
Adapt-cMolGPT: A Conditional Generative Pre-Trained Transformer with Adapter-Based Fine-Tuning for Target-Specific Molecular Generation


Abstract
:1. Introduction
2. Results and Discussion
2.1. Drug-like Compound Generation Results from Pre-Trained Model
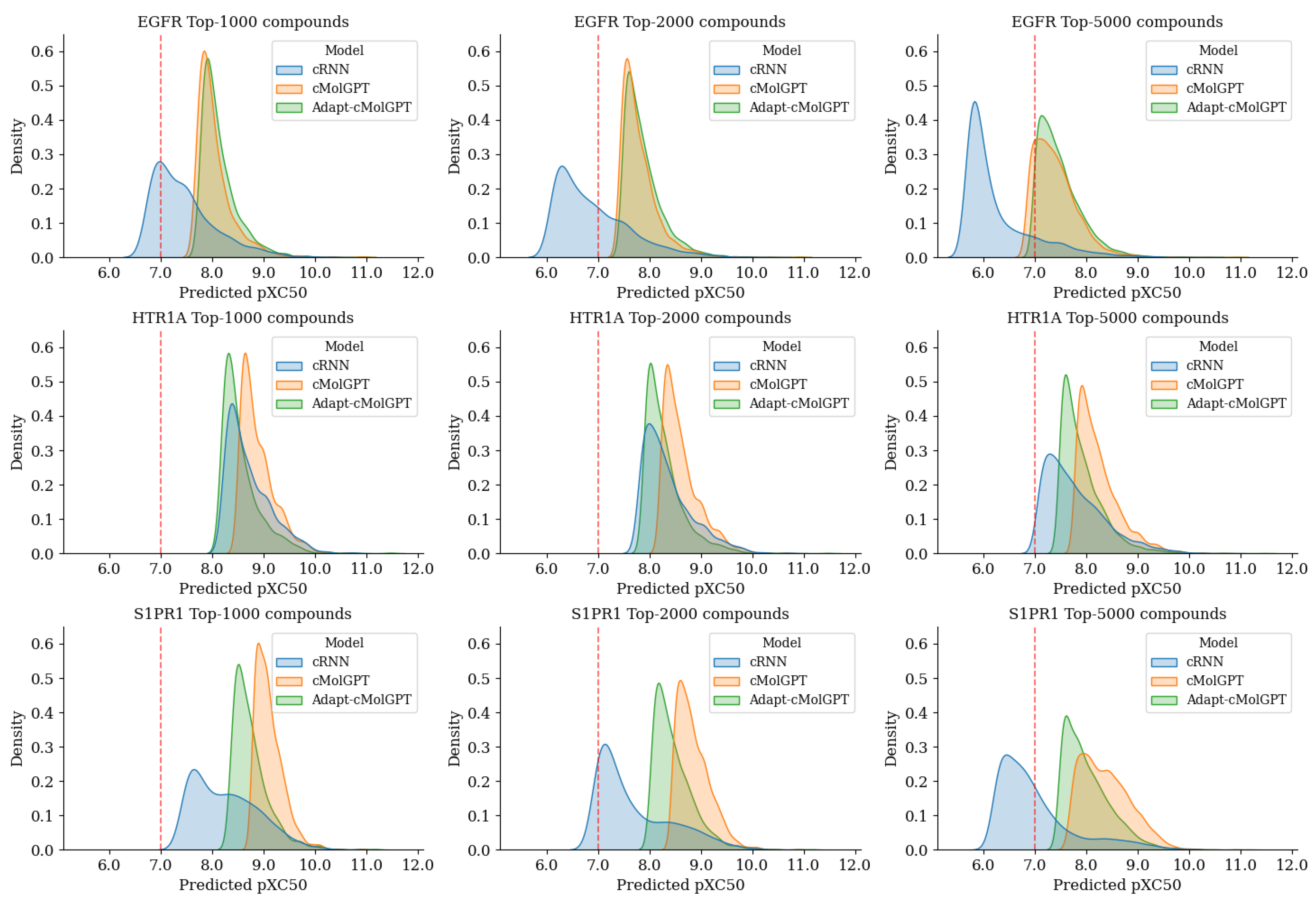
2.2. Target-Specific Compound Generation Results from Fine-Tuned Model
3. Methods and Materials
3.1. Dataset
3.2. Molecular Representation Methods
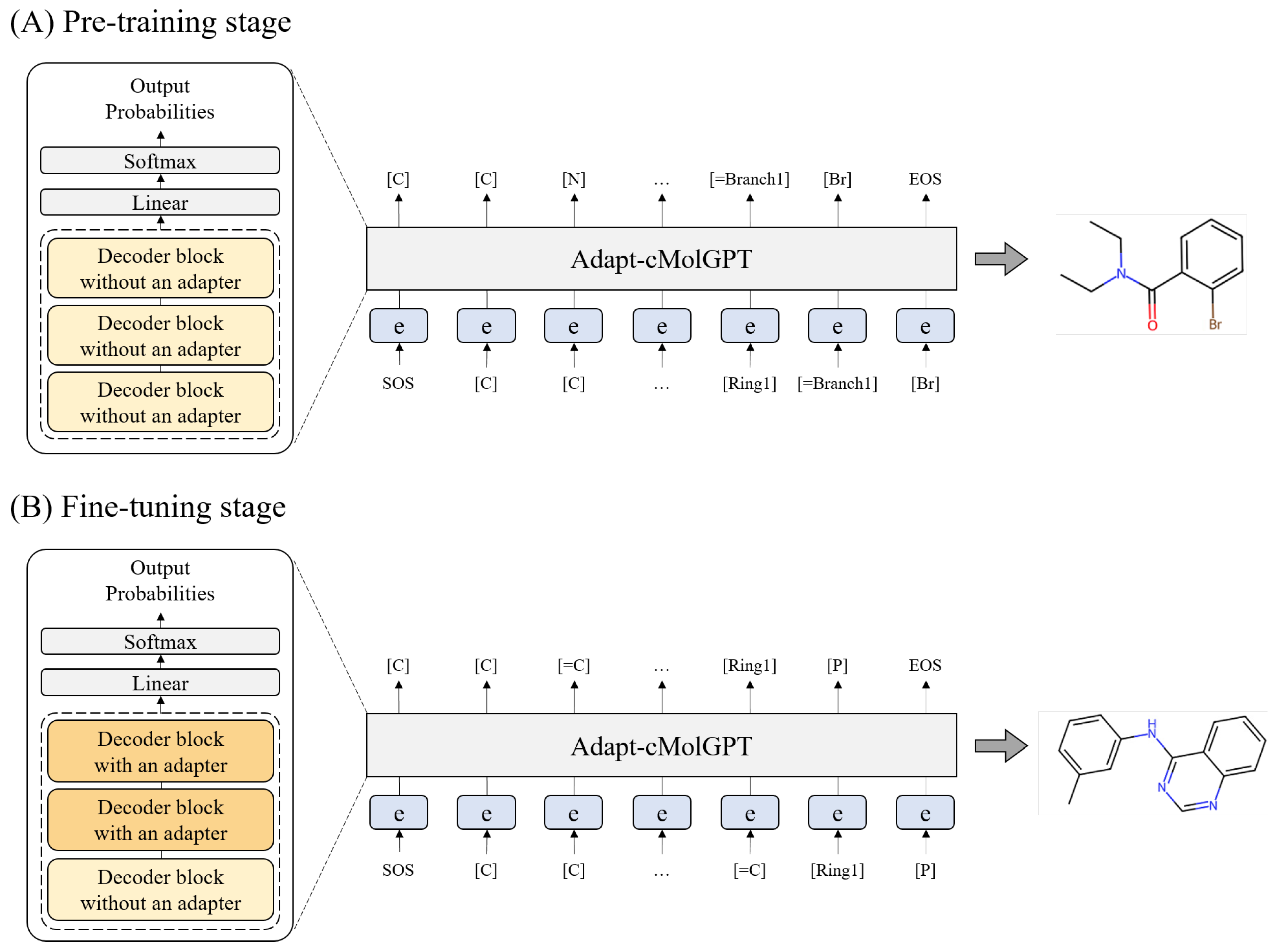
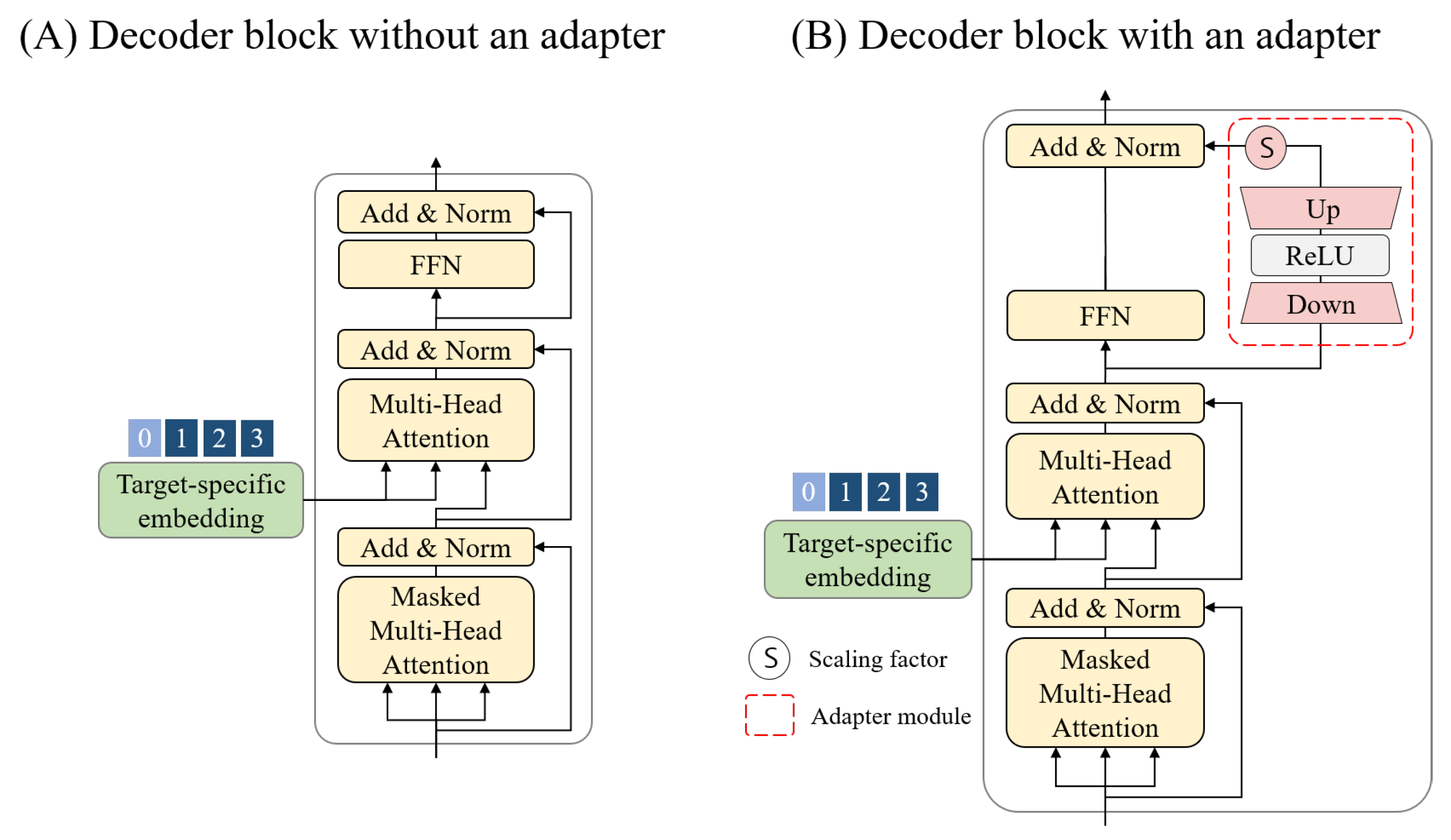
3.3. Generative Pre-Trained Transformer for Conditional Molecular Generation
3.4. Workflow for Training
3.5. Pre-Training Process
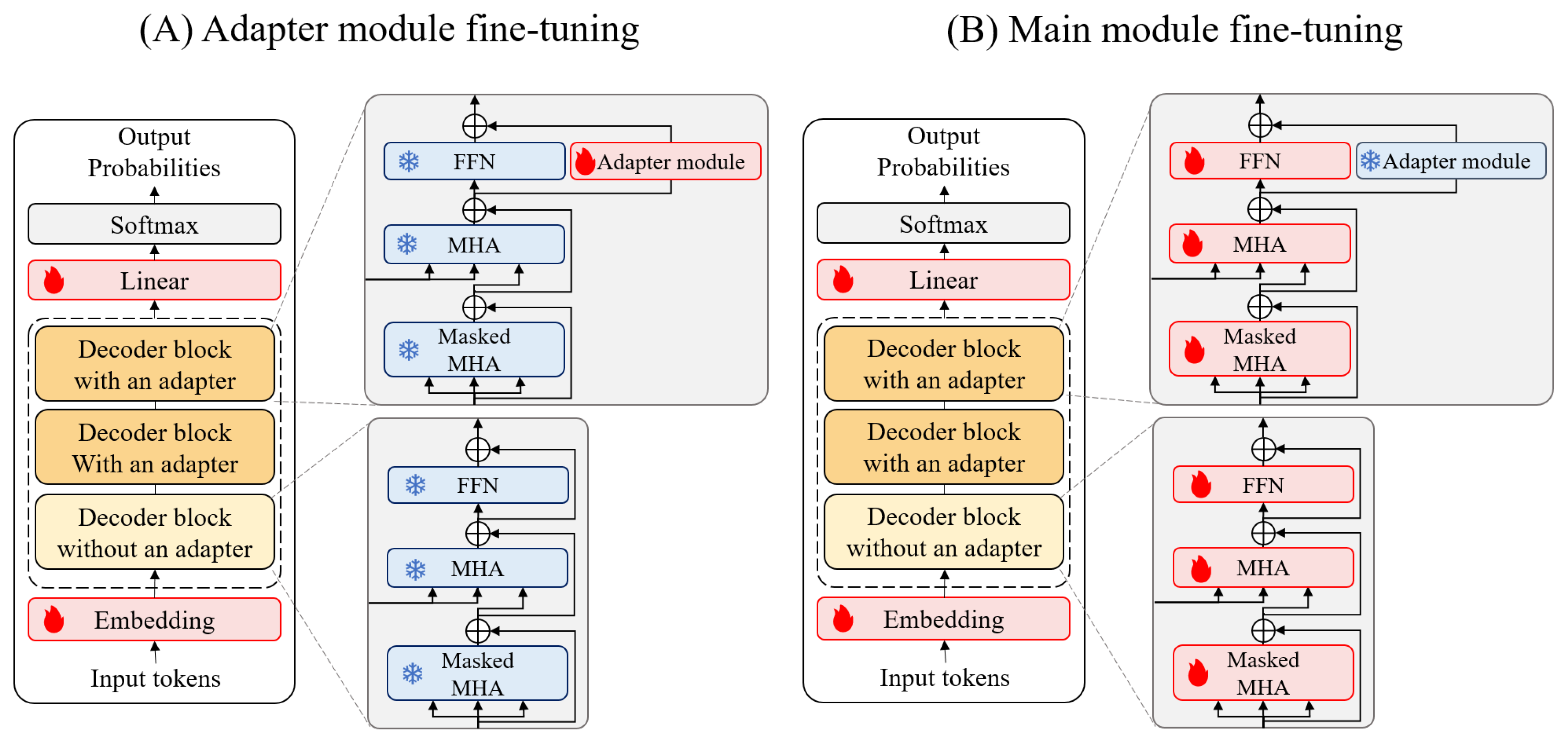
3.6. Fine-Tuning Process
3.7. Generation Process
3.8. ML-Based QSAR Model for Active Scoring
3.9. Performance Metrics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Polishchuk, P.G.; Madzhidov, T.I.; Varnek, A. Estimation of the size of drug-like chemical space based on GDB-17 data. J. Comput. Aided Mol. Des. 2013, 27, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on natural products for drug design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated research and development investment needed to bring a new medicine to market, 2009–2018. JAMA 2020, 323, 844–853. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [PubMed]
- Schlander, M.; Hernandez-Villafuerte, K.; Cheng, C.Y.; Mestre-Ferrandiz, J.; Baumann, M. How much does it cost to research and develop a new drug? A systematic review and assessment. Pharmacoeconomics 2021, 39, 1243–1269. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.B.M.A. Artificial intelligence effecting a paradigm shift in drug development. SLAS Technol. 2021, 26, 3–15. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A. Research and development costs of new drugs. JAMA 2020, 324, 517. [Google Scholar] [CrossRef]
- Scotti, L.; Scotti, M.T. Recent advancement in computer-aided drug design. Curr. Pharm. Des. 2020, 26, 1635–1636. [Google Scholar] [CrossRef]
- Ekins, S.; Mestres, J.; Testa, B. In silico pharmacology for drug discovery: Methods for virtual ligand screening and profiling. Br. J. Pharmacol. 2007, 152, 9–20. [Google Scholar] [CrossRef]
- Schneider, P.; Schneider, G. De novo design at the edge of chaos: Miniperspective. J. Med. Chem. 2016, 59, 4077–4086. [Google Scholar] [CrossRef]
- Devi, R.V.; Sathya, S.S.; Coumar, M.S. Evolutionary algorithms for de novo drug design—A survey. Appl. Soft Comput. 2015, 27, 543–552. [Google Scholar] [CrossRef]
- Gupta, A.; Müller, A.T.; Huisman, B.J.; Fuchs, J.A.; Schneider, P.; Schneider, G. Generative recurrent networks for de novo drug design. Mol. Inform. 2018, 37, 1700111. [Google Scholar] [CrossRef] [PubMed]
- Segler, M.H.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 2018, 4, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef]
- Kadurin, A.; Nikolenko, S.; Khrabrov, K.; Aliper, A.; Zhavoronkov, A. druGAN: An advanced generative adversarial autoencoder model for de novo generation of new molecules with desired molecular properties in silico. Mol. Pharm. 2017, 14, 3098–3104. [Google Scholar] [CrossRef]
- Prykhodko, O.; Johansson, S.V.; Kotsias, P.C.; Arús-Pous, J.; Bjerrum, E.J.; Engkvist, O.; Chen, H. A de novo molecular generation method using latent vector based generative adversarial network. J. Cheminform. 2019, 11, 74. [Google Scholar] [CrossRef] [PubMed]
- Olivecrona, M.; Blaschke, T.; Engkvist, O.; Chen, H. Molecular de novo design through deep reinforcement learning. J. Cheminform. 2017, 9, 48. [Google Scholar] [CrossRef]
- Liu, X.; Ye, K.; Van Vlijmen, H.W.; IJzerman, A.P.; Van Westen, G.J. An exploration strategy improves the diversity of de novo ligands using deep reinforcement learning: A case for the adenosine A2A receptor. J. Cheminform. 2019, 11, 35. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, H.; Sciabola, S.; Wang, W. cMolGPT: A conditional generative pre-trained transformer for target-specific de novo molecular generation. Molecules 2023, 28, 4430. [Google Scholar] [CrossRef]
- Radford, A.; Narasimhan, K.; Salimans, T.; Sutskever, I. Improving Language Understanding by Generative Pre-Training. 2018. Available online: https://cdn.openai.com/research-covers/language-unsupervised/language_understanding_paper.pdf (accessed on 14 May 2024).
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Krenn, M.; Häse, F.; Nigam, A.; Friederich, P.; Aspuru-Guzik, A. Self-referencing embedded strings (SELFIES): A 100% robust molecular string representation. Mach. Learn. Sci. Technol. 2020, 1, 045024. [Google Scholar] [CrossRef]
- Handa, K.; Thomas, M.C.; Kageyama, M.; Iijima, T.; Bender, A. On the difficulty of validating molecular generative models realistically: A case study on public and proprietary data. J. Cheminf. 2023, 15, 112. [Google Scholar] [CrossRef] [PubMed]
- Probst, D.; Reymond, J.L. A probabilistic molecular fingerprint for big data settings. J. Cheminform. 2018, 10, 66. [Google Scholar] [CrossRef] [PubMed]
- Probst, D.; Reymond, J.L. Visualization of very large high-dimensional data sets as minimum spanning trees. J. Cheminform. 2020, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Polykovskiy, D.; Zhebrak, A.; Sanchez-Lengeling, B.; Golovanov, S.; Tatanov, O.; Belyaev, S.; Kurbanov, R.; Artamonov, A.; Aladinskiy, V.; Veselov, M.; et al. Molecular sets (MOSES): A benchmarking platform for molecular generation models. Front. Pharmacol. 2020, 11, 565644. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Yüksel, A.; Ulusoy, E.; Ünlü, A.; Doğan, T. SELFormer: Molecular representation learning via SELFIES language models. Mach. Learn. Sci. Technol. 2023, 4, 025035. [Google Scholar] [CrossRef]
- Chen, S.; Ge, C.; Tong, Z.; Wang, J.; Song, Y.; Wang, J.; Luo, P. Adaptformer: Adapting vision transformers for scalable visual recognition. Adv. Neural Inf. Process Syst. 2022, 35, 16664–16678. [Google Scholar]
- Xu, L.; Xie, H.; Qin, S.Z.J.; Tao, X.; Wang, F.L. Parameter-efficient fine-tuning methods for pretrained language models: A critical review and assessment. arXiv 2023, arXiv:2312.12148. [Google Scholar]
- Sun, C.; Qiu, X.; Xu, Y.; Huang, X. How to fine-tune bert for text classification? In Proceedings of the 18th China National Conference on Computational Linguistics, Kunming, China, 18 October 2019; pp. 194–206. [Google Scholar]
- Howard, J.; Ruder, S. Universal language model fine-tuning for text classification. arXiv 2018, arXiv:1801.06146. [Google Scholar]
- Sun, J.; Jeliazkova, N.; Chupakhin, V.; Golib-Dzib, J.F.; Engkvist, O.; Carlsson, L.; Wegner, J.; Ceulemans, H.; Georgiev, I.; Jeliazkov, V.; et al. ExCAPE-DB: An integrated large scale dataset facilitating Big Data analysis in chemogenomics. J. Cheminf. 2017, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.Y. Lightgbm: A highly efficient gradient boosting decision tree. Adv. Neural Inf. Process Syst. 2017, 30, 3146–3154. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Valid | Unique@1k | Unique@10k | Novel |
|---|---|---|---|---|
| cMolGPT | 0.985 | 1.0 | 1.0 | 0.835 |
| Adapt-cMolGPT | 1.0 | 1.0 | 0.999 | 0.999 |
| Target | Model | Valid | Unique@10k | Novel |
|---|---|---|---|---|
| EGFR | cRNN | 0.926 | 0.862 | 0.946 |
| cMolGPT | 0.842 | 0.922 | 0.955 | |
| Adapt-cMolGPT | 1.0 | 0.944 | 1.0 | |
| HTR1A | cRNN | 0.922 | 0.845 | 0.782 |
| cMolGPT | 0.885 | 0.889 | 0.889 | |
| Adapt-cMolGPT | 1.0 | 0.962 | 1.0 | |
| S1PR1 | cRNN | 0.921 | 0.861 | 0.947 |
| cMolGPT | 0.905 | 0.869 | 0.912 | |
| Adapt-cMolGPT | 1.0 | 0.931 | 1.0 |
| MW ([200, 500]) | TPSA ([20, 130]) | LogP ([−1, 6]) | HBD ([0, 5]) | HBA ([0, 10]) | QED ([0.4, 1]) | SA ([1, 5]) | |
|---|---|---|---|---|---|---|---|
| EGFR | 71.68% | 93.95% | 89.63% | 99.20% | 99.16% | 51.66% | 80.64% |
| HTR1A | 89.30% | 93.12% | 97.51% | 99.89% | 99.92% | 78.28% | 87.91% |
| S1PR1 | 86.09% | 89.50% | 90.43% | 99.39% | 99.45% | 55.41% | 81.77% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoo, S.; Kim, J. Adapt-cMolGPT: A Conditional Generative Pre-Trained Transformer with Adapter-Based Fine-Tuning for Target-Specific Molecular Generation. Int. J. Mol. Sci. 2024, 25, 6641. https://doi.org/10.3390/ijms25126641
Yoo S, Kim J. Adapt-cMolGPT: A Conditional Generative Pre-Trained Transformer with Adapter-Based Fine-Tuning for Target-Specific Molecular Generation. International Journal of Molecular Sciences. 2024; 25(12):6641. https://doi.org/10.3390/ijms25126641
Chicago/Turabian StyleYoo, Soyoung, and Junghyun Kim. 2024. "Adapt-cMolGPT: A Conditional Generative Pre-Trained Transformer with Adapter-Based Fine-Tuning for Target-Specific Molecular Generation" International Journal of Molecular Sciences 25, no. 12: 6641. https://doi.org/10.3390/ijms25126641