
Downregulation of Protease Cathepsin D and Upregulation of Pathologic α-Synuclein Mediate Paucity of DNAJC6-Induced Degeneration of Dopaminergic Neurons
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
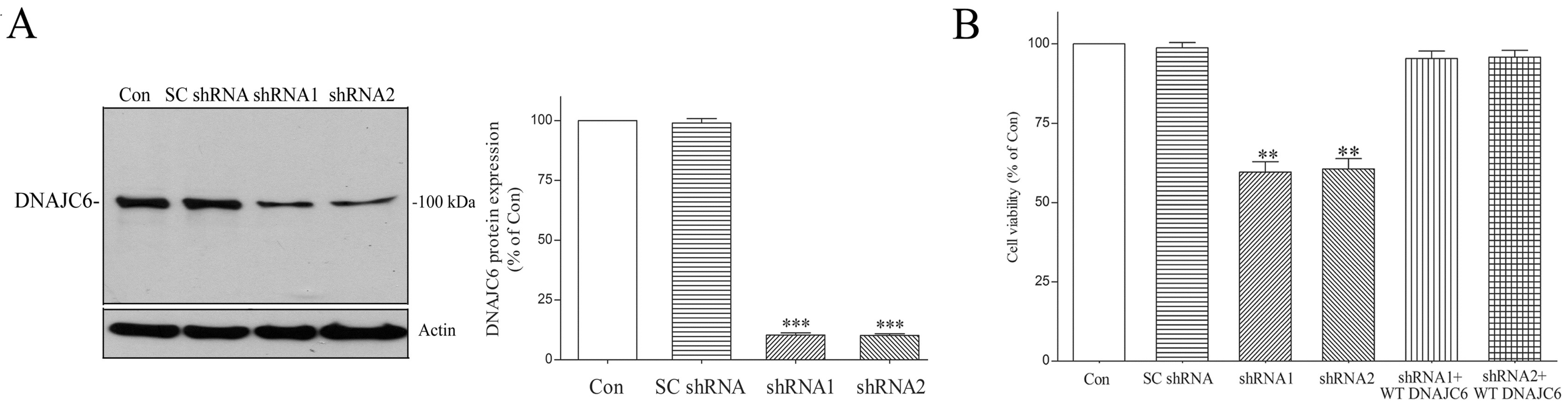
2.1. WT DNAJC6 Exerts a Neuroprotective Effect on Dopaminergic Neurons
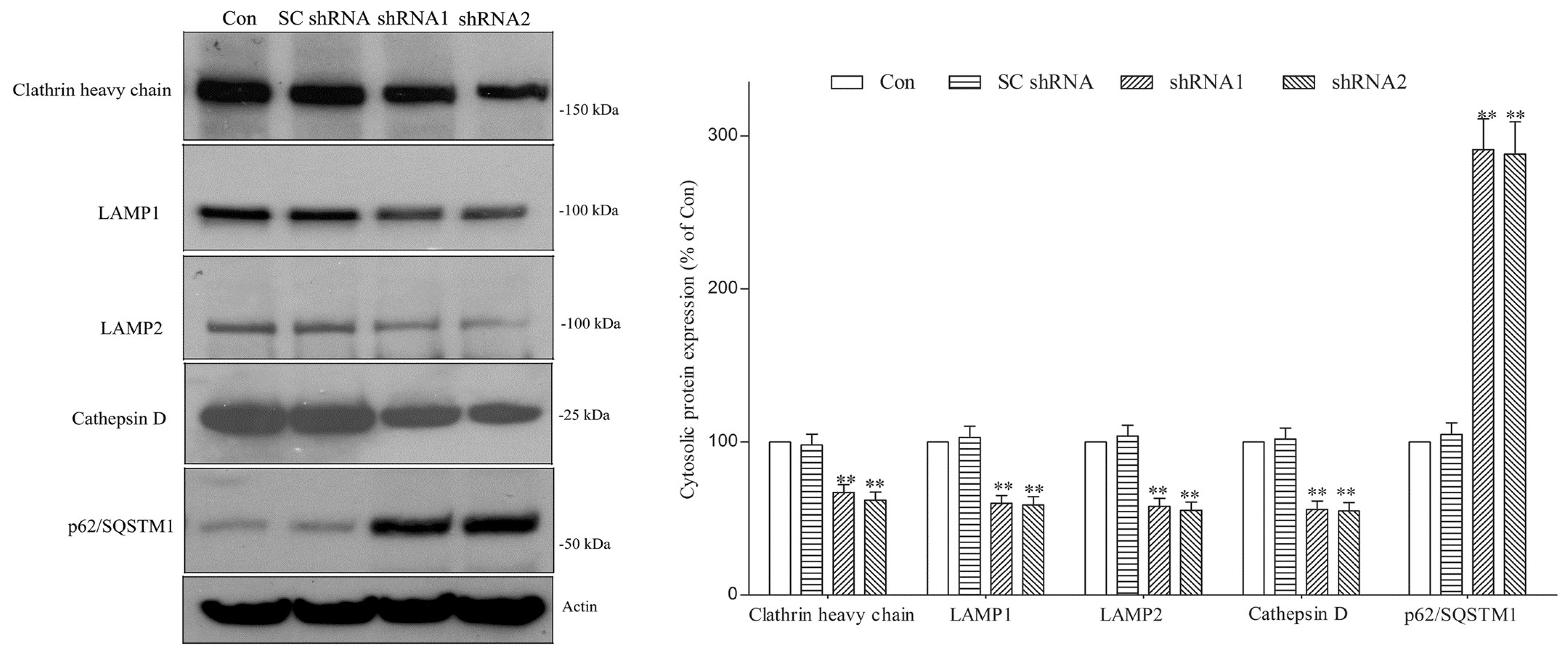
2.2. DNAJC6 Deficiency Reduces Protein Level of Intraneuronal Clathrin Heavy Chain and Number of Lysosomes in Dopaminergic Neurons
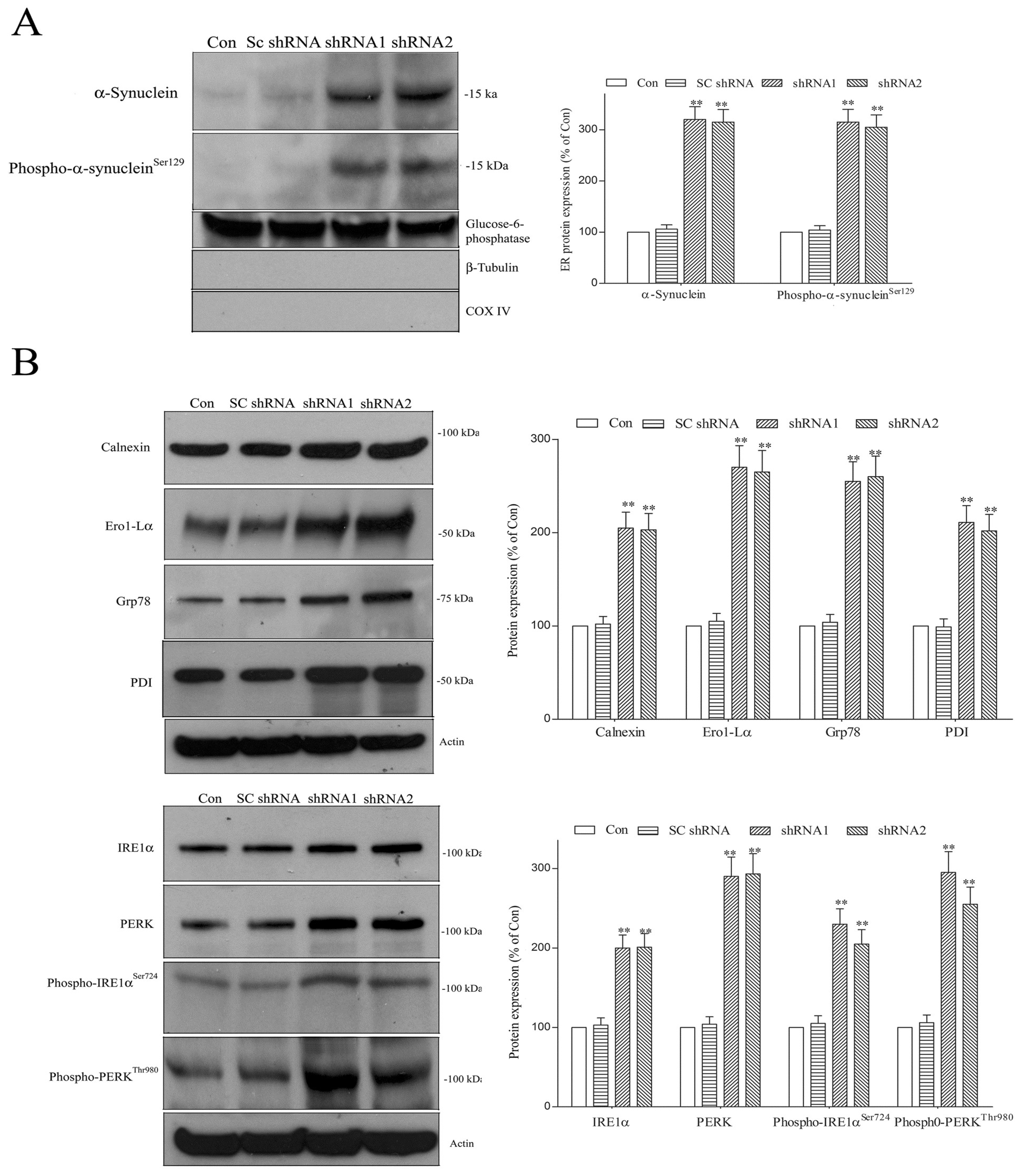
2.3. Paucity of DNAJC6 Causes Macroautophagy Impairment and Decreases Protein Level of Protease Cathepsin D, Resulting in Upregulation of Pathologic α-Synuclein or Phospho-α-Synucleinser129 within Dopaminergic Neurons
2.4. shRNA Targeting α-Synuclein or Overexpression of FLAG-Tagged Cathepsin D Blocks DNAJC6 Deficiency-Induced Degeneration of Dopaminergic Cells
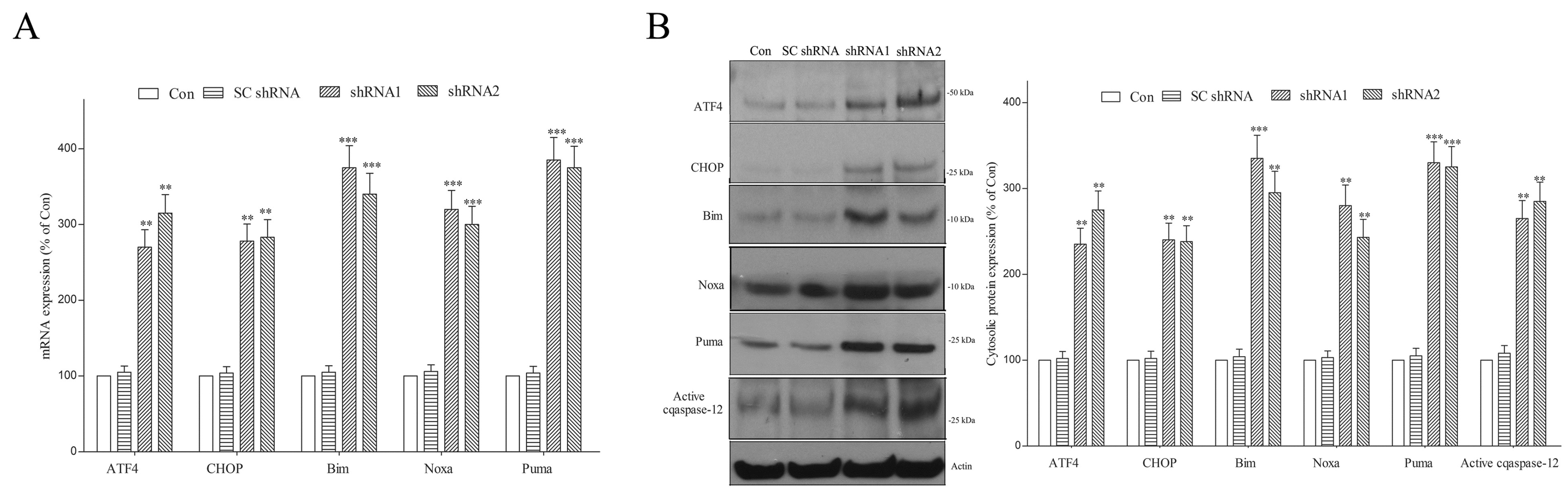
2.5. Lack of DNAJC6 Upregulates ER α-Synuclein or Phospho-α-Synucleinser129 and Activates ER Stress, UPR and ER Stress-Triggered Pro-Apoptotic Signaling within Dopaminergic Neurons
2.6. Paucity of DNAJC6 Increases Mitochondrial α-Synuclein or Phospho-α-Synucleinser129 and Induces Mitochondrial Impairment and Oxidative Stress in Dopaminergic Neurons
2.7. DNAJC6 Paucity Promotes Apoptotic Death of Dopaminergic Neurons via Activating Mitochondrial Apoptotic Signaling
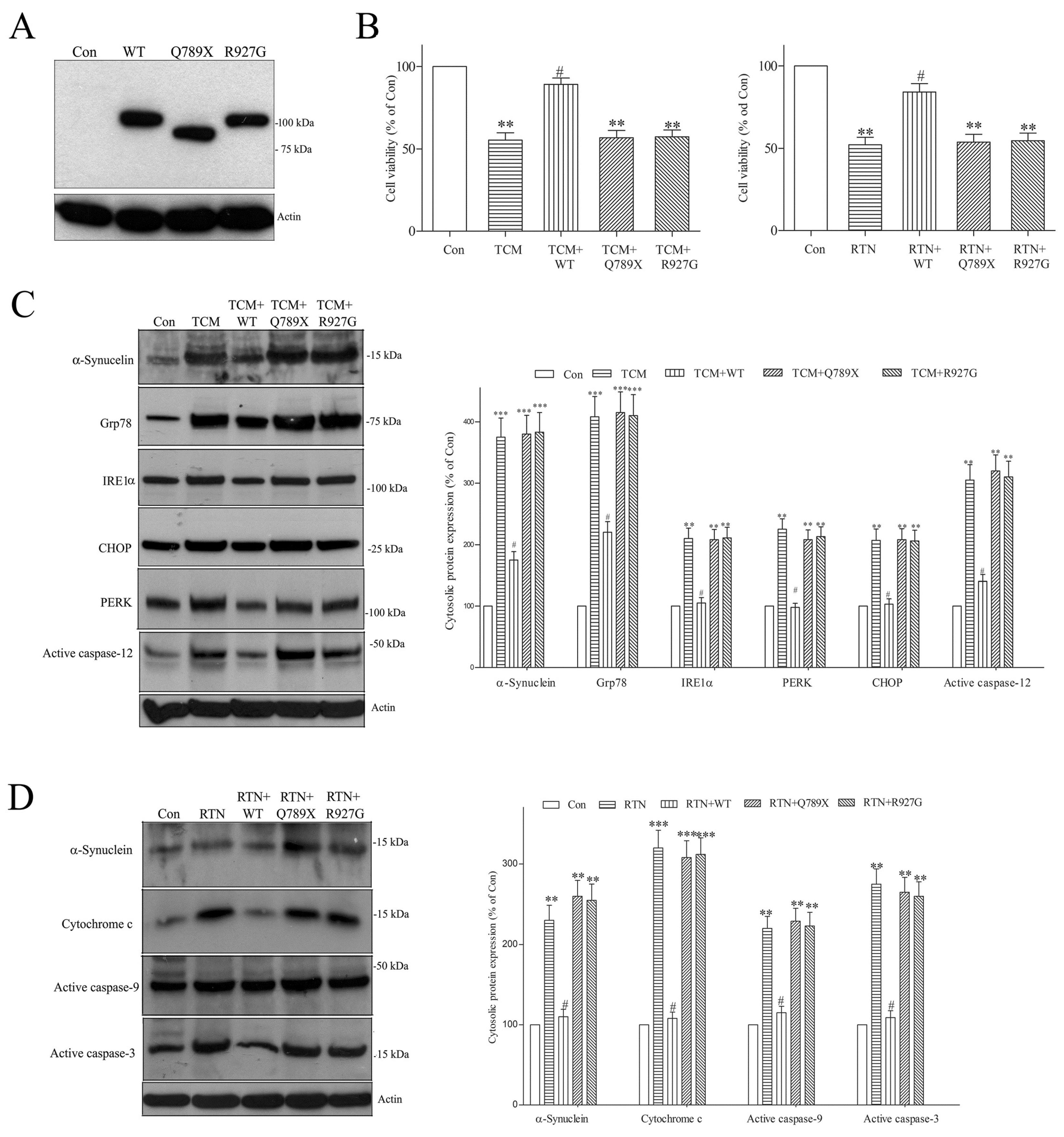
2.8. PARK19 DNAJC6 Mutant Fails to Reverse Tunicamycin- or Rotenone-Evoked Upregulation of Pathologic α-Synuclein and Stimulation of Apoptotic Pathway in Dopaminergic Neurons
3. Discussion
4. Materials and Methods
4.1. Cultured Differentiated SH-SY5Y Dopaminergic Neurons
4.2. Transfecting shRNAs to Differentiated SH-SY5Y Dopaminergic Neurons
4.3. Construction of cDNA Encoding PARK19 DNAJC6
4.4. Measurement of Neuronal Viability
4.5. Subcellularfractionation
4.6. Western Blot Analysis of Clathrin, p62/SQSTM1 and Lysosomal Proteins
4.7. Live Cell Staining of Lysosomes Using LysoTracker
4.8. Immunofluorescence Staining of Lysosomal Marker Protein LAMP2
4.9. Western Blot Analysis of α-Synuclein and Phospho-α-Synucleinser129
4.10. Immunoblotting Assays of ER Stress, UPR or ER Stress-Evoked Pro-Apoptotic Cascade
4.11. Quantitative Real-Time RT-PCR Analysis
4.12. Determination of Mitochondrial Superoxide and Mitochondrial Membrane Potential (ΔΨm)
4.13. Western Blot Assays of Mitochondria-Mediated Apoptotic Pathway
4.14. TUNEL Analysis of Dopaminergic Neurons
4.15. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hernandez, D.G.; Reed, X.; Singleton, A.B. Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J. Neurochem. 2016, 139 (Suppl. S1), 59–74. [Google Scholar] [CrossRef]
- Jia, F.; Fellner, A.; Raj Kumar, K. Monogenic Parkinson’s disease: Genotype, phenotype, pathophysiology, and genetic testing. Genes 2022, 13, 471. [Google Scholar] [CrossRef]
- Edvardson, S.; Cinnamon, Y.; TaShma, A.; Shaag, A.; Yim, Y.I.; Zenvirt, S.; Jalas, C.; Lesage, S.; Brice, A.; Taraboulos, A.; et al. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin is associated with juvenile parkinsonism. PLoS ONE 2012, 7, e36458. [Google Scholar] [CrossRef]
- Koroglu, C.; Baysal, L.; Cetinkaya, M.; Karasoy, H.; Tolun, A. DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Park. Relat. Disord. 2013, 19, 320–324. [Google Scholar] [CrossRef]
- Elsayed, L.E.O.; Drouet, V.; Usenko, T.; Mohammed, I.N.; Hamed, A.A.A.; Elseed, M.A.; Salih, M.A.; Koko, M.E.; Mohamed, A.Y.; Siddig, R.A.; et al. A novel nonsense mutation in DNAJC6 expands the phenotype of autosomal-recessive juvenile-onset Parkinson’s disease. Ann. Neurol. 2016, 79, 335–337. [Google Scholar] [CrossRef]
- Ng, J.; Cortès-Saladelafont, E.; Abela, L.; Termsarasab, P.; Mankad, K.; Sudhakar, S.; Gorman, K.M.; Heales, S.J.; Pope, S.; Biassoni, L.; et al. DNAJC6 mutations disrupt dopamine homeostasis in juvenile parkinsonism-dystonia. Mov. Disord. 2020, 35, 1357–1368. [Google Scholar] [CrossRef]
- Olgiati, S.; Quadri, M.; Fang, M.; Rood, J.P.M.; Saute, J.A.; Chien, H.F.; Bouwkamp, C.G.; Graafland, J.; Minneboo, M.; Breedveld, G.J.; et al. DNAJC6 mutations associated with early-onset Parkinson’s disease. Ann. Neurol. 2016, 79, 244–256. [Google Scholar] [CrossRef]
- Qiu, X.B.; Shao, Y.M.; Miao, S.; Wang, L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol. Life Sci. 2006, 63, 2560–2570. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Craig, E.A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Craig, E.A.; Marszalek, J. How do J-proteins get Hsp70 to do so many different things? Trends Biochem. Sci. 2017, 42, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Yim, Y.I.; Sun, T.; Wu, L.G.; Raimondi, A.; De Camilli, P.; Eisenberg, E.; Greene, L.E. Endocytosis and clathrin-uncoating defects at synapses of auxilin knockout mice. Proc. Nat. Acad. Sci. USA 2010, 107, 4412–4417. [Google Scholar] [CrossRef]
- Stricher, F.; Macri, C.; Ruff, M.; Muller, S. HSPA8/HSC70 chaperone protein: Structure, function, and chemical targeting. Autophagy 2013, 9, 1937–1954. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef]
- Roosen, D.A.; Blauwendraat, C.; Cookson, M.R.; Lewis, P.A. DNAJC proteins and pathways to parkinsonism. FEBS J. 2019, 286, 3080–3094. [Google Scholar] [CrossRef]
- Wong, Y.C.; Krainc, D. α-Synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef]
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. Alpha-synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Kirik, D.; Rosenblad, C.; Burger, C.; Lundberg, C.; Johansen, T.E.; Muzyczka, N.; Mandel, R.J.; Björklund, A. Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J. Neurosci. 2002, 22, 2780–2791. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O‘Brien, P.; Trojanowski, J.Q.; Lee, V.M.-Y. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Zheng, W.; Wang, X.; Chen, Z. Proteostasis of α-synuclein and its role in the pathogenesis of Parkinson’s disease. Front. Cell Neurosci. 2020, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Serratos, I.N.; Hernández-Pérez, E.; Campos, C.; Aschner, M.; Santamaría, A. An update on the critical role of α-synuclein in Parkinson’s disease and other synucleinopathies: From tissue to cellular and molecular levels. Mol. Neurobiol. 2022, 59, 620–642. [Google Scholar] [CrossRef] [PubMed]
- De Mattos, E.P.; Wentink, A.; Nussbaum-Krammer, C.; Hansen, C.; Bergink, S.; Melki, R.; Kampinga, H.H. Protein quality control pathways at the crossroad of synucleinopathies. J. Parkinsons Dis. 2020, 10, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Watzlawik, J.O.; Fiesel, F.C.; Springer, W. Autophagy in Parkinson’s disease. J. Mol. Biol. 2020, 432, 2651–2672. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Romero, A.; Montpeyó, M.; Martinez-Vicente, M. The Emerging role of the lysosome in Parkinson’s disease. Cells 2020, 9, 2399. [Google Scholar] [CrossRef] [PubMed]
- Fellner, L.; Gabassi, E.; Haybaeck, J.; Edenhofer, F. Autophagy in α-synucleinopathies-an overstrained system. Cells 2021, 10, 3143. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef]
- Abe, T.; Kuwahara, T. Targeting of lysosomal pathway genes for Parkinson’s disease modification: Insights from cellular and animal models. Front. Neurol. 2021, 12, 681369. [Google Scholar] [CrossRef]
- Udayar, V.; Chen, Y.; Sidransky, E.; Jagasia, R. Lysosomal dysfunction in neurodegeneration: Emerging concepts and methods. Trends Neurosci. 2022, 45, 184–199. [Google Scholar] [CrossRef]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, L. Development of research into autophagic lysosome reformation. Mol. Cells 2018, 41, 45–49. [Google Scholar] [PubMed]
- Yang, C.; Wang, X. Lysosome biogenesis: Regulation and functions. J. Cell Biol. 2021, 220, e202102001. [Google Scholar] [CrossRef] [PubMed]
- Nanayakkara, R.; Gurung, R.; Rodgers, S.J.; Eramo, M.J.; Ramm, G.; Mitchell, C.A.; McGrath, M.J. Autophagic lysosome reformation in health and disease. Autophagy 2023, 19, 1378–1395. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Liu, M.; Ma, L.; Du, W.; Zhang, H.; Tian, Y.; Cao, Z.; Li, Y.; Ren, H.; Zhang, C.; et al. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat. Cell Biol. 2012, 14, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Su, Q.P.; Chen, Y.; Zhu, Y.; Jiang, D.; Rong, Y.; Zhang, S.; Zhang, Y.; Ren, H.; Zhang, C.; et al. Kinesin 1 drives autolysosome tubulation. Dev. Cell 2016, 37, 326–336. [Google Scholar] [CrossRef] [PubMed]
- McMahon, H.T.; Boucrot, E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2011, 12, 517–533. [Google Scholar] [CrossRef] [PubMed]
- Vidoni, C.; Follo, C.; Savino, M.; Melone, M.A.B.; Isidoro, C. The role of cathepsin D in the pathogenesis of human neurodegenerative disorders. Med. Res. Rev. 2016, 36, 845–870. [Google Scholar] [CrossRef] [PubMed]
- Bluhm, A.; Schrempel, S.; von Hörsten, S.; Schulze, A.; Roßner, S. Proteolytic α-synuclein cleavage in health and disease. Int. J. Mol. Sci. 2021, 22, 5450. [Google Scholar] [CrossRef]
- Sevlever, D.; Jiang, P.; Yen, S.H.C. Cathepsin D is the main lysosomal enzyme involved in the degradation of alpha-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 2008, 47, 9678–9687. [Google Scholar] [CrossRef]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Hamamichi, S.; Caldwell, K.A.; Caldwell, G.A.; Yacoubian, T.A.; Wilson, S.; Xie, Z.-L.; Speake, L.D.; Parks, R.; Crabtree, D.; et al. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol. Brain 2008, 1, 17. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, C.; Yamaguchi, J.; Sanada, T.; Trejo, J.A.O.; Kakuta, S.; Shibata, M.; Tanida, I.; Uchiyama, Y. Lack of cathepsin D in the central nervous system results in microglia and astrocyte activation and the accumulation of proteinopathy-related proteins. Sci. Rep. 2022, 12, 11662. [Google Scholar] [CrossRef] [PubMed]
- Huarcaya, S.P.; Drobny, A.; Marques, A.R.A.; Di Spiezio, A.; Dobert, J.P.; Balta, D.; Werner, C.; Rizo, T.; Gallwitz, L.; Bub, S.; et al. Recombinant pro-CTSD (cathepsin D) enhances SNCA/α-synuclein degradation in α-synucleinopathy models. Autophagy 2022, 18, 1127–1151. [Google Scholar] [CrossRef] [PubMed]
- Robak, L.A.; Jansen, I.E.; van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; Heutink, P.; Shulman, J.M. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 2017, 140, 3191–3203. [Google Scholar] [CrossRef] [PubMed]
- Colla, E.; Jensen, P.H.; Pletnikova, O.; Troncoso, J.C.; Glabe, C.; Lee, M.K. Accumulation of toxic alpha-synuclein oligomer within endoplasmic reticulum occurs in alpha-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3301–3305. [Google Scholar] [CrossRef] [PubMed]
- Sugeno, N.; Takeda, A.; Hasegawa, T.; Kobayashi, M.; Kikuchi, A.; Mori, F.; Wakabayashi, K.; Itoyama, Y. Serine 129 phosphorylation of alpha-synuclein induces unfolded protein response-mediated cell death. J. Biol. Chem. 2008, 283, 23179–23188. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–436. [Google Scholar] [CrossRef]
- Colla, E. Linking the endoplasmic reticulum to Parkinson’s disease and alpha-synucleinopathy. Front. Neurosci. 2019, 13, 560. [Google Scholar] [CrossRef]
- da Costa, C.A.; El Manaa, W.; Duplan, E.; Checler, F. The endoplasmic reticulum stress/unfolded protein response and their contributions to Parkinson’s disease physiopathology. Cells 2020, 9, 2495. [Google Scholar] [CrossRef] [PubMed]
- Vicario, M.; Cieri, D.; Brini, M.; Calì, T. The close encounter between alpha-synuclein and mitochondria. Front. Neurosci. 2018, 12, 388. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.E.; Akther, M.; Azam, S.; Kim, I.S.; Lin, Y.; Lee, Y.H.; Choi, D.-K. Targeting α-synuclein aggregation and its role in mitochondrial dysfunction in Parkinson’s disease. Br. J. Pharmacol. 2022, 179, 23–45. [Google Scholar] [CrossRef] [PubMed]
- Sohrabi, T.; Mirzaei-Behbahani, B.; Zadali, R.; Pirhaghi, M.; Morozova-Roche, L.A.; Meratan, A.A. Common mechanisms underlying α-synuclein-induced mitochondrial dysfunction in Parkinson’s disease. J. Mol. Biol. 2023, 435, 167992. [Google Scholar] [CrossRef] [PubMed]
- Borsche, M.; Pereira, S.L.; Klein, C.; Grunewald, A. Mitochondria and Parkinson’s disease: Clinical, molecular, and translational aspects. J. Parkinson’s Dis. 2021, 11, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Trinh, D.; Israwi, A.R.; Arathoon, L.R.; Gleave, J.A.; Nash, J.E. The multi-faceted role of mitochondria in the pathology of Parkinson’s disease. J. Neurochem. 2021, 156, 715–752. [Google Scholar] [CrossRef] [PubMed]
- McMillan, C.R.; Sharma, R.; Ottenhof, T.; Niles, L.P. Modulation of tyrosine hydroxylase expression by melatonin in human SH-SY5Y neuroblastoma cells. Neurosci. Lett. 2007, 419, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, K.; Indrani, D.; Sowmithra, S.; Joshi, P.; Bhonde, R. Influence of 6-hydroxydopamine toxicity on α-synuclein phosphorylation, resting vesicle expression, and vesicular dopamine release. J. Cell Biochem. 2016, 117, 2719–2736. [Google Scholar] [CrossRef] [PubMed]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The SH-SY5Y cell line in Parkinson’s disease research: A systematic review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar] [CrossRef]
- Ahsan, A.; Zheng, Y.; Ma, S.; Liu, M.; Cao, M.; Li, Y.; Zheng, W.; Zhou, X.; Xin, M.; Hu, W.-W.; et al. Tomatidine protects against ischemic neuronal injury by improving lysosomal function. Eur. J. Pharmacol. 2020, 882, 173280. [Google Scholar] [CrossRef]
- Lu, S.; Sung, T.; Lin, N.; Abraham, R.T.; Jessen, B.A. Lysosomal adaptation: How cells respond to lysosomotropic compounds. PLoS ONE 2017, 12, e0173771. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.V.; Mills, J.; Lapierre, L.R. Selective autophagy receptor p62/SQSTM1, a pivotal player in stress and aging. Front. Cell Dev. Biol. 2022, 10, 793328. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Kitakaze, M. ER stress in cardiovascular disease. J. Mol. Cell Cardiol. 2010, 48, 1105–1110. [Google Scholar] [CrossRef]
- Zheng, J.H.; Follis, A.V.; Kriwacki, R.W.; Moldoveanu, T. Discoveries and controversies in BCL-2 protein-mediated apoptosis. FEBS J. 2016, 283, 2690–2700. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Qu, L.; Zhang, N.; Yu, X.; Xiao, Z.; Song, L.; Xie, J.; Xu, H. Ndfip1 prevents rotenone-induced neurotoxicity and upregulation of α-synuclein in SH-SY5Y cells. Front. Mol. Neurosci. 2021, 13, 613404. [Google Scholar] [CrossRef] [PubMed]
- da Silva, D.C.; Valentão, P.; Andrade, P.B.; Pereira, D.M. Endoplasmic reticulum stress signaling in cancer and neurodegenerative disorders: Tools and strategies to understand its complexity. Pharmacol. Res. 2020, 155, 104702. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Lee, S.; Blackstone, C. Spastic paraplegia proteins spastizin and spatacsin mediate autophagic lysosome reformation. J. Clin. Investig. 2014, 124, 5249–5262. [Google Scholar] [CrossRef] [PubMed]
- Khundadze, M.; Ribaudo, F.; Hussain, A.; Stahlberg, H.; Brocke-Ahmadinejad, N.; Franzka, P.; Varga, R.-E.; Zarkovic, M.; Pungsrinont, T.; Kokal, M.; et al. Mouse models for hereditary spastic paraplegia uncover a role of PI4K2A in autophagic lysosome reformation. Autophagy 2021, 17, 3690–3706. [Google Scholar] [CrossRef] [PubMed]
- Serra-Vinardell, J.; Sandler, M.B.; De Pace, R.; Manzella-Lapeira, J.; Cougnoux, A.; Keyvanfar, K.; Introne, W.J.; Brzostowski, J.A.; Ward, M.E.; Gahl, W.A.; et al. LYST deficiency impairs autophagic lysosome reformation in neurons and alters lysosome number and size. Cell Mol. Life Sci. 2023, 80, 53. [Google Scholar] [CrossRef]
- Calcagni, A.; Staiano, L.; Zampelli, N.; Minopoli, N.; Herz, N.J.; Di Tullio, G.; Huynh, T.; Monfregola, J.; Esposito, A.; Cirillo, C.; et al. Loss of the batten disease protein CLN3 leads to mis-trafficking of M6PR and defective autophagic-lysosomal reformation. Nat. Commun. 2023, 14, 3911. [Google Scholar] [CrossRef]
- Parnetti, L.; Paciotti, S.; Eusebi, P.; Dardis, A.; Zampieri, S.; Chiasserini, D.; Tasegian, A.; Tambasco, N.; Bembi, B.; Calabresi, P.; et al. Cerebrospinal fluid β-glucocerebrosidase activity is reduced in Parkinson’s disease patients. Mov. Disord. 2017, 32, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Kim, J.W.; Heo, H.; Lee, J.; Park, K.Y.; Yoon, J.H.; Chang, J. Identification of BAG2 and cathepsin D as plasma biomarkers for Parkinson’s disease. Clin. Transl. Sci. 2021, 14, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.W.; Kordower, J.H. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to alpha-synuclein inclusions. Neurobiol. Dis. 2009, 35, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Moors, T.E.; Paciotti, S.; Ingrassia, A.; Quadri, M.; Breedveld, G.; Tasegian, A.; Chiasserini, D.; Eusebi, P.; Duran-Pacheco, G.; Kremer, T.; et al. Characterization of brain lysosomal activities in GBA-related and sporadic Parkinson’s disease and dementia with Lewy bodies. Mol. Neurobiol. 2019, 56, 1344–1355. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.C.; Weng, Y.H.; Yeh, T.H.; Lu, J.C.; Chen, W.S.; Li, A.H.R.; Chen, Y.-L.; Wei, K.-C.; Wang, H.-L. Deficiency of RAB39B activates ER stress-induced pro-apoptotic pathway and causes mitochondrial dysfunction and oxidative stress in dopaminergic neurons by impairing autophagy and upregulating α-synuclein. Mol. Neurobiol. 2023, 60, 2706–2728. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139, 216–231. [Google Scholar] [CrossRef]
- Jiang, J.; Taylor, A.B.; Prasad, K.; Ishikawa-Brush, Y.; Hart, P.J.; Lafer, E.M.; Sousa, R. Structure-function analysis of the auxilin J-domain reveals an extended Hsc70 interaction interface. Biochemistry 2003, 42, 5748–5753. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiu, C.-C.; Chen, Y.-L.; Weng, Y.-H.; Liu, S.-Y.; Li, H.-L.; Yeh, T.-H.; Wang, H.-L. Downregulation of Protease Cathepsin D and Upregulation of Pathologic α-Synuclein Mediate Paucity of DNAJC6-Induced Degeneration of Dopaminergic Neurons. Int. J. Mol. Sci. 2024, 25, 6711. https://doi.org/10.3390/ijms25126711
Chiu C-C, Chen Y-L, Weng Y-H, Liu S-Y, Li H-L, Yeh T-H, Wang H-L. Downregulation of Protease Cathepsin D and Upregulation of Pathologic α-Synuclein Mediate Paucity of DNAJC6-Induced Degeneration of Dopaminergic Neurons. International Journal of Molecular Sciences. 2024; 25(12):6711. https://doi.org/10.3390/ijms25126711
Chicago/Turabian StyleChiu, Ching-Chi, Ying-Ling Chen, Yi-Hsin Weng, Shu-Yu Liu, Hon-Lun Li, Tu-Hsueh Yeh, and Hung-Li Wang. 2024. "Downregulation of Protease Cathepsin D and Upregulation of Pathologic α-Synuclein Mediate Paucity of DNAJC6-Induced Degeneration of Dopaminergic Neurons" International Journal of Molecular Sciences 25, no. 12: 6711. https://doi.org/10.3390/ijms25126711