
A Review of CAR-T Combination Therapies for Treatment of Gynecological Cancers


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Challenges of Using CAR-T Therapy for Solid Tumors
2.1. Fast T-Cell Exhaustion
2.2. Stromal Barrier
2.3. Antigen-Shedding
3. Strategies to Enhance CAR-T Therapy in Solid Tumors
3.1. Combination with Checkpoint Inhibitors
3.2. Common Immune-Related Adverse Effects of Checkpoint Inhibitors
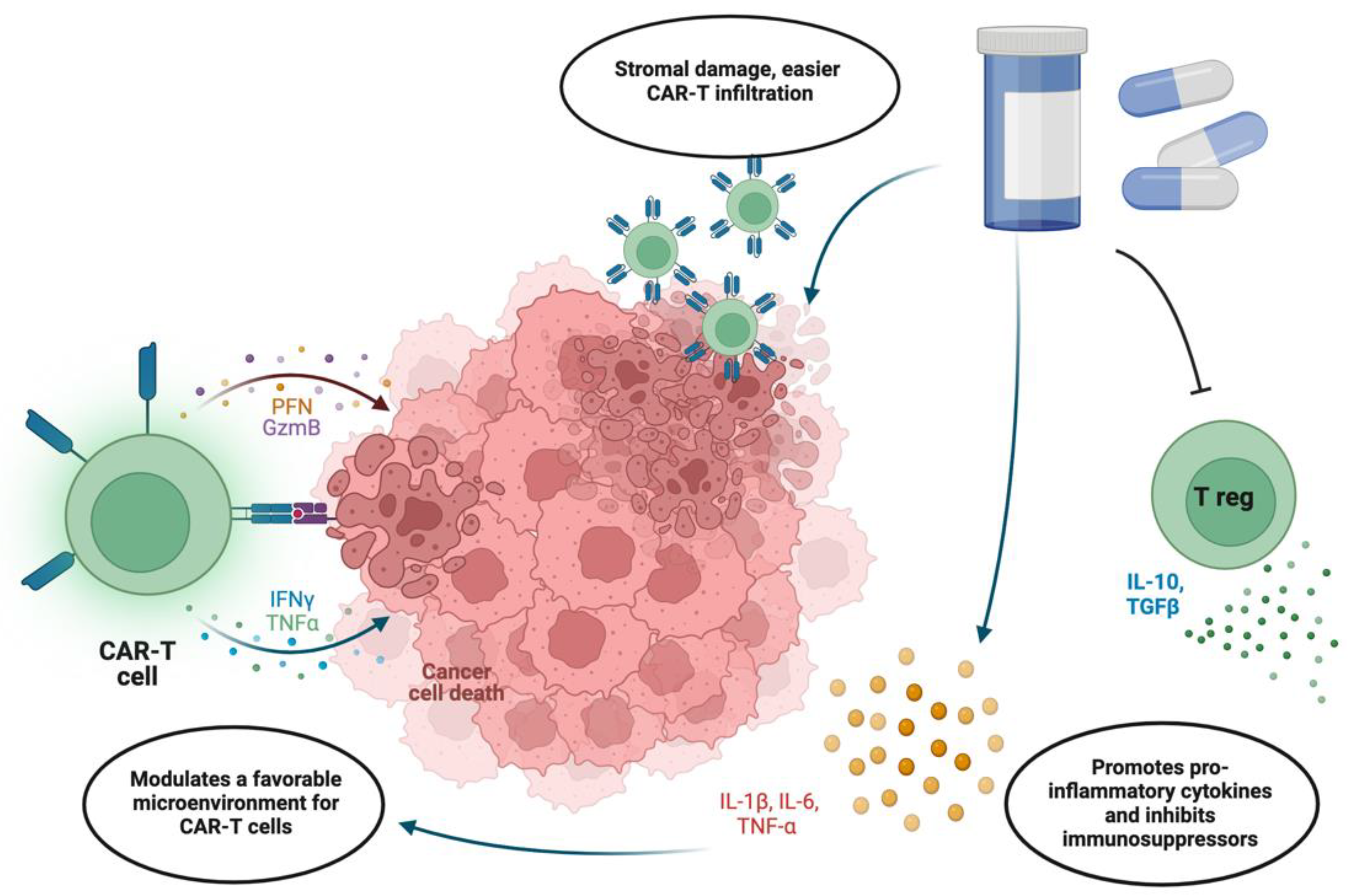
3.3. Integration with Chemotherapy
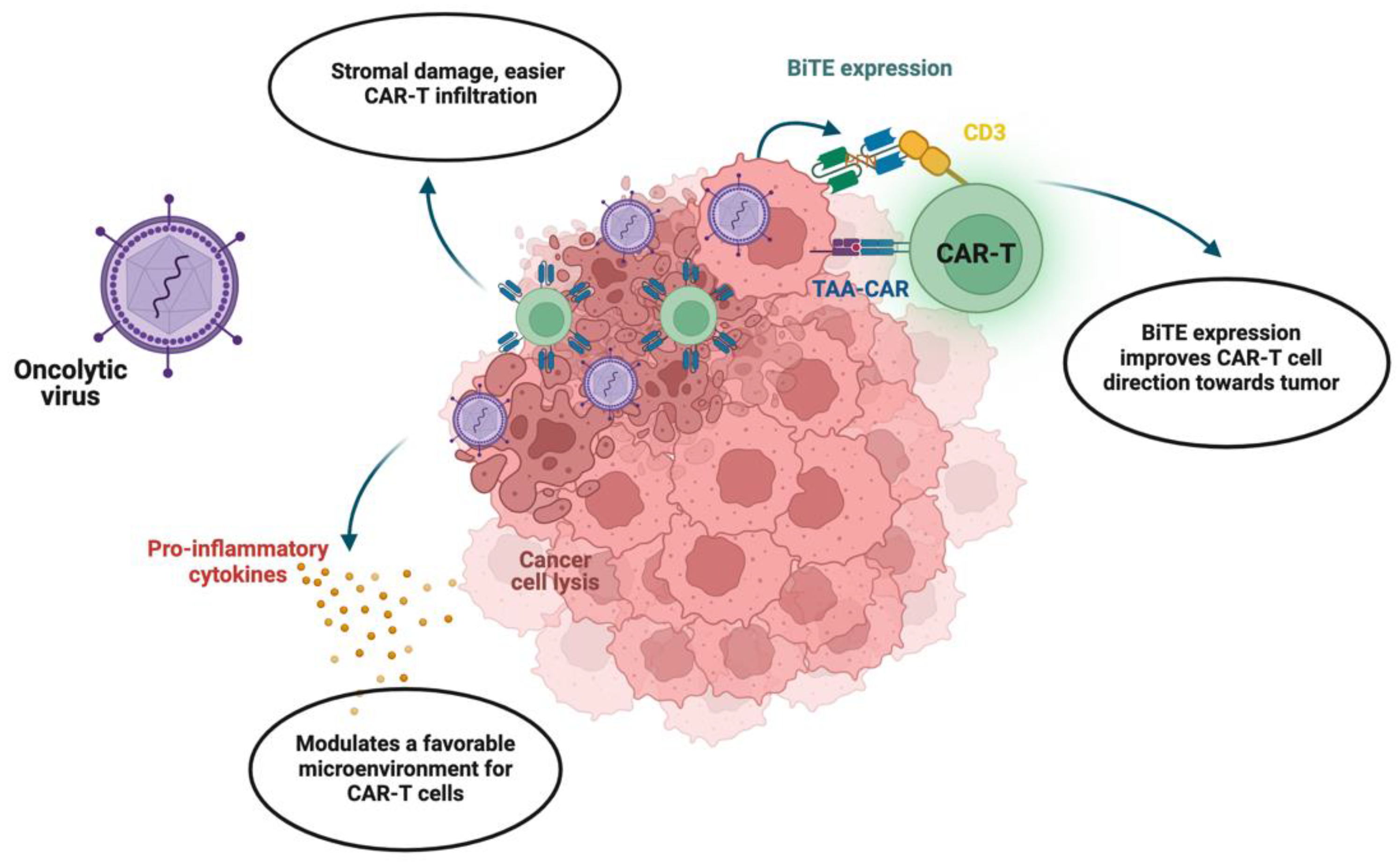
4. Novel Oncolytic Viral Therapy Combined with CAR-T
4.1. Prospects of Viral Therapy Combination
4.2. Challenges with Viral Therapy Combination
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pignata, S.; Pisano, C.; di Napoli, M.; Cecere, S.C.; Tambaro, R.; Attademo, L. Treatment of recurrent epithelial ovarian cancer. Cancer 2019, 125, 4609–4615. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Peng, H.; Qi, X.; Wu, M.; Zhao, X. Targeted therapies in gynecological cancers: A comprehensive review of clinical evidence. Signal Transduct. Target. Ther. 2020, 5, 137. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Dickinson, M.J.; Dreyling, M.H.; Martinez, J.; Kolstad, A.; Butler, J.P.; Ghosh, M.; Popplewell, L.; Chavez, J.C.; Bachy, E.; et al. Efficacy and safety of tisagenlecleucel (Tisa-cel) in adult patients (Pts) with relapsed/refractory follicular lymphoma (r/r FL): Primary analysis of the phase 2 Elara trial. J. Clin. Oncol. 2021, 39, 7508. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Dimitri, A.; Wang, W.; Jung, I.-Y.; Ott, C.J.; Fasolino, M.; Wang, Y.; Kulikovskaya, I.; Gupta, M.; Yoder, T.; et al. BET bromodomain protein inhibition reverses chimeric antigen receptor extinction and reinvigorates exhausted T cells in chronic lymphocytic leukemia. J. Clin. Investig. 2021, 131, e145459. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Shah, B.D.; Ghobadi, A.; O Oluwole, O.; Logan, A.C.; Boissel, N.; Cassaday, R.D.; Leguay, T.; Bishop, M.R.; Topp, M.S.; Tzachanis, D.; et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: Phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet 2021, 398, 491–502. [Google Scholar] [CrossRef]
- He, J.; Munir, F.; Ragoonanan, D.; Zaky, W.; Khazal, S.J.; Tewari, P.; Fueyo, J.; Gomez-Manzano, C.; Jiang, H. Combining CAR T Cell Therapy and Oncolytic Virotherapy for Pediatric Solid Tumors: A Promising Option. Immuno 2023, 3, 37–56. [Google Scholar] [CrossRef]
- Wang, Y.; Drum, D.L.; Sun, R.; Zhang, Y.; Chen, F.; Sun, F.; Dal, E.; Yu, L.; Jia, J.; Arya, S.; et al. Stressed target cancer cells drive nongenetic reprogramming of CAR T cells and solid tumor microenvironment. Nat. Commun. 2023, 14, 5727. [Google Scholar] [CrossRef]
- Watowich, M.B.; Gilbert, M.R.; Larion, M. T cell exhaustion in malignant gliomas. Trends Cancer 2023, 9, 270–292. [Google Scholar] [CrossRef] [PubMed]
- Schurich, A.; Magalhaes, I.; Mattsson, J. Metabolic regulation of CAR T cell function by the hypoxic microenvironment in solid tumors. Immunotherapy 2019, 11, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Harrer, D.C.; Bezler, V.; Hartley, J.; Herr, W.; Abken, H. IRF4 downregulation improves sensitivity and endurance of CAR T cell functional capacities. Front. Immunol. 2023, 14, 1185618. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.C.; Dündar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S.; et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Teng, X.; Guo, X.; Zhang, H.; Huang, Y.; Cui, J.; Si, X.; Ding, L.; Wang, X.; Li, X.; et al. Inhibition of Calcium Signaling Prevents Exhaustion and Enhances Anti-Leukemia Efficacy of CAR-T Cells via SOCE-Calcineurin-NFAT and Glycolysis Pathways. Adv. Sci. 2022, 9, 2103508. [Google Scholar] [CrossRef] [PubMed]
- Tsui, C.; Kretschmer, L.; Rapelius, S.; Gabriel, S.S.; Chisanga, D.; Knöpper, K.; Utzschneider, D.T.; Nüssing, S.; Liao, Y.; Mason, T.; et al. MYB orchestrates T cell exhaustion and response to checkpoint inhibition. Nature 2022, 609, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Du, Y.; Li, G.; Yu, M.; Hu, H.; Pan, C.; Wang, D.; Shi, Z.; Yan, X.; Li, X.; et al. Trogocytosis of CAR molecule regulates CAR-T cell dysfunction and tumor antigen escape. Signal Transduct. Target. Ther. 2023, 8, 457. [Google Scholar] [CrossRef] [PubMed]
- Frisbie, L.; Buckanovich, R.J.; Coffman, L. Carcinoma-Associated Mesenchymal Stem/Stromal Cells: Architects of the Pro-tumorigenic Tumor Microenvironment. Stem Cells 2022, 40, 705–715. [Google Scholar] [CrossRef]
- Song, M.; He, J.; Pan, Q.; Yang, J.; Zhao, J.; Zhang, Y.; Huang, Y.; Tang, Y.; Wang, Q.; He, J.; et al. Cancer-Associated Fibroblast-Mediated Cellular Crosstalk Supports Hepatocellular Carcinoma Progression. Hepatology 2021, 73, 1717–1735. [Google Scholar] [CrossRef]
- Nissen, N.I.; Johansen, A.Z.; Chen, I.; Johansen, J.S.; Pedersen, R.S.; Hansen, C.P.; Karsdal, M.A.; Willumsen, N. Collagen Biomarkers Quantify Fibroblast Activity In Vitro and Predict Survival in Patients with Pancreatic Ductal Adenocarcinoma. Cancers 2022, 14, 819. [Google Scholar] [CrossRef]
- Ying, L.; Yazdani, M.; Koya, R.; Zhao, R. Engineering tumor stromal mechanics for improved T cell therapy. Biochim. Biophys. Acta Gen. Subj. 2022, 1866, 130095. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, M.; Soeda, S.; Sasaki, M.; Handa, H.; Imai, Y.; Tanaka, N.; Tanosaki, S.; Ito, S.; Odajima, T.; Sugimori, H.; et al. Clinical impact of serum soluble SLAMF7 in multiple myeloma. Oncotarget 2018, 9, 34784–34793. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Gao, F.; Gao, Z.; Ao, L.; Ma, S.; Jia, M.; Li, N.; Lu, P.; Sun, B.; Ho, M.; et al. Shed antigen-induced blocking effect on CAR-T cells targeting Glypican-3 in Hepatocellular Carcinoma. J. Immunother. Cancer 2021, 9, e001875. [Google Scholar] [CrossRef] [PubMed]
- Raffaghello, L.; Prigione, I.; Airoldi, I.; Camoriano, M.; Levreri, I.; Gambini, C.; Pende, D.; Steinle, A.; Ferrone, S.; Pistoia, V. Downregulation and/or release of NKG2D ligands as immune evasion strategy of human neuroblastoma. Neoplasia 2004, 6, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Maurer, S.; Kropp, K.N.; Klein, G.; Steinle, A.; Haen, S.P.; Walz, J.S.; Hinterleitner, C.; Märklin, M.; Kopp, H.-G.; Salih, H.R. Platelet-mediated shedding of NKG2D ligands impairs NK cell immune-surveillance of tumor cells. OncoImmunology 2018, 7, e1364827. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef] [PubMed]
- John, L.B.; Devaud, C.; Duong, C.P.M.; Yong, C.S.; Beavis, P.A.; Haynes, N.M.; Chow, M.T.; Smyth, M.J.; Kershaw, M.H.; Darcy, P.K. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 2013, 19, 5636–5646. [Google Scholar] [CrossRef] [PubMed]
- Suarez, E.R.; de Chang, K.; Sun, J.; Sui, J.; Freeman, G.J.; Signoretti, S.; Zhu, Q.; Marasco, W.A. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget 2016, 7, 34341–34355. [Google Scholar] [CrossRef]
- Created with BioRender Software. Available online: https://www.biorender.com/ (accessed on 2 April 2024).
- Li, Z.; Li, Y.; Gao, J.; Fu, Y.; Hua, P.; Jing, Y.; Cai, M.; Wang, H.; Tong, T. The role of CD47-SIRPα immune checkpoint in tumor immune evasion and innate immunotherapy. Life Sci. 2021, 273, 119150. [Google Scholar] [CrossRef]
- Dacek, M.M.; Kurtz, K.G.; Wallisch, P.; Pierre, S.A.; Khayat, S.; Bourne, C.M.; Gardner, T.J.; Vogt, K.C.; Aquino, N.; Younes, A.; et al. Potentiating antibody-dependent killing of cancers with CAR T cells secreting CD47-SIRPα checkpoint blocker. Blood 2023, 141, 2003–2015. [Google Scholar] [CrossRef]
- Zhao, H.; Song, S.; Ma, J.; Yan, Z.; Xie, H.; Feng, Y.; Che, S. CD47 as a promising therapeutic target in oncology. Front. Immunol. 2022, 13, 757480. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yang, Y.; Deng, Y.; Wei, F.; Zhao, Q.; Liu, Y.; Liu, Z.; Yu, B.; Huang, Z. Delivery of CD47 blocker SIRPα-Fc by CAR-T cells enhances antitumor efficacy. J. Immunother. Cancer 2022, 10, e003737. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.C.; Xu, Y.; Chen, X.C.; Zheng, L. Advances in the application of immune checkpoint inhibitors in gynecological tumors. Int. Immunopharmacol. 2023, 117, 109774. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Liang, H.; Hu, J.; Liu, S.; Hao, X.; Wong, M.S.K.; Li, X.; Hu, L. PD-L1 expression correlates with tumor infiltrating lymphocytes and response to neoadjuvant chemotherapy in cervical cancer. J. Cancer 2018, 9, 2938–2945. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Shapira-Frommer, R.; Santin, A.D.; Lisyanskaya, A.S.; Pignata, S.; Vergote, I.; Raspagliesi, F.; Sonke, G.S.; Birrer, M.; Provencher, D.M.; et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: Results from the phase II KEYNOTE-100 study. Ann. Oncol. 2019, 30, 1080–1087. [Google Scholar] [CrossRef]
- Zsiros, E.; Lynam, S.; Attwood, K.M.; Wang, C.; Chilakapati, S.; Gomez, E.C.; Liu, S.; Akers, S.; Lele, S.; Frederick, P.J.; et al. Efficacy and Safety of Pembrolizumab in Combination with Bevacizumab and Oral Metronomic Cyclophosphamide in the Treatment of Recurrent Ovarian Cancer A Phase 2 Nonrandomized Clinical Trial. JAMA Oncol. 2021, 7, 78–85. [Google Scholar] [CrossRef]
- Zheng, L.; Yang, X.; Wei, Y.; You, J.; Li, H.; Liao, J.; Yi, C. Advanced materials for management of immune-related adverse events induced by immune checkpoint inhibitors. Mater. Des. 2022, 219, 110738. [Google Scholar] [CrossRef]
- Abid, M.B. Early immunomodulators with CAR T-cell immunotherapy in the COVID-19 era. Lancet Oncol. 2022, 23, 16–18. [Google Scholar] [CrossRef]
- Morgado, M.; Plácido, A.; Morgado, S.; Roque, F. Management of the adverse effects of immune checkpoint inhibitors. Vaccines 2020, 8, 575. [Google Scholar] [CrossRef]
- Gu, S.L.; Nath, S.; Markova, A. Safety of Immunomodulatory Systemic Therapies Used in the Management of Immune-Related Cutaneous Adverse Events. Pharmaceuticals 2023, 16, 1610. [Google Scholar] [CrossRef]
- Rozenberg, J.M.; Zvereva, S.; Dalina, A.; Blatov, I.; Zubarev, I.; Luppov, D.; Bessmertnyi, A.; Romanishin, A.; Alsoulaiman, L.; Kumeiko, V.; et al. The p53 family member p73 in the regulation of cell stress response. Biol. Direct 2021, 16, 23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, L.; Nguyen, D.; Lu, H. TP53 mutations in epithelial ovarian cancer. Transl. Cancer Res. 2016, 5, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Rada, M.; Vasileva, E.; Lezina, L.; Marouco, D.; Antonov, A.V.; Macip, S.; Melino, G.; A Barlev, N. Human EHMT2/G9a activates p53 through methylation-independent mechanism. Oncogene 2017, 36, 922–932. [Google Scholar] [CrossRef] [PubMed]
- Lezina, L.; Aksenova, V.; Fedorova, O.; Malikova, D.; Shuvalov, O.; Antonov, A.V.; Tentler, D.; Garabadgiu, A.V.; Melino, G.; Barlev, N.A. KMT Set7/9 affects genotoxic stress response via the Mdm2 axis. Oncotarget 2015, 6, 25843–25855. [Google Scholar] [CrossRef] [PubMed]
- Sailer, C.; Offensperger, F.; Julier, A.; Kammer, K.-M.; Walker-Gray, R.; Gold, M.G.; Scheffner, M.; Stengel, F. Structural dynamics of the E6AP/UBE3A-E6-p53 enzyme-substrate complex. Nat. Commun. 2018, 9, 4441. [Google Scholar] [CrossRef]
- Wallis, B.; Bowman, K.R.; Lu, P.; Lim, C.S. The Challenges and Prospects of p53-Based Therapies in Ovarian Cancer. Biomolecules 2023, 13, 159. [Google Scholar] [CrossRef] [PubMed]
- Hassin, O.; Oren, M. Drugging p53 in cancer: One protein, many targets. Nat. Rev. Drug Discov. 2023, 22, 127–144. [Google Scholar] [CrossRef]
- Rahaman, J.; Steiner, N.; Hayes, M.P.; Chuang, L.; Fishman, D.; Iii, H.G. Chemotherapy for gynecologic cancers. Mt. Sinai J. Med. A J. Transl. Pers. Med. 2009, 76, 577–588. [Google Scholar] [CrossRef]
- Denkert, C.; von Minckwitz, G.; Brase, J.C.; Sinn, B.V.; Gade, S.; Kronenwett, R.; Pfitzner, B.M.; Salat, C.; Loi, S.; Schmitt, W.D.; et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2–positive and triple-negative primary breast cancers. J. Clin. Oncol. 2015, 33, 983–991. [Google Scholar] [CrossRef]
- Suryadevara, C.M.; Desai, R.; Abel, M.L.; Riccione, K.A.; Batich, K.A.; Shen, S.H.; Chongsathidkiet, P.; Gedeon, P.C.; Elsamadicy, A.A.; Snyder, D.J.; et al. Temozolomide lymphodepletion enhances CAR abundance and correlates with antitumor efficacy against established glioblastoma. OncoImmunology 2018, 7, e1434464. [Google Scholar] [CrossRef]
- Wang, A.X.; Ong, X.J.; D’souza, C.; Neeson, P.J.; Zhu, J.J. Combining chemotherapy with CAR-T cell therapy in treating solid tumors. Front. Immunol. 2023, 14, 1140541. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Hudecek, M.; Pender, B.; Robinson, E.; Hawkins, R.; Chaney, C.; Cherian, S.; Chen, X.; et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8 + and CD4 + CD19-specific chimeric antigen receptor–modified T cells. Sci. Transl. Med. 2016, 8, 355ra116. [Google Scholar] [CrossRef]
- Sesques, P.; Ferrant, E.; Safar, V.; Wallet, F.; Tordo, J.; Dhomps, A.; Karlin, L.; Brisou, G.; Vercasson, M.; Hospital-Gustem, C.; et al. Commercial anti-CD19 CAR cell therapy for patients with relapsed/refractory aggressive B cell lymphoma in a European center. Am. J. Hematol. 2020, 95, 1324–1333. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, R.; Musteanu, M.; Garcia-Garcia, E.; Lopez-Casas, P.P.; Megias, D.; Guerra, C.; Muñoz, M.; Quijano, Y.; Cubillo, A.; Rodriguez-Pascual, J.; et al. Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. Br. J. Cancer 2013, 109, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; He, Q.; Mao, D.; Wang, C.; Huang, L.; Wang, M.; Zhang, J. Efficacy and adverse reaction management of oncolytic viral intervention combined with chemotherapy in patients with liver metastasis of gastrointestinal malignancy. Front. Oncol. 2023, 13, 1159802. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Luo, Y.; Da, T.; Guedan, S.; Ruella, M.; Scholler, J.; Keith, B.; Young, R.M.; Engels, B.; Sorsa, S.; et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight 2018, 3, e99573. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, E.; Milenova, I.; Wenthe, J.; Ståhle, M.; Leja-Jarblad, J.; Ullenhag, G.; Dimberg, A.; Moreno, R.; Alemany, R.; Loskog, A. Shaping the tumor stroma and sparking immune activation by CD40 and 4-1BB signaling induced by an armed oncolytic virus. Clin. Cancer Res. 2017, 23, 5846–5857. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.K.; Wang, L.-C.S.; Bekdache, K.; Lynn, R.C.; Lo, A.; Thorne, S.H.; Albelda, S.M. Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. OncoImmunology 2018, 7, e1395997. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I study of DNX-2401 (delta-24-RGD) oncolytic adenovirus: Replication and immunotherapeutic effects in recurrent malignant glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef]
- Russell, S.J.; Federspiel, M.J.; Peng, K.-W.; Tong, C.; Dingli, D.; Morice, W.G.; Lowe, V.; O’Connor, M.K.; Kyle, R.A.; Leung, N.; et al. Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clin. Proc. 2014, 89, 926–933. [Google Scholar] [CrossRef]
- Wing, A.; Fajardo, C.A.; Posey, A.D.; Shaw, C.; Da, T.; Young, R.M.; Alemany, R.; June, C.H.; Guedan, S. Improving CART-cell therapy of solid tumors with oncolytic virus–driven production of a bispecific T-cell engager. Cancer Immunol. Res. 2018, 6, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xiao, F.; Zhang, A.; Zhang, D.; Nie, W.; Xu, T.; Han, B.; Seth, P.; Wang, H.; Yang, Y.; et al. Oncolytic adenovirus targeting TGF-β enhances anti-tumor responses of mesothelin-targeted chimeric antigen receptor T cell therapy against breast cancer. Cell. Immunol. 2020, 348, 104041. [Google Scholar] [CrossRef] [PubMed]
- Rosewell Shaw, A.; Porter, C.E.; Watanabe, N.; Tanoue, K.; Sikora, A.; Gottschalk, S.; Brenner, M.K.; Suzuki, M. Adenovirotherapy Delivering Cytokine and Checkpoint Inhibitor Augments CAR T Cells against Metastatic Head and Neck Cancer. Mol. Ther. 2017, 25, 2440–2451. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; McKenna, M.K.; Shaw, A.R.; Suzuki, M. Clinical CAR-T Cell and Oncolytic Virotherapy for Cancer Treatment. Mol. Ther. 2021, 29, 505–520. [Google Scholar] [CrossRef] [PubMed]
- Benencia, F.; Courreges, M.C.; Conejo-García, J.R.; Buckanovich, R.J.; Zhang, L.; Carroll, R.H.; Morgan, M.A.; Coukos, G. Oncolytic HSV exerts direct antiangiogenic activity in ovarian carcinoma. Hum. Gene Ther. 2005, 16, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, J.-H.; Choi, K.-J.; Kim, P.-H.; Yun, C.-O. E1A- and E1B-double mutant replicating adenovirus elicits enhanced oncolytic and antitumor effects. Hum. Gene Ther. 2007, 18, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Evgin, L.; Huff, A.L.; Wongthida, P.; Thompson, J.; Kottke, T.; Tonne, J.; Schuelke, M.; Ayasoufi, K.; Driscoll, C.B.; Shim, K.G.; et al. Oncolytic virus-derived type I interferon restricts CAR T cell therapy. Nat. Commun. 2020, 11, 3187. [Google Scholar] [CrossRef]
- Zheng, N.; Long, Y.; Bai, Z.; Li, J.; Wang, H.; Song, D.-D.; Liu, H.-L.; Shi, J.-H.; Zhao, S. Melatonin as an immunomodulator in CD19-targeting CAR-T cell therapy: Managing cytokine release syndrome. J. Transl. Med. 2024, 22, 58. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olifirenko, V.; Barlev, N.A. A Review of CAR-T Combination Therapies for Treatment of Gynecological Cancers. Int. J. Mol. Sci. 2024, 25, 6595. https://doi.org/10.3390/ijms25126595
Olifirenko V, Barlev NA. A Review of CAR-T Combination Therapies for Treatment of Gynecological Cancers. International Journal of Molecular Sciences. 2024; 25(12):6595. https://doi.org/10.3390/ijms25126595
Chicago/Turabian StyleOlifirenko, Valentina, and Nikolai A. Barlev. 2024. "A Review of CAR-T Combination Therapies for Treatment of Gynecological Cancers" International Journal of Molecular Sciences 25, no. 12: 6595. https://doi.org/10.3390/ijms25126595
APA StyleOlifirenko, V., & Barlev, N. A. (2024). A Review of CAR-T Combination Therapies for Treatment of Gynecological Cancers. International Journal of Molecular Sciences, 25(12), 6595. https://doi.org/10.3390/ijms25126595