
Machine Learning-Driven Classification of Urease Inhibitors Leveraging Physicochemical Properties as Effective Filter Criteria

 , ,
, ,  , ,
, ,
Abstract
:1. Introduction
1.1. Urease Enzyme and Its Implications in the Human Context
1.2. Machine Learning in the New Era of Computer-Aided Drug Discovery
2. Results and Discussion
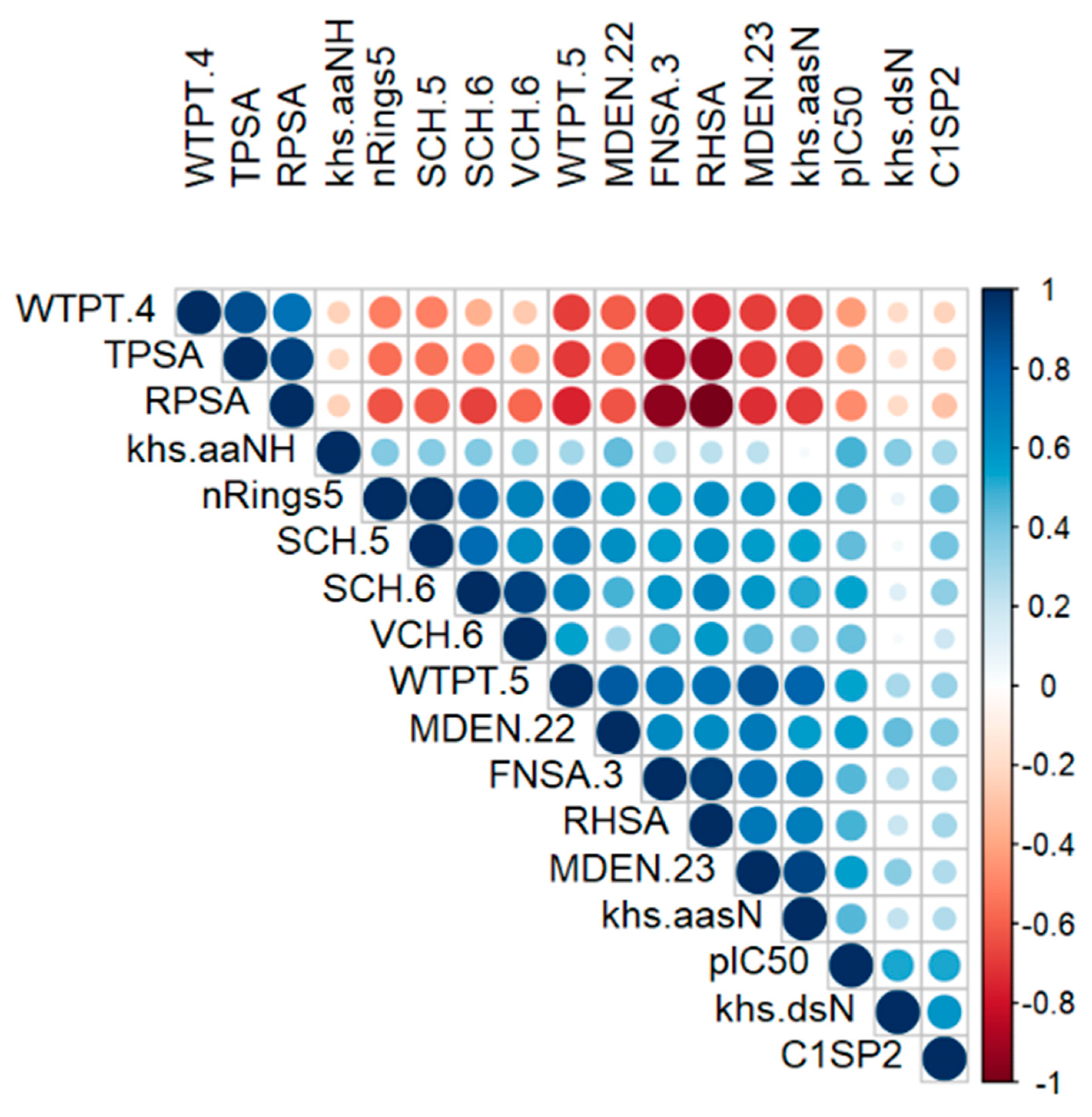
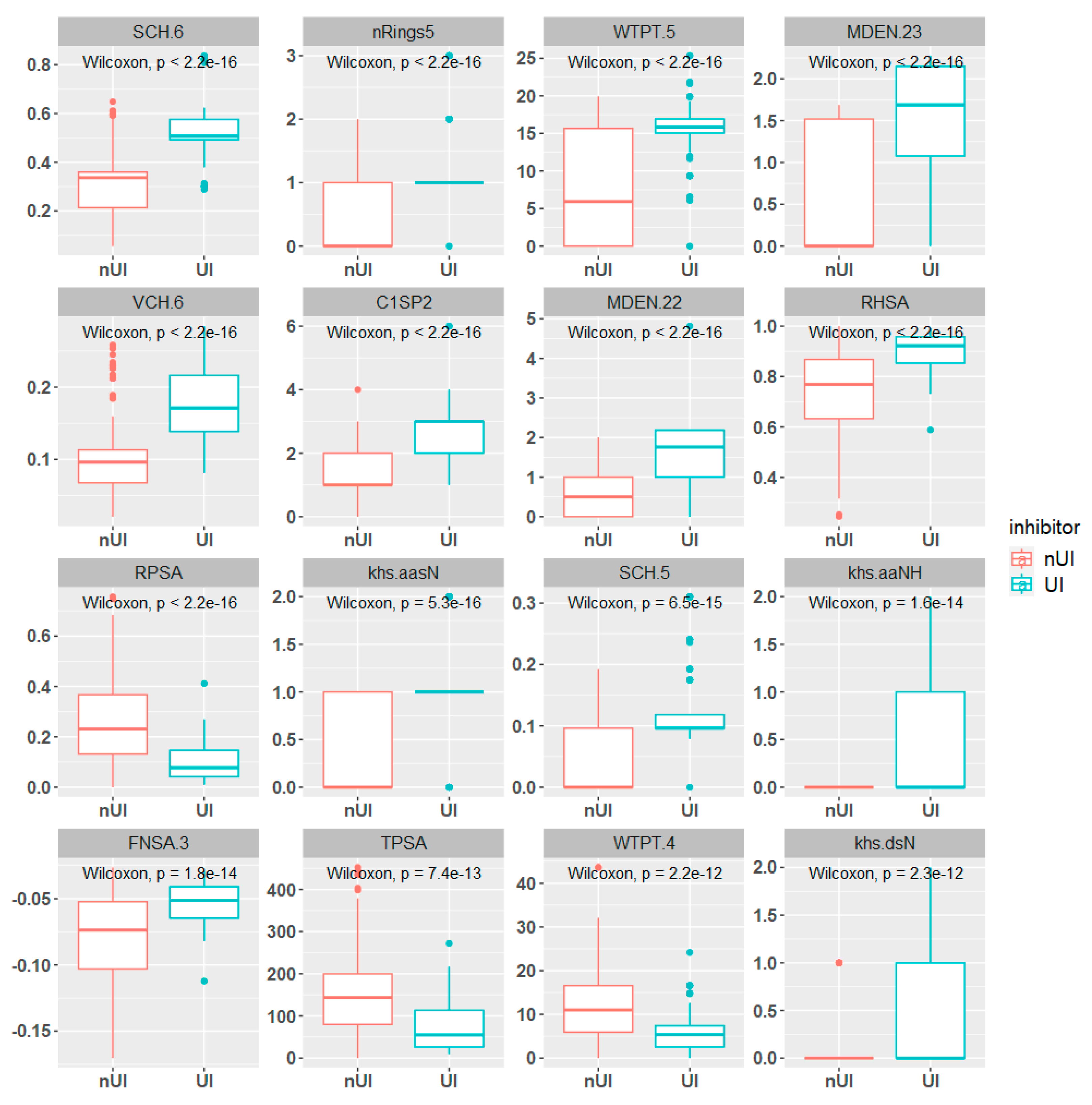
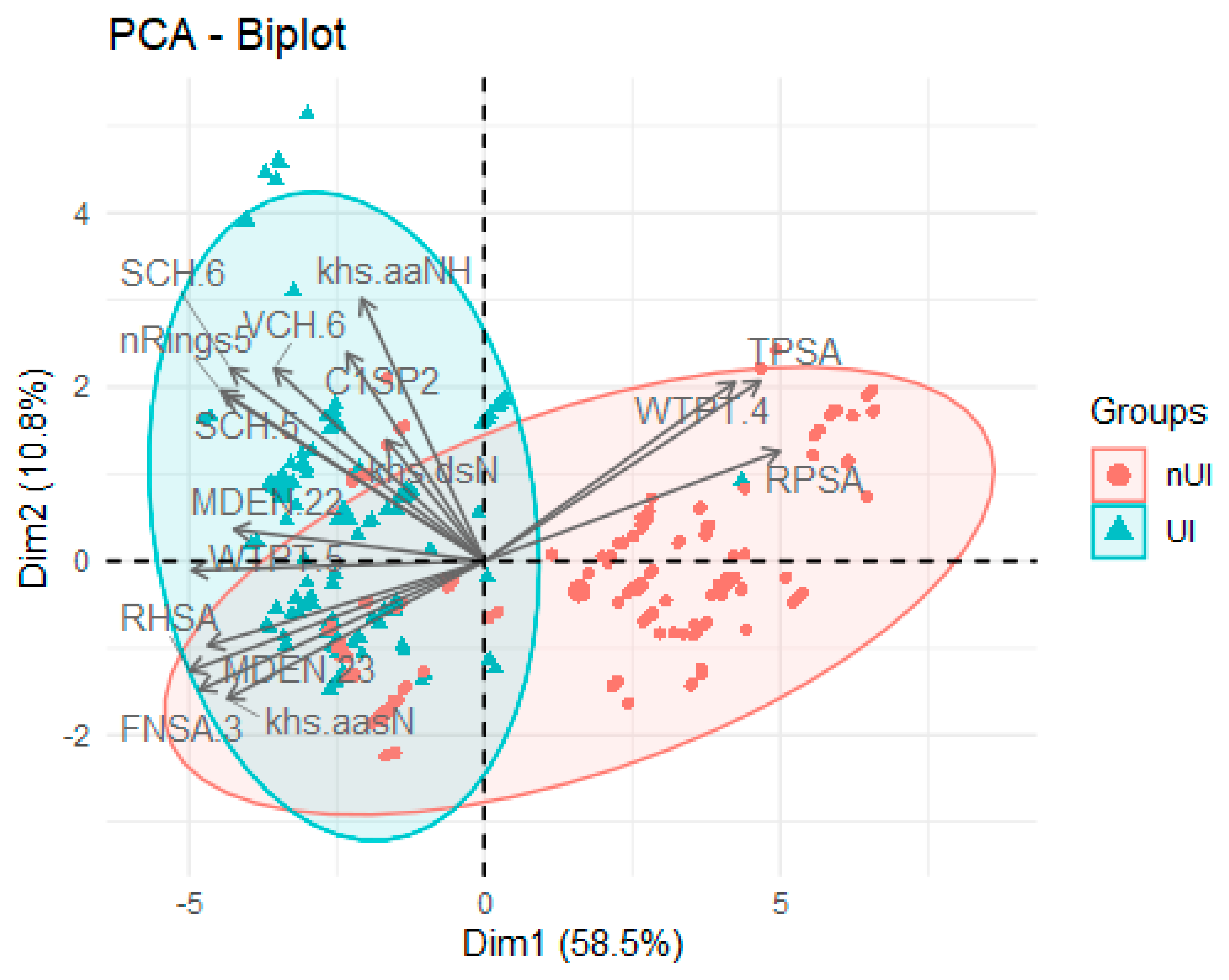
2.1. Exploratory Data Analysis
2.2. Machine Learning Models
3. Materials and Methods
3.1. Data Collection
3.2. Characterization and Preprocessing
3.3. Exploratory Data Analysis
3.4. Strategies for Bioactivity Categorization (Data Splitting)
3.5. Training and Testing of Inhibitory Classification Models
4. Conclusions
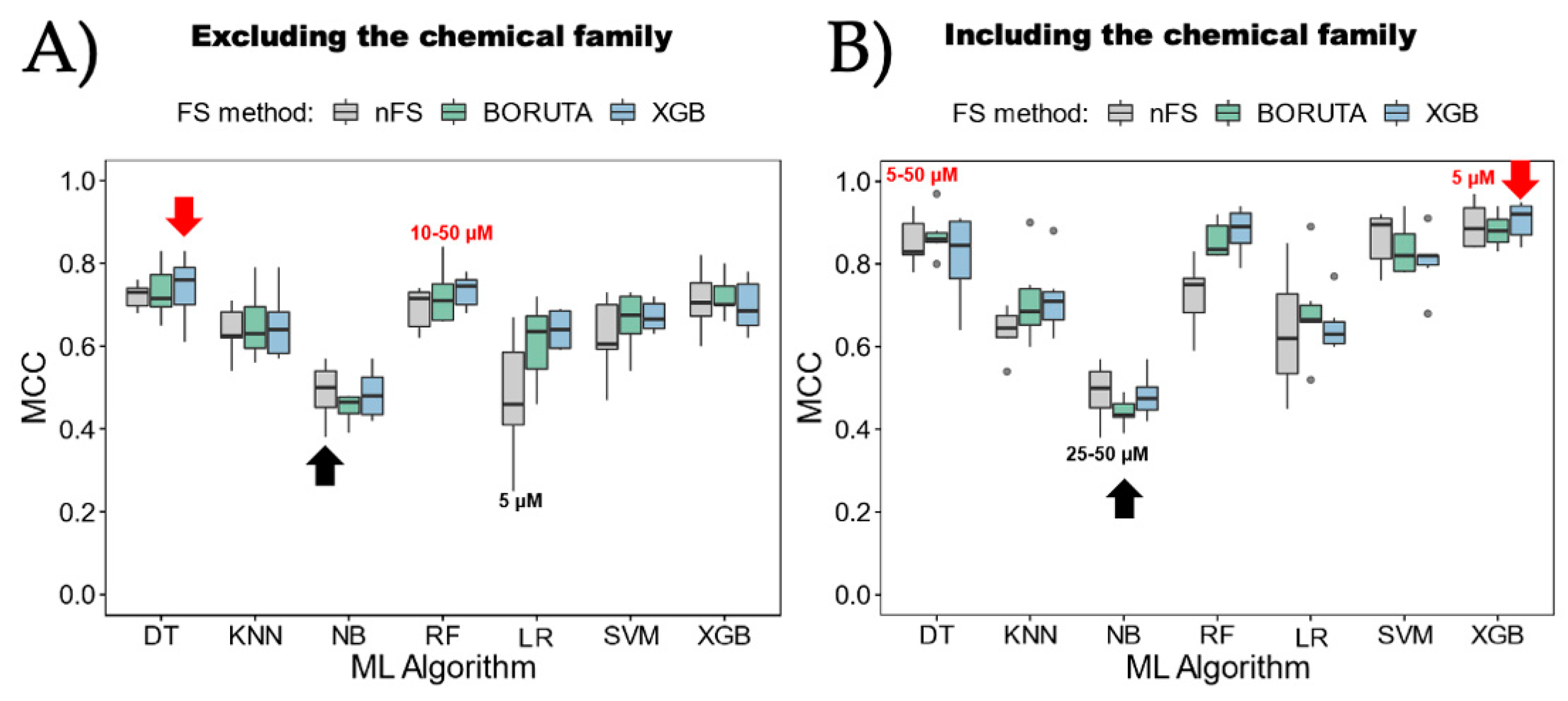
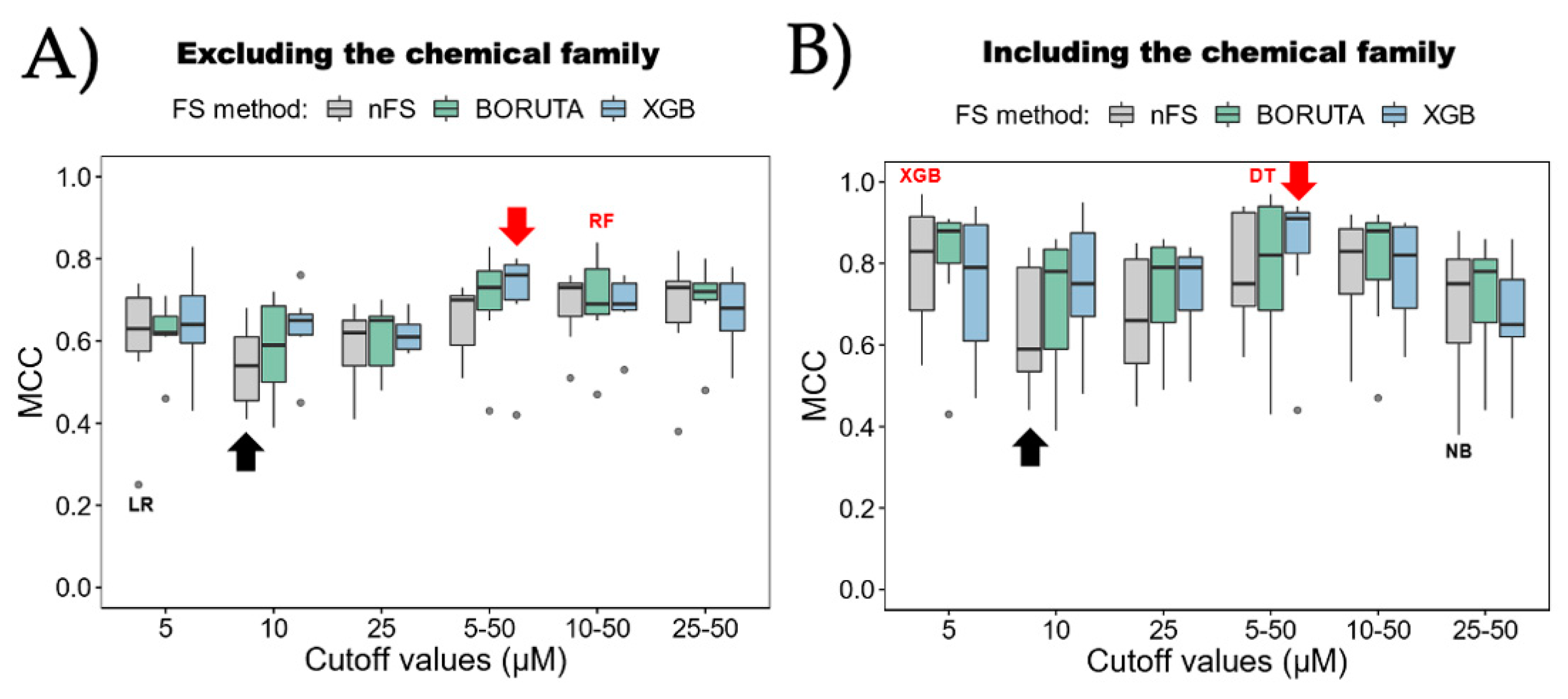
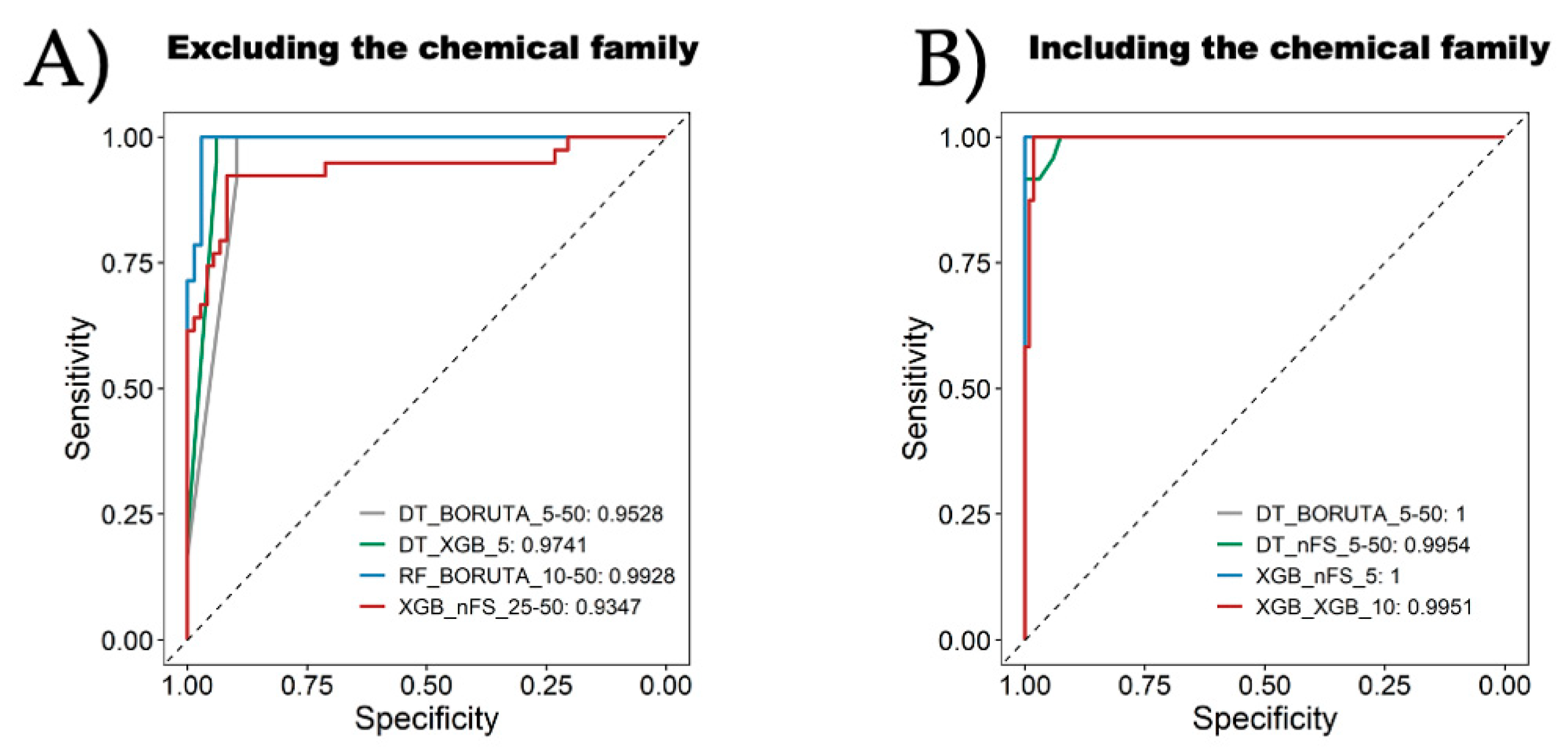
- Algorithm Preference: The study recommends favoring tree-based methods, including random forest (RF), decision tree (DT), and eXtreme Gradient Boosting (XGB), over other algorithms like k-nearest neighbor (KNN), support vector machine (SVM), naive Bayes (NB), or logistic regression (LR) for inhibitor classification.
- Attribute Selection Influence: While attribute selection methods could potentially improve model performance, their influence varies based on the ML algorithm chosen. There is not a clear preference for one method over another, suggesting their implementation should be algorithm-specific.
- Effective Categorization Strategies: The exploratory data analysis and ML analysis recommend employing strategies that involve a gray zone, utilizing two cutoffs for categorizing urease UIs and nUIs. These strategies tend to yield better model performance, offering improved accuracy in classification tasks, reaching almost 10 percent over one-cutoff strategies in our models. By delineating these boundaries, we can effectively train our models to distinguish between active and inactive compounds, thus enhancing the accuracy of our predictions. Moreover, understanding the implications of these cutoffs is critical for optimizing model performance. Nevertheless, strategies with a gray zone can lead to better performance, and it is crucial to consider the biological implications. Expanding the gray zone for categorization may result in the loss of important information about inhibitors.
- Consideration of Chemical Family: The inclusion of the chemical family attribute significantly enhances the classification models. However, obtaining this attribute might require manual annotation or inspection as automatic extraction from databases like ChemBL might not be straightforward. Despite the effort required, incorporating this attribute contributes to the models’ effectiveness.
- State of Art in Urease Inhibitors: To the best of our knowledge, in the context of drug discovery targeting Hp infection through urease inhibition, our study stands out as the most comprehensive and systematic evaluation of optimal conditions for developing predictive models of bioactivity for potential inhibitor candidates. By rigorously testing various attribute selection methods, machine learning algorithms, and bioactivity categorization strategies, we provide a robust framework that could significantly accelerate the identification and development of novel urease inhibitors. The elucidation of the structure–activity relationship (SAR) is crucial for rational drug design as it provides valuable information about how changes in the chemical structure of compounds affect their biological activity. Our investigation contributes to this understanding by identifying molecular features that correlate with UIs. By analyzing these relationships, researchers can gain insights into the chemical properties that are essentials for designing potent UIs.
- Practical Significance for Drug Design: Our approach serves as a practical guide applicable not only to urease but also to other proteins in drug design, potentially impacting the field with its systematic methodology and comprehensive evaluation. This intersection between computational modeling and biological relevance highlights the significance of our findings in advancing both drug discovery efforts and our understanding of urease inhibition mechanisms. Developing predictive models that accurately classify compounds based on their inhibitory activity against any relevant clinical target, as demonstrated in our study, enables the efficient screening of large compound libraries to identify promising drug candidates. This can significantly accelerate the drug discovery process by prioritizing compounds with the highest likelihood of exhibiting inhibitory activity.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Minkara, M.S.; Weaver, M.N.; Merz, K.M. Effect of 10.5 M Aqueous Urea on Helicobacter pylori Urease: A Molecular Dynamics Study. Biochemistry 2015, 54, 4121–4130. [Google Scholar] [CrossRef] [PubMed]
- Kafarski, P.; Talma, M. Recent Advances in Design of New Urease Inhibitors: A Review. J. Adv. Res. 2018, 13, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Meng, X.; Li, Y.; Zhao, C.N.; Tang, G.Y.; Li, S.; Gan, R.Y.; Li, H. Bin Natural Products for the Prevention and Management of Helicobacter pylori Infection. Compr. Rev. Food Sci. Food Saf. 2018, 17, 937–952. [Google Scholar] [CrossRef] [PubMed]
- Bravo, D.; Hoare, A.; Soto, C.; Valenzuela, M.A.; Quest, A.F. Helicobacter pylori in Human Health and Disease: Mechanisms for Local Gastric and Systemic Effects. World J. Gastroenterol. 2018, 24, 3071–3089. [Google Scholar] [CrossRef] [PubMed]
- González, I.; Araya, P.; Roj, A. Helicobacter pylori Infection and Lung Cancer: New Insights and Future Challenges. Chin. J. Lung Cancer 2018, 21, 658–662. [Google Scholar] [CrossRef]
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef]
- Vaira, D.; Holton, J.; Miglioli, M.; Menegatti, M.; Mule, P.; Barbara, L. Peptic Ulcer Disease and Helicobacter pylori Infection. Curr. Opin. Gastroenterol. 1994, 10, 98–104. [Google Scholar] [CrossRef]
- Kavitt, R.T.; Lipowska, A.M.; Anyane-Yeboa, A.; Gralnek, I.M. Diagnosis and Treatment of Peptic Ulcer Disease. Am. J. Med. 2019, 132, 447–456. [Google Scholar] [CrossRef]
- Wijarnpreecha, K.; Thongprayoon, C.; Nissaisorakarn, P.; Jaruvongvanich, V.; Nakkala, K.; Rajapakse, R.; Cheungpasitporn, W. Association of Helicobacter pylori with Chronic Kidney Diseases: A Meta-Analysis. Dig. Dis. Sci. 2017, 62, 2045–2052. [Google Scholar] [CrossRef]
- Kim, T.H.; Cheung, D.Y. Helicobacter pylori Eradication, a Gordian Knot for Idiopathic Thrombocytopenic Purpura? Gut Liver 2016, 10, 323–324. [Google Scholar] [CrossRef]
- Kim, B.J.; Kim, H.S.; Jang, H.J.; Kim, J.H. Helicobacter pylori Eradication in Idiopathic Thrombocytopenic Purpura: A Meta-Analysis of Randomized Trials. Gastroenterol. Res. Pract. 2018, 2018, 6090878. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Osaki, T.; Kamiya, S.; Zhang, X.S.; Blaser, M.J. Helicobacter pylori SabA Gene Is Associated with Iron Deficiency Anemia in Childhood and Adolescence. PLoS ONE 2017, 12, e0184046. [Google Scholar] [CrossRef]
- Doorakkers, E.; Lagergren, J.; Engstrand, L.; Brusselaers, N. Helicobacter pylori Eradication Treatment and the Risk of Gastric Adenocarcinoma in a Western Population. Gut 2018, 67, 2092–2096. [Google Scholar] [CrossRef] [PubMed]
- Shapla, U.M.; Raihan, J.; Islam, A.; Alam, F.; Solayman, N.; Gan, S.H.; Hossen, S.; Khalil, I. Propolis: The Future Therapy against Helicobacter pylori-Mediated Gastrointestinal Diseases. J. Appl. Biomed. 2018, 16, 81–99. [Google Scholar] [CrossRef]
- Floch, P.; Mégraud, F.; Lehours, P. Helicobacter pylori Strains and Gastric MALT Lymphoma. Toxins 2017, 9, 132. [Google Scholar] [CrossRef]
- Violeta Filip, P.; Cuciureanu, D.; Sorina Diaconu, L.; Maria Vladareanu, A.; Silvia Pop, C. MALT Lymphoma: Epidemiology, Clinical Diagnosis and Treatment. J. Med. Life 2018, 11, 187–193. [Google Scholar] [CrossRef]
- Salar, A. Gastric MALT Lymphoma and Helicobacter pylori. Med. Clin. 2019, 152, 65–71. [Google Scholar] [CrossRef]
- Flores-Treviño, S.; Mendoza-Olazarán, S.; Bocanegra-Ibarias, P.; Maldonado-Garza, H.J.; Garza-González, E. Helicobacter pylori Drug Resistance: Therapy Changes and Challenges. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer Statistics for the Year 2020: An Overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Malfertheiner, P.; Megraud, F.; O’Morain, C.; Gisbert, J.P.; Kuipers, E.J.; Axon, A.; Bazzoli, F.; Gasbarrini, A.; Atherton, J.; Graham, D.Y.; et al. Management of Helicobacter pylori Infection-the Maastricht V/Florence Consensus Report. Gut 2017, 66, 6–30. [Google Scholar] [CrossRef]
- Liou, J.M.; Fang, Y.J.; Chen, C.C.; Bair, M.J.; Chang, C.Y.; Lee, Y.C.; Chen, M.J.; Chen, C.C.; Tseng, C.H.; Hsu, Y.C.; et al. Concomitant, Bismuth Quadruple, and 14-Day Triple Therapy in the First-Line Treatment of Helicobacter pylori: A Multicentre, Open-Label, Randomised Trial. Lancet 2016, 388, 2355–2365. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.T.; Chang, W.L.; Wu, C.T.; Yang, H.B.; Kuo, H.Y.; Lin, M.Y.; Cheng, H.C.; Tsai, Y.C.; Sheu, B.S. Optimizing the MIC Breakpoints of Amoxicillin and Tetracycline for Antibiotic Selection in the Rescue Therapy of H. Pylori with Bismuth Quadruple Regimen. Eur. J. Clin. Pharmacol. 2020, 76, 1581–1589. [Google Scholar] [CrossRef] [PubMed]
- Horie, R.; Handa, O.; Ando, T.; Ose, T.; Murakami, T.; Suzuki, N.; Sendo, R.; Imamoto, E.; Itoh, Y. Helicobacter pylori Eradication Therapy Outcome according to Clarithromycin Susceptibility Testing in Japan. Helicobacter 2020, 25, e12698. [Google Scholar] [CrossRef] [PubMed]
- Alomari, M.; Taha, M.; Imran, S.; Jamil, W.; Selvaraj, M.; Uddin, N.; Rahim, F. Design, Synthesis, in Vitro Evaluation, Molecular Docking and ADME Properties Studies of Hybrid Bis-Coumarin with Thiadiazole as a New Inhibitor of Urease. Bioorg. Chem. 2019, 92, 103235. [Google Scholar] [CrossRef]
- Kazmi, M.; Khan, I.; Khan, A.; Halim, S.A.; Saeed, A.; Mehsud, S.; Al-Harrasi, A.; Ibrar, A. Developing New Hybrid Scaffold for Urease Inhibition Based on Carbazole-Chalcone Conjugates: Synthesis, Assessment of Therapeutic Potential and Computational Docking Analysis. Bioorg. Med. Chem. 2019, 27, 115123. [Google Scholar] [CrossRef] [PubMed]
- Modolo, L.V.; da-Silva, C.J.; Brandão, D.S.; Chaves, I.S. A Minireview on What We Have Learned about Urease Inhibitors of Agricultural Interest since Mid-2000s. J. Adv. Res. 2018, 13, 29–37. [Google Scholar] [CrossRef]
- Liu, H.; Wang, Y.; Lv, M.; Luo, Y.; Liu, B.M.; Huang, Y.; Wang, M.; Wang, J. Flavonoid Analogues as Urease Inhibitors: Synthesis, Biological Evaluation, Molecular Docking Studies and In-Silico ADME Evaluation. Bioorg. Chem. 2020, 105, 104370. [Google Scholar] [CrossRef] [PubMed]
- Chandra Babu, T.M.; Rajesh, S.S.; Bhaskar, B.V.; Devi, S.; Rammohan, A.; Sivaraman, T.; Rajendra, W. Molecular Docking, Molecular Dynamics Simulation, Biological Evaluation and 2D QSAR Analysis of Flavonoids from Syzygium alternifolium as Potent Anti-Helicobacter pylori Agents. RSC Adv. 2017, 7, 18277–18292. [Google Scholar] [CrossRef]
- Yener, I.; Kocakaya, S.O.; Ertas, A.; Erhan, B.; Kaplaner, E.; Oral, E.V.; Yilmaz-Ozden, T.; Yilmaz, M.A.; Ozturk, M.; Kolak, U. Selective In Vitro and In Silico Enzymes Inhibitory Activities of Phenolic Acids and Flavonoids of Food Plants: Relations with Oxidative Stress. Food Chem. 2020, 327, 127045. [Google Scholar] [CrossRef]
- Taha, M.; Rahim, F.; Khan, A.A.; Anouar, E.H.; Ahmed, N.; Shah, S.A.A.; Ibrahim, M.; Zakari, Z.A. Synthesis of Diindolylmethane (DIM) Bearing Thiadiazole Derivatives as a Potent Urease Inhibitor. Sci. Rep. 2020, 10, 7969. [Google Scholar] [CrossRef]
- Khan, I.; Khan, A.; Ahsan Halim, S.; Saeed, A.; Mehsud, S.; Csuk, R.; Al-Harrasi, A.; Ibrar, A. Exploring Biological Efficacy of Coumarin Clubbed Thiazolo[3,2–b][1,2,4]triazoles as Efficient Inhibitors of Urease: A Biochemical and In Silico Approach. Int. J. Biol. Macromol. 2020, 142, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Sokmen, B.B.; Gumrukcuoglu, N.; Ugras, S.; Ugras, H.I.; Yanardag, R. Synthesis, Antibacterial, Antielastase, Antiurease and Antioxidant Activities of New Methoxy Substitued Bis-1,2,4-Triazole Derivatives. J. Enzym. Inhib. Med. Chem. 2013, 28, 72–77. [Google Scholar] [CrossRef]
- Khan, I.; Ali, S.; Hameed, S.; Rama, N.H.; Hussain, M.T.; Wadood, A.; Uddin, R.; Ul-Haq, Z.; Khan, A.; Ali, S.; et al. Synthesis, Antioxidant Activities and Urease Inhibition of Some New 1,2,4-Triazole and 1,3,4-Thiadiazole Derivatives. Eur. J. Med. Chem. 2010, 45, 5200–5207. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.; Goli-Garmroodi, F.; Allahyari-Devin, M.; Pilali, H.; Hassanzadeh, M.; Mahernia, S.; Mahdavi, M.; Firoozpour, L.; Amanlou, M.; Foroumadi, A. Synthesis, Evaluation, and Molecular Docking Studies of Aryl Urea-Triazole-Based Derivatives as Anti-Urease Agents. Arch. Pharm. 2018, 351, 2–9. [Google Scholar] [CrossRef]
- Salehi Ashani, R.; Azizian, H.; Sadeghi Alavijeh, N.; Fathi Vavsari, V.; Mahernia, S.; Sheysi, N.; Biglar, M.; Amanlou, M.; Balalaie, S. Synthesis, Biological Evaluation and Molecular Docking of Deferasirox and Substituted 1,2,4-Triazole Derivatives as Novel Potent Urease Inhibitors: Proposing Repositioning Candidate. Chem. Biodivers. 2020, 17, e1900710. [Google Scholar] [CrossRef] [PubMed]
- Menteşe, E.; Akyüz, G.; Emirik, M.; Baltaş, N. Synthesis, in Vitro Urease Inhibition and Molecular Docking Studies of Some Novel Quinazolin-4(3H)-One Derivatives Containing Triazole, Thiadiazole and Thiosemicarbazide Functionalities. Bioorg. Chem. 2019, 83, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Salar, U.; Nizamani, A.; Arshad, F.; Khan, K.M.; Fakhri, M.I.; Perveen, S.; Ahmed, N.; Choudhary, M.I. Bis-Coumarins; Non-Cytotoxic Selective Urease Inhibitors and Antiglycation Agents. Bioorg. Chem. 2019, 91, 103170. [Google Scholar] [CrossRef]
- Rashid, U.; Rahim, F.; Taha, M.; Arshad, M.; Ullah, H.; Mahmood, T.; Ali, M. Synthesis of 2-Acylated and Sulfonated 4-Hydroxycoumarins: In Vitro Urease Inhibition and Molecular Docking Studies. Bioorg. Chem. 2016, 66, 111–116. [Google Scholar] [CrossRef]
- Naz, F.; Kanwal; Latif, M.; Salar, U.; Khan, K.M.; Al-Rashida, M.; Ali, I.; Ali, B.; Taha, M.; Perveen, S. 4-Oxycoumarinyl Linked Acetohydrazide Schiff Bases as Potent Urease Inhibitors. Bioorg. Chem. 2020, 105, 104365. [Google Scholar] [CrossRef]
- Khan, K.M.; Iqbal, S.; Lodhi, M.A.; Maharvi, G.M.; Zia-Ullah; Choudhary, M.I.; Atta-ur-Rahman; Perveen, S. Biscoumarin: New Class of Urease Inhibitors; Economical Synthesis and Activity. Bioorg. Med. Chem. 2004, 12, 1963–1968. [Google Scholar] [CrossRef]
- Faisal, M.; Saeed, A.; Shahzad, D.; Fattah, T.A.; Lal, B.; Channar, P.A.; Mahar, J.; Saeed, S.; Mahesar, P.A.; Larik, F.A. Enzyme Inhibitory Activities an Insight into the Structure–Activity Relationship of Biscoumarin Derivatives. Eur. J. Med. Chem. 2017, 141, 386–403. [Google Scholar] [CrossRef] [PubMed]
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009–2018. JAMA 2020, 323, 844–853. [Google Scholar] [CrossRef]
- Tang, Y.; Zhu, W.; Chen, K.; Jiang, H. New Technologies in Computer-Aided Drug Design: Toward Target Identification and New Chemical Entity Discovery. Drug Discov. Today Technol. 2006, 3, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Gertrudes, J.C.; Maltarollo, V.G.; Silva, R.A.; Oliveira, P.R.; Honorio, K.M.; da Silva, A.B.F. Machine Learning Techniques and Drug Design. Curr. Med. Chem. 2012, 19, 4289–4297. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine Learning in Chemoinformatics and Drug Discovery. Drug Discov. Today 2018, 23, 1538. [Google Scholar] [CrossRef]
- Trofymchuk, O.S.; Ortega, D.E.; Cortés-Arriagada, D.; Pereira, A.; Daniliuc, C.G.; Klitzke, C.F.; Santos, L.S.; Rojas, R.S. Neutral and Cationic Methallyl Nickel Complexes in Alkene Activation: A Combined DFT, ESI-MS and Chemometric Approach. Catal. Sci. Technol. 2021, 11, 7475–7485. [Google Scholar] [CrossRef]
- Pereira, A.; Albornoz, C.; Trofymchuk, O.S. Data-Driven Analysis of Reactions Catalyzed by [CoCp*(CO)I2]. Organometallics 2022, 41, 1158–1166. [Google Scholar] [CrossRef]
- Pereira, A.; Trofymchuk, O.S. Machine Learning Prediction of High-Yield Cobalt- and Nickel-Catalyzed Borylations. J. Phys. Chem. C 2023, 127, 12983–12994. [Google Scholar] [CrossRef]
- Patel, L.; Shukla, T.; Huang, X.; Ussery, D.W.; Wang, S. Machine Learning Methods in Drug Discovery. Molecules 2020, 25, 5277. [Google Scholar] [CrossRef]
- Adeshina, Y.O.; Deeds, E.J.; Karanicolas, J. Machine Learning Classification Can Reduce False Positives in Structure-Based Virtual Screening. Proc. Natl. Acad. Sci. USA 2020, 117, 18477–18488. [Google Scholar] [CrossRef]
- Liu, H.X.; Zhang, R.S.; Yao, X.J.; Liu, M.C.; Hu, Z.D.; Fan, B.T. QSAR and Classification Models of a Novel Series of COX-2 Selective Inhibitors: 1,5-Diarylimidazoles Based on Support Vector Machines. J. Comput. Aided Mol. Des. 2004, 18, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.K.; Patra, N. Prediction of COMT Inhibitors Using Machine Learning and Molecular Dynamics Methods. J. Phys. Chem. B 2021, 126, 3477–3492. [Google Scholar] [CrossRef]
- Tinivella, A.; Pinzi, L.; Rastelli, G. Prediction of Activity and Selectivity Profiles of Human Carbonic Anhydrase Inhibitors Using Machine Learning Classification Models. J. Cheminform. 2021, 13, 18. [Google Scholar] [CrossRef]
- Mermer, A. Design, Synthesize and Antiurease Activity of Novel Thiazole Derivatives: Machine Learning, Molecular Docking and Biological Investigation. J. Mol. Struct. 2020, 1222, 128860. [Google Scholar] [CrossRef]
- Aniceto, N.; Albuquerque, T.S.; Bonifácio, V.D.B.; Guedes, R.C.; Martinho, N. Using Machine Learning and Molecular Docking to Leverage Urease Inhibition Data for Virtual Screening. Int. J. Mol. Sci. 2023, 24, 8180. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2021-1; Maestro, Schrödinger, LCC: New York, NY, USA, 2024.
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A Software Program for PKa Prediction and Protonation State Generation for Drug-like Molecules. J. Comput.-Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Guha, R. Chemical Informatics Functionality in R. J. Stat. Softw. 2007, 18, 1–16. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria; Available online: https://www.R-project.org (accessed on 7 April 2024).
- van Buuren, S.; Groothuis-Oudshoorn, K. Mice: Multivariate Imputation by Chained Equations in R. J. Stat. Softw. 2011, 45, 1–67. [Google Scholar] [CrossRef]
- Kuhn, M. Building Predictive Models in R Using the Caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Medina-Franco, J.L.; Chávez-Hernández, A.L.; López-López, E.; Saldívar-González, F.I. Chemical Multiverse: An Expanded View of Chemical Space. Mol. Inform. 2022, 41, e2200116. [Google Scholar] [CrossRef]
- Cihan Sorkun, M.; Mullaj, D.; Vianney, A.; Koelman, J.M.; Er, S. ChemPlot, a Python Library for Chemical Space Visualization. ChemRxiv 2022. [Google Scholar] [CrossRef]
- Kursa, M.B.; Rudnicki, W.R. Feature Selection with the Boruta Package. J. Stat. Softw. 2010, 36, 1–13. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; Association for Computing Machinery: New York, NY, USA; pp. 785–794. [Google Scholar]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. SMOTE: Synthetic Minority Over-Sampling Technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| One-Cutoff Strategies | Two-Cutoff Strategies |
|---|---|
| UIs < 5 µM < nUIs | UIs < 5 µM < gray zone < 50 µM < nUIs |
| UIs < 10 µM < nUIs | UIs < 10 µM < gray zone < 50 µM < nUIs |
| UIs < 25 µM < nUIs | UIs < 25 µM < gray zone < 50 µM < nUIs |
| IC50: 5 µM | IC50: 10 µM | IC50: 25 µM | ||||
|---|---|---|---|---|---|---|
| N° | % | N° | % | N° | % | |
| UIs | 119 | 18 | 145 | 22 | 221 | 33 |
| nUIs | 558 | 82 | 532 | 78 | 456 | 67 |
| IC50: 5 µM and 50 µM | IC50: 10 µM and 50 µM | IC50: 25 µM and 50 µM | ||||
|---|---|---|---|---|---|---|
| N° | % | N° | % | N° | % | |
| UIs | 119 | 26 | 145 | 30 | 221 | 39 |
| nUIs | 341 | 74 | 341 | 70 | 341 | 61 |
| 1-Cutoff Strategies | 2-Cutoff Strategies | ||||
|---|---|---|---|---|---|
| IC50 | Training | Testing | IC50 | Training | Testing |
| IC50: 5 µM | 542 | 135 | IC50: 5 µM and 50 µM | 368 | 92 |
| IC50: 10 µM | 542 | 135 | IC50: 10 µM and 50 µM | 389 | 97 |
| IC50: 25 µM | 542 | 135 | IC50: 25 µM and 50 µM | 450 | 112 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales, N.; Valdés-Muñoz, E.; González, J.; Valenzuela-Hormazábal, P.; Palma, J.M.; Galarza, C.; Catagua-González, Á.; Yáñez, O.; Pereira, A.; Bustos, D. Machine Learning-Driven Classification of Urease Inhibitors Leveraging Physicochemical Properties as Effective Filter Criteria. Int. J. Mol. Sci. 2024, 25, 4303. https://doi.org/10.3390/ijms25084303
Morales N, Valdés-Muñoz E, González J, Valenzuela-Hormazábal P, Palma JM, Galarza C, Catagua-González Á, Yáñez O, Pereira A, Bustos D. Machine Learning-Driven Classification of Urease Inhibitors Leveraging Physicochemical Properties as Effective Filter Criteria. International Journal of Molecular Sciences. 2024; 25(8):4303. https://doi.org/10.3390/ijms25084303
Chicago/Turabian StyleMorales, Natalia, Elizabeth Valdés-Muñoz, Jaime González, Paulina Valenzuela-Hormazábal, Jonathan M. Palma, Christian Galarza, Ángel Catagua-González, Osvaldo Yáñez, Alfredo Pereira, and Daniel Bustos. 2024. "Machine Learning-Driven Classification of Urease Inhibitors Leveraging Physicochemical Properties as Effective Filter Criteria" International Journal of Molecular Sciences 25, no. 8: 4303. https://doi.org/10.3390/ijms25084303