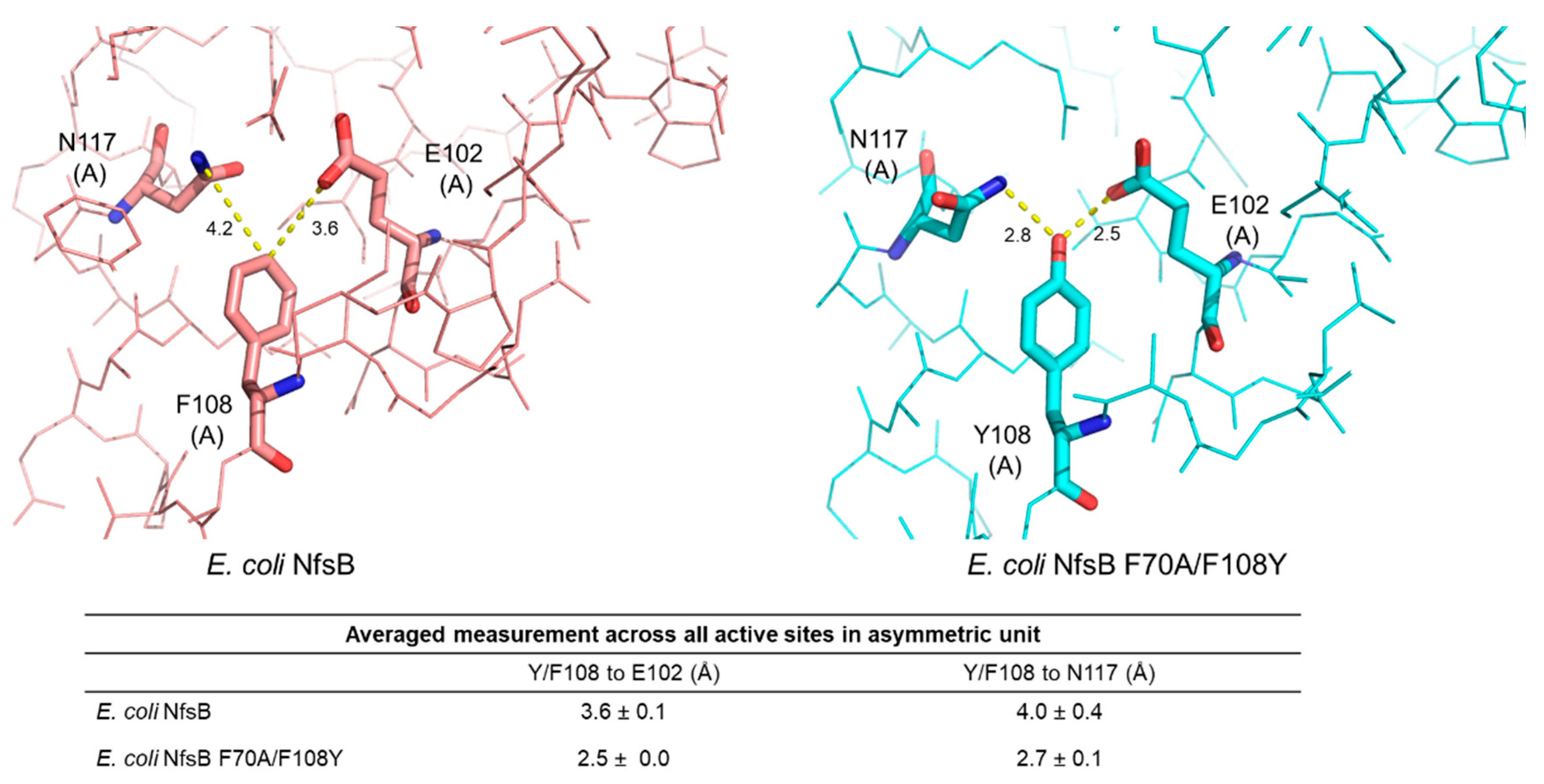
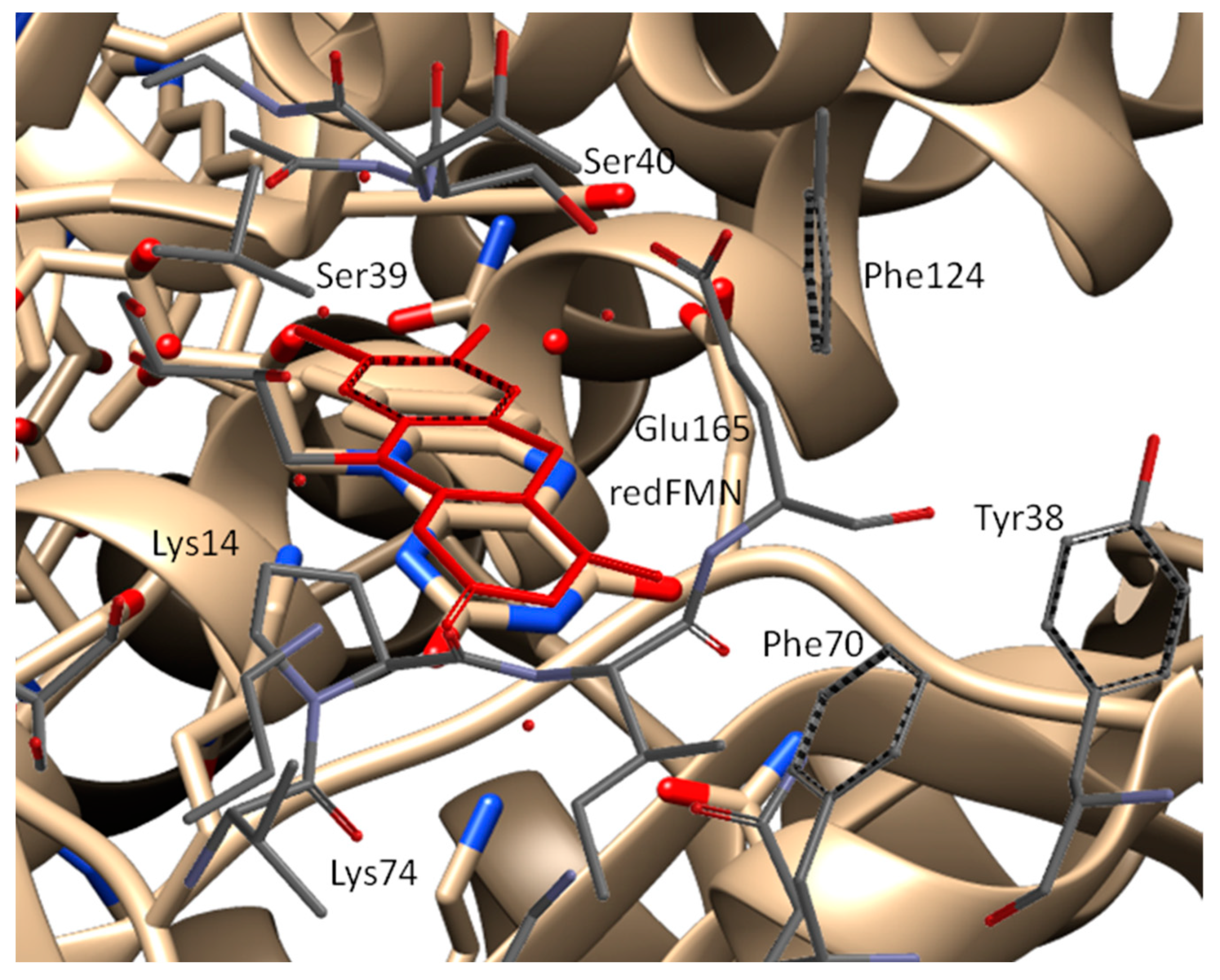
2.2. Structural Basis for the Improved Activity
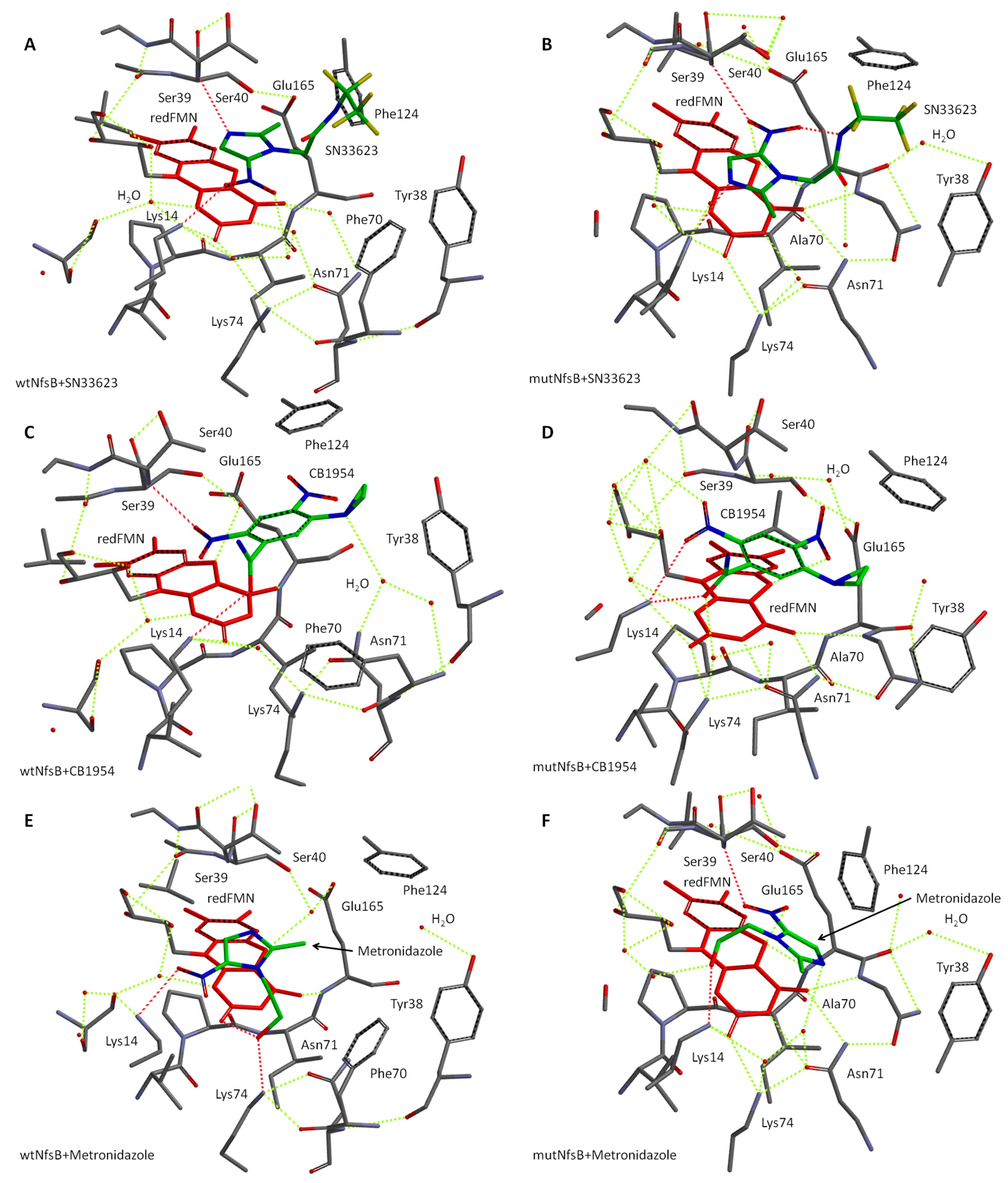
2.3. Molecular Modeling of Prodrug Binding—E. coli NfsB F70A/F108Y vs. Wild Type
3. Materials and Methods
3.1. Bacterial Strains, Media, and Growth Conditions
3.2. Cloning, Expression, and Purification of E. coli NfsB F70A/F108Y
3.3. Crystallization
3.4. X-ray Data Collection
3.5. Structure Solution and Refinement
3.6. Theoretical Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, J.; Kale, V.; Chen, M. Gene-directed enzyme prodrug therapy. AAPS J. 2015, 17, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Franzyk, H.; Christensen, S.B. Targeting toxins toward tumors. Molecules 2021, 26, 1292. [Google Scholar] [CrossRef] [PubMed]
- Dhankhar, R.; Kawatra, A.; Mohanty, A.; Gulati, P. Microbial enzymes used in prodrug activation for cancer therapy: Insights and future perspectives. Curr. Protein Pept. Sci. 2021, 22, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Rainov, N.G. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum. Gene Ther. 2000, 11, 2389–2401. [Google Scholar] [CrossRef] [PubMed]
- Sandmair, A.M.; Loimas, S.; Puranen, P.; Immonen, A.; Kossila, M.; Puranen, M.; Hurskainen, H.; Tyynelä, K.; Turunen, M.; Vanninen, R.; et al. Thymidine kinase gene therapy for human malignant glioma, using replication-deficient retroviruses or adenoviruses. Hum. Gene Ther. 2000, 11, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- Nemunaitis, J.; Cunningham, C.; Senzer, N.; Kuhn, J.; Cramm, J.; Litz, C.; Cavagnolo, R.; Cahill, A.; Clairmont, C.; Sznol, M. Pilot trial of genetically modified, attenuated Salmonella expressing the E. coli cytosine deaminase gene in refractory cancer patients. Cancer Gene Ther. 2003, 10, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Pandha, H.S.; Martin, L.A.; Rigg, A.; Hurst, H.C.; Stamp, G.W.; Sikora, K.; Lemoine, N.R. Genetic prodrug activation therapy for breast cancer: A phase I clinical trial of erbB-2-directed suicide gene expression. J. Clin. Oncol. 1999, 17, 2180–2189. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.H.; Mautner, V.; Mirza, D.; Oliff, S.; Gerritsen, W.; Van der Sijp, J.R.; Hubscher, S.; Reynolds, G.; Bonney, S.; Rajaratnam, R. Virus-directed enzyme prodrug therapy: Intratumoral administration of a replication-deficient adenovirus encoding nitroreductase to patients with resectable liver cancer. J. Clin. Oncol. 2004, 22, 1546–1552. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Young, J.G.; Mautner, V.; Ashdown, D.; Bonney, S.; Pineda, R.G.; Collins, S.I.; Searle, P.F.; Hull, D.; Peers, E. A phase I/II clinical trial in localized prostate cancer of an adenovirus expressing nitroreductase with CB1954. Mol. Ther. 2009, 17, 1292–1299. [Google Scholar] [CrossRef]
- Denny, W.A. Nitroreductase-based GDEPT. Curr. Pharm. Des. 2002, 8, 1349–1361. [Google Scholar] [CrossRef]
- Schellmann, N.; Deckert, P.M.; Bachran, D.; Fuchs, H.; Bachran, C. Targeted enzyme prodrug therapies. Mini Rev. Med. Chem. 2010, 10, 887–904. [Google Scholar] [CrossRef]
- Ruiz de Garibay, G.; García de Jalón, E.; Stigen, E.; Lund, K.B.; Popa, M.; Davidson, B.; Safont, M.M.; Rygh, C.B.; Espedal, H.; Barrett, T.M.; et al. Repurposing (18)F-FMISO as a PET tracer for translational imaging of nitroreductase-based gene directed enzyme prodrug therapy. Theranostics 2021, 11, 6044–6057. [Google Scholar] [CrossRef]
- Copp, J.N.; Mowday, A.M.; Williams, E.M.; Guise, C.P.; Ashoorzadeh, A.; Sharrock, A.V.; Flanagan, J.U.; Smaill, J.B.; Patterson, A.V.; Ackerley, D.F. Engineering a multifunctional nitroreductase for improved activation of prodrugs and PET probes for cancer gene therapy. Cell Chem. Biol. 2017, 24, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Sekar, T.V.; Paulmurugan, R. Theranostic imaging of cancer gene therapy. Methods Mol. Biol. 2016, 1461, 241–254. [Google Scholar] [CrossRef]
- Bhaumik, S. Advances in imaging gene-directed enzyme prodrug therapy. Curr. Pharm. Biotechnol. 2011, 12, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Mowday, A.M.; Copp, J.N.; Syddall, S.P.; Dubois, L.J.; Wang, J.; Lieuwes, N.G.; Biemans, R.; Ashoorzadeh, A.; Abbattista, M.R.; Williams, E.M.; et al. E. coli nitroreductase NfsA is a reporter gene for non-invasive PET imaging in cancer gene therapy applications. Theranostics 2020, 10, 10548–10562. [Google Scholar] [CrossRef]
- Williams, E.M.; Rich, M.H.; Mowday, A.M.; Ashoorzadeh, A.; Copp, J.N.; Guise, C.P.; Anderson, R.F.; Flanagan, J.U.; Smaill, J.B.; Patterson, A.V.; et al. Engineering Escherichia coli NfsB to activate a hypoxia-resistant analogue of the PET probe EF5 to enable non-invasive imaging during enzyme prodrug therapy. Biochemistry 2019, 58, 3700–3710. [Google Scholar] [CrossRef]
- Sharrock, A.V.; Mulligan, T.S.; Hall, K.R.; Williams, E.M.; White, D.T.; Zhang, L.; Emmerich, K.; Matthews, F.; Nimmagadda, S.; Washington, S.; et al. NTR 2.0: A rationally engineered prodrug-converting enzyme with substantially enhanced efficacy for targeted cell ablation. Nat. Methods 2022, 19, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Akiva, E.; Copp, J.N.; Tokuriki, N.; Babbitt, P.C. Evolutionary and molecular foundations of multiple contemporary functions of the nitroreductase superfamily. Proc. Natl. Acad. Sci. USA 2017, 114, E9549–E9558. [Google Scholar] [CrossRef]
- Williams, E.M.; Little, R.F.; Mowday, A.M.; Rich, M.H.; Chan-Hyams, J.V.; Copp, J.N.; Smaill, J.B.; Patterson, A.V.; Ackerley, D.F. Nitroreductase gene-directed enzyme prodrug therapy: Insights and advances toward clinical utility. Biochem. J. 2015, 471, 131–153. [Google Scholar] [CrossRef]
- Pitsawong, W.; Hoben, J.P.; Miller, A.-F. Understanding the broad substrate repertoire of nitroreductase based on its kinetic mechanism. J. Biol. Chem. 2014, 289, 15203–15214. [Google Scholar] [CrossRef]
- Parkinson, G.N.; Skelly, J.V.; Neidle, S. Crystal structure of FMN-dependent nitroreductase from Escherichia coli B: A prodrug-activating enzyme. J. Med. Chem. 2000, 43, 3624–3631. [Google Scholar] [CrossRef]
- Johansson, E.; Parkinson, G.N.; Denny, W.A.; Neidle, S. Studies on the nitroreductase prodrug-activating system. Crystal structures of complexes with the inhibitor dicoumarol and dinitrobenzamide prodrugs and of the enzyme active form. J. Med. Chem. 2003, 46, 4009–4020. [Google Scholar] [CrossRef]
- Race, P.R.; Lovering, A.L.; White, S.A.; Grove, J.I.; Searle, P.F.; Wrighton, C.W.; Hyde, E. Kinetic and structural characterisation of Escherichia coli nitroreductase mutants showing improved efficacy for the prodrug substrate CB1954. J. Mol. Biol. 2007, 368, 481–492. [Google Scholar] [CrossRef]
- LinWu, S.-W.; Wu, C.-A.; Peng, F.-C.; Wang, A.H.-J. Structure-based development of bacterial nitroreductase against nitrobenzodiazepine-induced hypnosis. Biochem. Pharmacol. 2012, 83, 1690–1699. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Zhou, Y.; Chen, Q.; Yang, Q.; Yang, J. Altering the regioselectivity of a nitroreductase in the synthesis of arylhydroxylamines by structure-based engineering. ChemBioChem 2015, 16, 1219–1225. [Google Scholar] [CrossRef] [PubMed]
- Estarellas, C.; Frontera, A.; Quiñonero, D.; Deyà, P.M. Anion-π interactions in flavoproteins. Chem. Asian J. 2011, 6, 2316–2318. [Google Scholar] [CrossRef]
- Race, P.R.; Lovering, A.L.; Green, R.M.; Ossor, A.; White, S.A.; Searle, P.F.; Wrighton, C.J.; Hyde, E.I. Structural and mechanistic studies of Escherichia coli nitroreductase with the antibiotic nitrofurazone: Reversed binding orientations in different redox states of the enzyme. J. Biol. Chem. 2005, 280, 13256–13264. [Google Scholar] [CrossRef] [PubMed]
- Valiauga, B.; Williams, E.M.; Ackerley, D.F.; Čėnas, N. Reduction of quinones and nitroaromatic compounds by Escherichia coli nitroreductase A (NfsA): Characterization of kinetics and substrate specificity. Arch. Biochem. Biophys. 2017, 614, 14–22. [Google Scholar] [CrossRef]
- Koder, R.L.; Haynes, C.A.; Rodgers, M.E.; Rodgers, D.W.; Miller, A.-F. Flavin thermodynamics explain the oxygen insensitivity of enteric nitroreductases. Biochemistry 2002, 41, 14197–14205. [Google Scholar] [CrossRef]
- Day, M.A.; Christofferson, A.J.; Anderson, J.L.R.; Vass, S.O.; Evans, A.; Searle, P.F.; White, S.A.; Hyde, E.I. Structure and dynamics of three Escherichia coli NfsB nitro-reductase mutants selected for enhanced activity with the cancer prodrug CB1954. Int. J. Mol. Sci. 2023, 24, 5987. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Pitsawong, W.; Haynes, C.A.; Koder, R.L., Jr.; Rodgers, D.W.; Miller, A.F. Mechanism-informed refinement reveals altered substrate-binding mode for catalytically competent nitroreductase. Structure 2017, 25, 978–987.e974. [Google Scholar] [CrossRef]
- Jaberipour, M.; Vass, S.O.; Guise, C.P.; Grove, J.I.; Knox, R.J.; Hu, L.; Hyde, E.I.; Searle, P.F. Testing double mutants of the enzyme nitroreductase for enhanced cell sensitisation to prodrugs: Effects of combining beneficial single mutations. Biochem. Pharmacol. 2010, 79, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Sharrock, A.V.; Mumm, J.S.; Bagdžiūnas, G.; Čėnas, N.; Arcus, V.L.; Ackerley, D.F. The crystal structure of engineered nitroreductase NTR 2.0 and impact of F70A and F108Y substitutions on substrate specificity. Int. J. Mol. Sci. 2023, 24, 6633. [Google Scholar] [CrossRef] [PubMed]
- Valiauga, B.; Bagdžiūnas, G.; Sharrock, A.V.; Ackerley, D.F.; Čėnas, N. The catalysis mechanism of E. coli nitroreductase A, a candidate for gene-directed prodrug therapy: Potentiometric and substrate specificity studies. Int. J. Mol. Sci. 2024, 25, 4413. [Google Scholar] [CrossRef] [PubMed]
- Bagdžiūnas, G. Theoretical design of molecularly imprinted polymers based on polyaniline and polypyrrole for detection of tryptophan. Mol. Syst. Des. Eng. 2020, 5, 1504–1512. [Google Scholar] [CrossRef]
- Vass, S.O.; Jarrom, D.; Wilson, W.R.; Hyde, E.I.; Searle, P.F. E. coli NfsA: An alternative nitroreductase for prodrug activation gene therapy in combination with CB1954. Br. J. Cancer 2009, 100, 1903–1911. [Google Scholar] [CrossRef]
- Collaborative Computational Project, N. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994, 50 Pt 5, 760–763. [Google Scholar] [CrossRef]
- Leslie, A.G.; Powell, H.R. Processing diffraction data with mosflm. In Evolving Methods for Macromolecular Crystallography; Read, R.J., Sussman, J.L., Eds.; NATO Science Series; Springer: Dordrecht, The Netherlands, 2007; Volume 245, pp. 41–51. [Google Scholar]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef]
- Vagin, A.; Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 1, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60 Pt 12, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67 Pt 4, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Radveikienė, I.; Palinauskas, D.; Ragauskaitė, E.; Bagdžiūnas, G. Self-assembled cyclodextrins-based nanostructures on indium-tin-oxide for a detection of catecholamine neurotransmitters. Appl. Surf. Sci. 2022, 600, 154170. [Google Scholar] [CrossRef]
- Bagdžiūnas, G.; Ramanavičius, A. Towards direct enzyme wiring: A theoretical investigation of charge carrier transfer mechanisms between glucose oxidase and organic semiconductors. Phys. Chem. Chem. Phys. 2019, 21, 2968–2976. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Application of the PM6 method to modeling proteins. J. Mol. Model. 2009, 15, 765–805. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wavelength (Å) | 0.9537 |
| Space group | P 21 21 21 |
| Unit-cell parameters (Å, °) | a = 87.22, b = 95.59, c = 112.31, α/β/γ = 90/90/90 |
| Resolution range (Å) | 1.98–87.22 (1.98–2.10) |
| Measured reflections | 469,649 (31,985) |
| Unique reflections | 66,067 (4399) |
| Multiplicity | 7.1 |
| Temperature (K) | 100 |
| Matthews coefficient (Å3 Da−1) | 2.45 |
| Solvent content (%) | 49.79 |
| No. of molecules in ASU | 4 |
| Completeness (%) | 100 (100) |
| Mean I/σ(I) | 9.9 (2.5) |
| Rmerge † (%) | 15.1 (81.4) |
| Rwork | 0.171 |
| CC(1/2) | 0.9615 |
| Rfree | 0.219 |
| Protein atoms | 6652 |
| Other ions/molecules | 14 |
| Number of waters | 516 |
| B factors (proteins) | 18.164/18.404/16.636/16.618 |
| B factors (waters) | 26.403 |
| RMSD | |
| Bond angles (°) | 1.5953 |
| Bond lengths (Å) | 0.0103 |
| Entry | Enzyme+Substrate | Intermolecular Interactions | d(N5···O) a, Å | −Ein, kJ mol−1 | IC50, μM |
|---|---|---|---|---|---|
| 1 | wtNfsB+SN33623 | Lys14···NO2, Ser39-NH···N of imidazole | 6.6 | 63.3 | >400 |
| 2 | mutNfsB+SN33623 | Lys14··· N of imidazole, Ser39-NH··· NO2 | 3.0 | 50.6 | 41 ± 1 |
| 3 | wtNfsB+CB1954 | Lys14···HNH-CO, Ser39-NH···2-NO2 | 4.8 | 59.5 | 380 ± 6 |
| 4 | mutNfsB+CB1954 | Lys14···HNH-CO, Lys14···2-NO2 | 3.0 | 43.5 | 110 ± 13 |
| 5 | wtNfsB+metronidazole | Lys14···NO2, Lys74···HO-, FMN C=O···HO- | 6.9 | 85.7 | 230 ± 39 |
| 6 | mutNfsB+metronidazole | Ser39-NH··· NO2, Lys14···HO- | 3.0 | 20.4 | 8 ± 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharrock, A.V.; Mumm, J.S.; Williams, E.M.; Čėnas, N.; Smaill, J.B.; Patterson, A.V.; Ackerley, D.F.; Bagdžiūnas, G.; Arcus, V.L. Structural Evaluation of a Nitroreductase Engineered for Improved Activation of the 5-Nitroimidazole PET Probe SN33623. Int. J. Mol. Sci. 2024, 25, 6593. https://doi.org/10.3390/ijms25126593
Sharrock AV, Mumm JS, Williams EM, Čėnas N, Smaill JB, Patterson AV, Ackerley DF, Bagdžiūnas G, Arcus VL. Structural Evaluation of a Nitroreductase Engineered for Improved Activation of the 5-Nitroimidazole PET Probe SN33623. International Journal of Molecular Sciences. 2024; 25(12):6593. https://doi.org/10.3390/ijms25126593
Chicago/Turabian StyleSharrock, Abigail V., Jeff S. Mumm, Elsie M. Williams, Narimantas Čėnas, Jeff B. Smaill, Adam V. Patterson, David F. Ackerley, Gintautas Bagdžiūnas, and Vickery L. Arcus. 2024. "Structural Evaluation of a Nitroreductase Engineered for Improved Activation of the 5-Nitroimidazole PET Probe SN33623" International Journal of Molecular Sciences 25, no. 12: 6593. https://doi.org/10.3390/ijms25126593