

Exploring the Structurally Conserved Regions and Functional Significance in Bacterial N-Terminal Nucleophile (Ntn) Amide-Hydrolases

 , , and
, , and 
Abstract
:
1. Introduction
2. Results
2.1. Structural Analysis
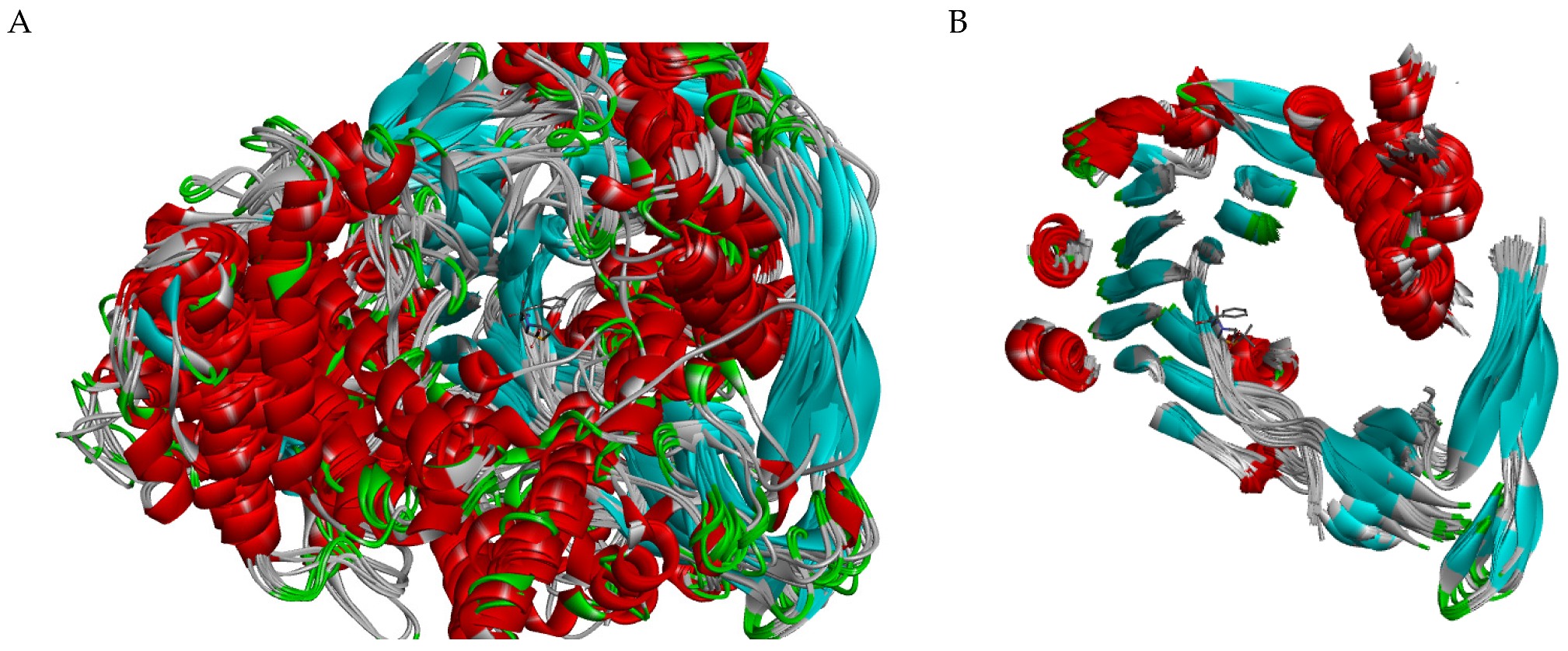
2.1.1. Chain A
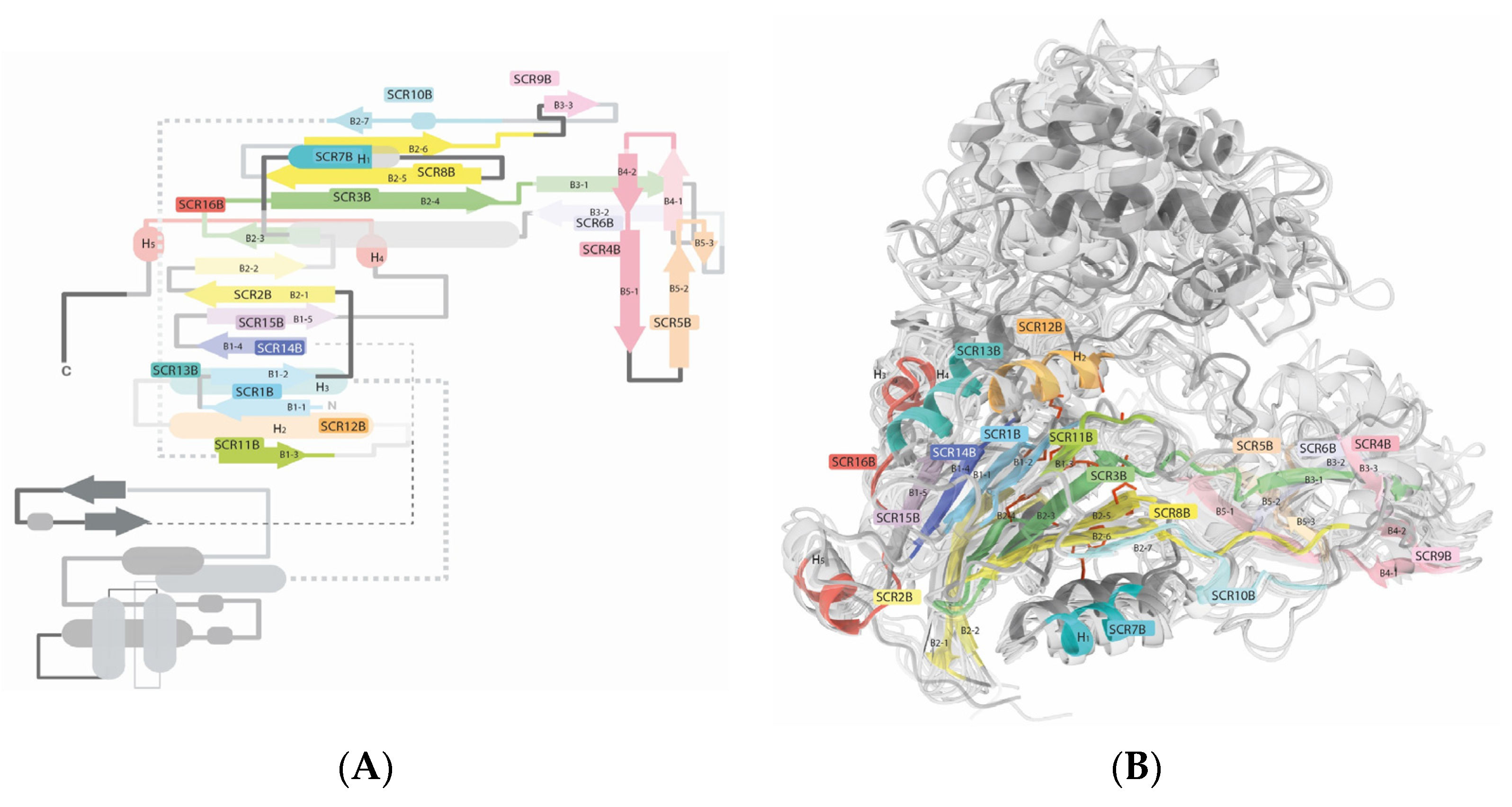
2.1.2. Chain B
2.1.3. Active Site
Variable Regions on the Active Site
3. Discussion
4. Materials and Methods
4.1. Data
4.2. Sequences
4.3. Structural Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Buchholz, K. A Breakthrough in Enzyme Technology to Fight Penicillin Resistance—Industrial Application of Penicillin Amidase. Appl. Microbiol. Biotechnol. 2016, 100, 3825–3839. [Google Scholar] [CrossRef]
- Wu, Z.; Liu, C.; Zhang, Z.; Zheng, R.; Zheng, Y. Amidase as a Versatile Tool in Amide-Bond Cleavage: From Molecular Features to Biotechnological Applications. Biotechnol. Adv. 2020, 43, 107574. [Google Scholar] [CrossRef]
- Meghwanshi, G.K.; Kaur, N.; Verma, S.; Dabi, N.K.; Vashishtha, A.; Charan, P.D.; Purohit, P.; Bhandari, H.S.; Bhojak, N.; Kumar, R. Enzymes for Pharmaceutical and Therapeutic Applications. Biotechnol. Appl. Biochem. 2020, 67, 586–601. [Google Scholar] [CrossRef]
- Umaru, I.J.; Adam, B.R.; Habibu, B.; Umaru, K.I.; Chizaram, B.C. Biochemical Impact of Microorganism and Enzymatic Activities in Food and Pharmaceutical Industries. Int. J. Adv. Biochem. Res. 2021, 5, 42–57. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Waller, M.; Barrett, A.J.; Bateman, A. MEROPS: The Database of Proteolytic Enzymes, Their Substrates and Inhibitors. Nucleic Acids Res. 2014, 42, D503–D509. [Google Scholar] [CrossRef]
- Jiang, W.; Fang, B. Synthesizing Chiral Drug Intermediates by Biocatalysis. Appl. Biochem. Biotechnol. 2020, 192, 146–179. [Google Scholar] [CrossRef]
- Youshko, M.I.; Moody, H.M.; Bukhanov, A.L.; Boosten, W.H.J.; Švedas, V.K. Penicillin Acylase-Catalyzed Synthesis of β-Lactam Antibiotics in Highly Condensed Aqueous Systems: Beneficial Impact of Kinetic Substrate Supersaturation: Beneficial Impact of Kinetic Substrate Supersaturation. Biotechnol. Bioeng. 2004, 85, 323–329. [Google Scholar] [CrossRef]
- Papamichael, E.M.; Stergiou, P.-Y. Enzyme Immobilization Strategies and Bioprocessing Applications. In Biomass, Biofuels, Biochemicals; Elsevier: Amsterdam, The Netherlands, 2020; pp. 217–241. [Google Scholar]
- Marešová, H.; Plačková, M.; Grulich, M.; Kyslík, P. Current State and Perspectives of Penicillin G Acylase-Based Biocatalyses. Appl. Microbiol. Biotechnol. 2014, 98, 2867–2879. [Google Scholar] [CrossRef]
- Grulich, M.; Štěpánek, V.; Kyslík, P. Perspectives and Industrial Potential of PGA Selectivity and Promiscuity. Biotechnol. Adv. 2013, 31, 1458–1472. [Google Scholar] [CrossRef]
- Galmés, M.À.; Nödling, A.R.; He, K.; Luk, L.Y.P.; Świderek, K.; Moliner, V. Computational Design of an Amidase by Combining the Best Electrostatic Features of Two Promiscuous Hydrolases. Chem. Sci. 2022, 13, 4779–4787. [Google Scholar] [CrossRef]
- Barends, T.R.M.; Yoshida, H.; Dijkstra, B.W. Three-Dimensional Structures of Enzymes Useful for β-Lactam Antibiotic Production. Curr. Opin. Biotechnol. 2004, 15, 356–363. [Google Scholar] [CrossRef]
- Chandel, A.K.; Rao, L.V.; Narasu, M.L.; Singh, O. V The Realm of Penicillin G Acylase in β-Lactam Antibiotics. Enzym. Microb. Technol. 2008, 42, 199–207. [Google Scholar] [CrossRef]
- Linhorst, A.; Lübke, T. The Human Ntn-Hydrolase Superfamily: Structure, Functions and Perspectives. Cells 2022, 11, 1592. [Google Scholar] [CrossRef]
- Oh, C.; Kim, T.D.; Kim, K.K. Carboxylic Ester Hydrolases in Bacteria: Active Site, Structure, Function and Application. Crystals 2019, 9, 597. [Google Scholar] [CrossRef]
- Wang, H.; Feng, Y.; Lu, H. Low-Level Cefepime Exposure Induces High-Level Resistance in Environmental Bacteria: Molecular Mechanism and Evolutionary Dynamics. Environ. Sci. Technol. 2022, 56, 15074–15083. [Google Scholar] [CrossRef]
- Chatonnet, A.; Perochon, M.; Velluet, E.; Marchot, P. The ESTHER Database on Alpha/Beta Hydrolase Fold Proteins-An Overview of Recent Developments. Chem. Biol. Interact. 2023, 383, 110671. [Google Scholar] [CrossRef]
- Oinonen, C.; Rouvinen, J. Structural Comparison of Ntn-Hydrolases. Protein Sci. 2000, 9, 2329–2337. [Google Scholar] [CrossRef]
- Tishkov, V.I.; Savin, S.S.; Yasnaya, A.S. Protein Engineering of Penicillin Acylase. Acta Naturae 2010, 2, 47–61. [Google Scholar] [CrossRef]
- Kasche, V.; Lummer, K.; Nurk, A.; Piotraschke, E.; Rieks, A.; Stoeva, S.; Voelter, W. Intramolecular Autoproteolysis Initiates the Maturation of Penicillin Amidase from Escherichia Coli. Biochim. Et Biophys. Acta BBA-Protein Struct. Mol. Enzymol. 1999, 1433, 76–86. [Google Scholar] [CrossRef]
- Alkema, W.B.L.; Dijkhuis, A.-J.; De Vries, E.; Janssen, D.B. The Role of Hydrophobic Active-Site Residues in Substrate Specificity and Acyl Transfer Activity of Penicillin Acylase. Eur. J. Biochem. 2002, 269, 2093–2100. [Google Scholar] [CrossRef]
- Buller, A.R.; Labonte, J.W.; Freeman, M.F.; Wright, N.T.; Schildbach, J.F.; Townsend, C.A. Autoproteolytic Activation of ThnT Results in Structural Reorganization Necessary for Substrate Binding and Catalysis. J. Mol. Biol. 2012, 422, 508–518. [Google Scholar] [CrossRef]
- Kirilin, E.M.; Bochkova, A.A.; Panin, N.V.; Pochinok, I.V.; Švedas, V. Molecular Modeling of Penicillin Acylase Binding with a Penicillin Nucleus by High Performance Computing: Can Enzyme or Its Mutants Possess β-Lactamase Activity? Supercomput. Front. Innov. 2022, 9, 68–78. [Google Scholar] [CrossRef]
- Done, S.H.; Brannigan, J.A.; Moody, P.C.E.; Hubbard, R.E. Ligand-Induced Conformational Change in Penicillin Acylase. J. Mol. Biol. 1998, 284, 463–475. [Google Scholar] [CrossRef]
- Salahuddin, P.; Kumar, A.; Khan, A.U. Structure, Function of Serine and Metallo-β-Lactamases and Their Inhibitors. Curr. Protein Pept. Sci. 2018, 19, 130–144. [Google Scholar] [CrossRef]
- Chandra, M.P. Studies on the Catalysis and Post Translational Processing of Penicillin V Acylase; University of Pune: Pune, India, 2005. [Google Scholar]
- Suplatov, D.; Panin, N.; Kirilin, E.; Shcherbakova, T.; Kudryavtsev, P.; Švedas, V. Computational Design of a PH Stable Enzyme: Understanding Molecular Mechanism of Penicillin Acylase’s Adaptation to Alkaline Conditions. PLoS ONE 2014, 9, e100643. [Google Scholar] [CrossRef]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and Sequence Analysis Tools Services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, 50, W276–W279. [Google Scholar] [CrossRef]
- Webb, B.M.; Braberg, H.; Tjioe, E.; Pieper, U.; Sali, A.; Madhusudhan, M.S. SALIGN: A Web Server for Alignment of Multiple Protein Sequences and Structures. Bioinformatics 2012, 28, 2072–2073. [Google Scholar] [CrossRef]
- Grigorenko, B.L.; Khrenova, M.G.; Nilov, D.K.; Nemukhin, A.V.; Svedas, V.K. Catalytic Cycle of Penicillin Acylase from Escherichia Coli: QM/MM Modeling of Chemical Transformations in the Enzyme Active Site upon Penicillin G Hydrolysis. ACS Catal. 2014, 4, 2521–2529. [Google Scholar] [CrossRef]
- Alkema, W.B.L.; Hensgens, C.M.H.; Kroezinga, E.H.; de Vries, E.; Floris, R.; van der Laan, J.-M.; Dijkstra, B.W.; Janssen, D.B. Characterization of the β-Lactam Binding Site of Penicillin Acylase of Escherichia Coli by Structural and Site-Directed Mutagenesis Studies. Protein Eng. 2000, 13, 857–863. [Google Scholar] [CrossRef]
- Aghajari, N.; Roth, M.; Haser, R. Crystallographic Evidence of a Transglycosylation Reaction: Ternary Complexes of a Psychrophilic α-Amylase. Biochemistry 2002, 41, 4273–4280. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, S.; Earnest, T.N.; Hol, W.G.J. Precursor Structure of Cephalosporin Acylase: Insights into Autoproteolytic Activation in a New N-Terminal Hydrolase Family. J. Biol. Chem. 2002, 277, 2823–2829. [Google Scholar] [CrossRef]
- Kim, J.K.; Yang, I.S.; Shin, H.J.; Cho, K.J.; Ryu, E.K.; Kim, S.H.; Park, S.S.; Kim, K.H. Insight into Autoproteolytic Activation from the Structure of Cephalosporin Acylase: A Protein with Two Proteolytic Chemistries. Proc. Natl. Acad. Sci. USA 2006, 103, 1732–1737. [Google Scholar] [CrossRef]
- Kim, J.K.; Yang, I.S.; Rhee, S.; Dauter, Z.; Lee, Y.S.; Park, S.S.; Kim, K.H. Crystal Structures of Glutaryl 7-Aminocephalosporanic Acid Acylase: Insight into Autoproteolytic Activation. Biochemistry 2003, 42, 4084–4093. [Google Scholar] [CrossRef]
- McVey, C.E.; Walsh, M.A.; Dodson, G.G.; Wilson, K.S.; Brannigan, J.A. Crystal Structures of Penicillin Acylase Enzyme-Substrate Complexes: Structural Insights into the Catalytic Mechanism. J. Mol. Biol. 2001, 313, 139–150. [Google Scholar] [CrossRef]
- Priya, R. Structural and Biophysical Characterization of Penicillin V Acylase from Bacillus Subtilis and Comparison with Related Hydrolases as Well as Study of Selected Proteins from Malarial Parasite; University of Pune: Pune, India, 2006. [Google Scholar]
- Kwon, T.H.; Rhee, S.; Lee, Y.S.; Park, S.S.; Kim, K.H. Crystallization and Preliminary X-ray Diffraction Analysis of Glutaryl-7-Aminocephalosporanic Acid Acylase from Pseudomonas Sp. GK16. J. Struct. Biol. 2000, 131, 79–81. [Google Scholar] [CrossRef]
- Mayer, J.; Pippel, J.; Günther, G.; Müller, C.; Lauermann, A.; Knuuti, T.; Blankenfeldt, W.; Jahn, D.; Biedendieck, R. Crystal Structures and Protein Engineering of Three Different Penicillin G Acylases from Gram-Positive Bacteria with Different Thermostability. Appl. Microbiol. Biotechnol. 2019, 103, 7537–7552. [Google Scholar] [CrossRef]
- Panigrahi, P.; Chand, D.; Mukherji, R.; Ramasamy, S.; Suresh, C.G. Sequence and Structure-Based Comparative Analysis to Assess, Identify and Improve the Thermostability of Penicillin G Acylases. J. Ind. Microbiol. Biotechnol. 2015, 42, 1493–1506. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Hirano, S.; Tanaka, K.; Ohnishi, Y.; Horinouchi, S. Conditionally Positive Effect of the TetR-Family Transcriptional Regulator AtrA on Streptomycin Production by Streptomyces Griseus. Microbiology 2008, 154, 905–914. [Google Scholar] [CrossRef]
- Funabashi, M.; Funa, N.; Horinouchi, S. Phenolic Lipids Synthesized by Type III Polyketide Synthase Confer Penicillin Resistance on Streptomyces Griseus. J. Biol. Chem. 2008, 283, 13983–13991. [Google Scholar] [CrossRef]
- Ohnishi, Y.; Ishikawa, J.; Hara, H.; Suzuki, H.; Ikenoya, M.; Ikeda, H.; Yamashita, A.; Hattori, M.; Horinouchi, S. Genome Sequence of the Streptomycin-Producing Microorganism Streptomyces Griseus IFO 13350. J. Bacteriol. 2008, 190, 4050–4060. [Google Scholar] [CrossRef]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2009, 37, D26–D31. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Rossmann, M.G.; Argos, P. Exploring Structural Homology of Proteins. J. Mol. Biol. 1976, 105, 75–95. [Google Scholar] [CrossRef]
- Smith, T.F.; Waterman, M.S. Identification of Common Molecular Subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolym. Orig. Res. Biomol. 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Russell, R.B.; Barton, G.J. Multiple Protein Sequence Alignment from Tertiary Structure Comparison: Assignment of Global and Residue Confidence Levels. Proteins Struct. Funct. Bioinform. 1992, 14, 309–323. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1FXH | 1FM2 | 1KEH | 2WYB | 3K3W | 4HSR | 4YF9 | 6NVW | 7EA4 | 7REO |
|---|---|---|---|---|---|---|---|---|---|
| A-M142 | A-M145 | A-M145 | A-L146 | A-M143 | A-G162 | A-N160 | A-M145 | A-G181 | A-M697 |
| A-R145 | A-L148 | A-L148 | A-E149 | A-R146 | A-L164 | A-A162 | A-Y147 | No fit | A-R700 |
| A-F146 | A-Y149 | A-Y149 | A-G150 | A-F147 | A-M165 | A-G163 | A-F148 | A-E182 | A-F701 |
| Β-S1 | Β-S170 | Β-A170 | Β-S1 | Β-S1 | Β-S1 | Β-S1 | Β-S1 | Β-S282 | Β-S1 |
| Β-P22 | Β-P191 | Β-P191 | Β-P22 | Β-P22 | Β-P22 | Β-P22 | Β-P22 | Β-P303 | Β-P22 |
| Β-Q23 | Β-H192 | Β-H192 | Β-H23 | Β-Q23 | Β-H23 | Β-H23 | Β-Q23 | Β-H304 | Β-Q23 |
| Β-F24 | Β-L193 | Β-L193 | Β-F24 | Β-F24 | Β-R24 | Β-W24 | Β-V24 | Β-R305 | Β-F24 |
| Β-V56 | Β-R226 | Β-R226 | Β-N57 | Β-L56 | Β-P56 | Β-Q57 | Β-M56 | Β-S337 | Β-L56 |
| Β-F57 | Β-F227 | Β-F227 | Β-I58 | Β-F57 | Β-F57 | Β-I58 | Β-F57 | Β-I338 | Β-F57 |
| Β-T68 | Β-T238 | Β-T238 | Β-T69 | Β-T69 | Β-T69 | Β-T69 | Β-T68 | Β-T349 | Β-T68 |
| Β-A69 | Β-V239 | Β-V239 | Β-V70 | Β-A70 | Β-H70 | Β-V70 | Β-A69 | Β-R350 | Β-A69 |
| Β-F71 | Β-G241 | B-G241 | B-T72 | B-P72 | B-F72 | B-T72 | B-Y71 | B-Y352 | B-A71 |
| Β-W154 | Β-Y322 | Β-Y322 | Β-W162 | Β-W154 | Β-L154 | Β-W165 | Β-Y158 | Β-S436 | Β-W154 |
| Β-I177 | Β-F346 | Β-F346 | Β-V187 | Β-I177 | Β-H178 | Β-V190 | Β-L181 | Β-E460 | Β-I177 |
| Β-N178 | Β-N347 | Β-N347 | Β-N189 | Β-N178 | Β-N179 | Β-N191 | Β-N182 | Β-N461 | Β-N178 |
| Β-A241 | Β-N413 | Β-N413 | Β-N269 | Β-N241 | Β-N242 | Β-N278 | Β-N245 | Β-N523 | Β-N241 |
| Β-R263 | Β-R443 | Β-R443 | Β-R297 | Β-R261 | Β-R263 | Β-R308 | Β-R266 | Β-R547 | Β-R263 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quiroga, I.; Hernández-González, J.A.; Bautista-Rodríguez, E.; Benítez-Rojas, A.C. Exploring the Structurally Conserved Regions and Functional Significance in Bacterial N-Terminal Nucleophile (Ntn) Amide-Hydrolases. Int. J. Mol. Sci. 2024, 25, 6850. https://doi.org/10.3390/ijms25136850
Quiroga I, Hernández-González JA, Bautista-Rodríguez E, Benítez-Rojas AC. Exploring the Structurally Conserved Regions and Functional Significance in Bacterial N-Terminal Nucleophile (Ntn) Amide-Hydrolases. International Journal of Molecular Sciences. 2024; 25(13):6850. https://doi.org/10.3390/ijms25136850
Chicago/Turabian StyleQuiroga, Israel, Juan Andrés Hernández-González, Elizabeth Bautista-Rodríguez, and Alfredo C. Benítez-Rojas. 2024. "Exploring the Structurally Conserved Regions and Functional Significance in Bacterial N-Terminal Nucleophile (Ntn) Amide-Hydrolases" International Journal of Molecular Sciences 25, no. 13: 6850. https://doi.org/10.3390/ijms25136850
APA StyleQuiroga, I., Hernández-González, J. A., Bautista-Rodríguez, E., & Benítez-Rojas, A. C. (2024). Exploring the Structurally Conserved Regions and Functional Significance in Bacterial N-Terminal Nucleophile (Ntn) Amide-Hydrolases. International Journal of Molecular Sciences, 25(13), 6850. https://doi.org/10.3390/ijms25136850