

Structure-Based Analysis of Cefaclor Pharmacokinetic Diversity According to Human Peptide Transporter-1 Genetic Polymorphism


Abstract
:
1. Introduction
2. Results and Discussion
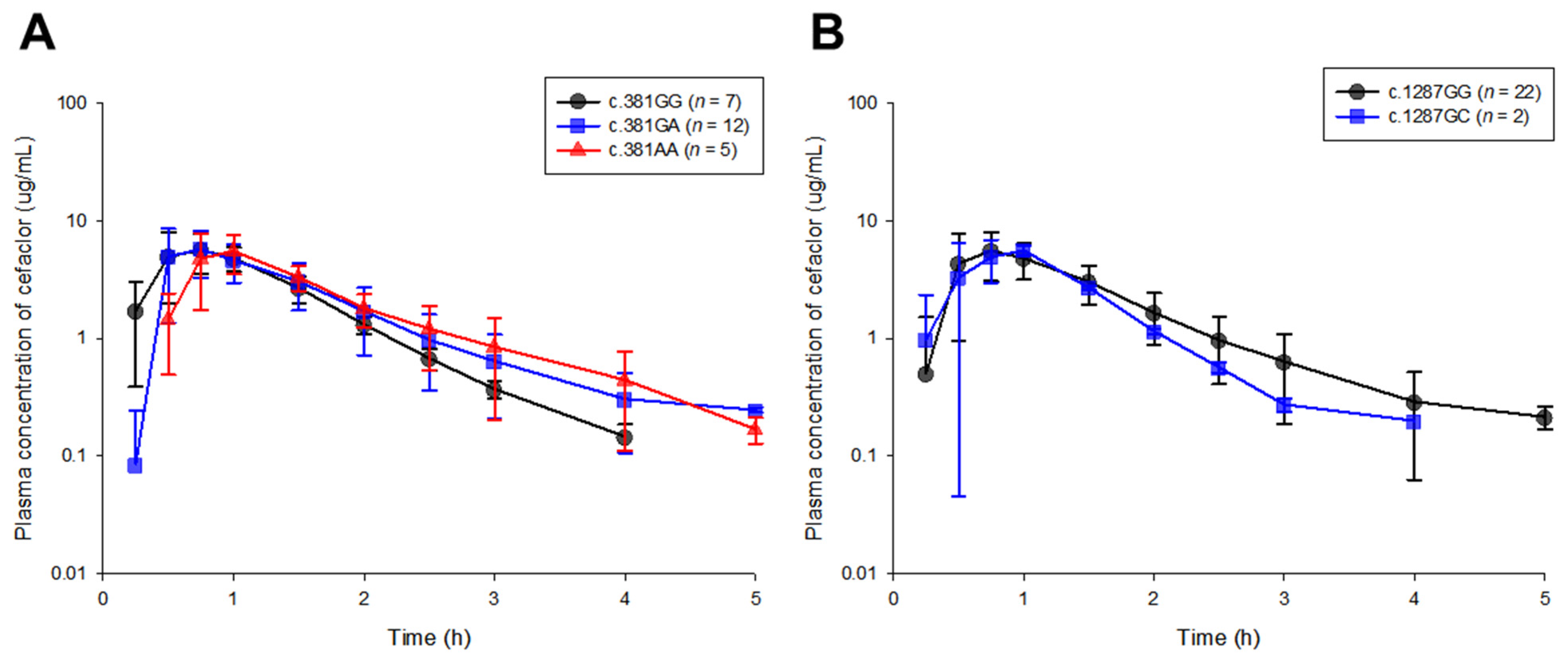
2.1. Pharmacokinetic and Modeling-Based Covariate Analysis
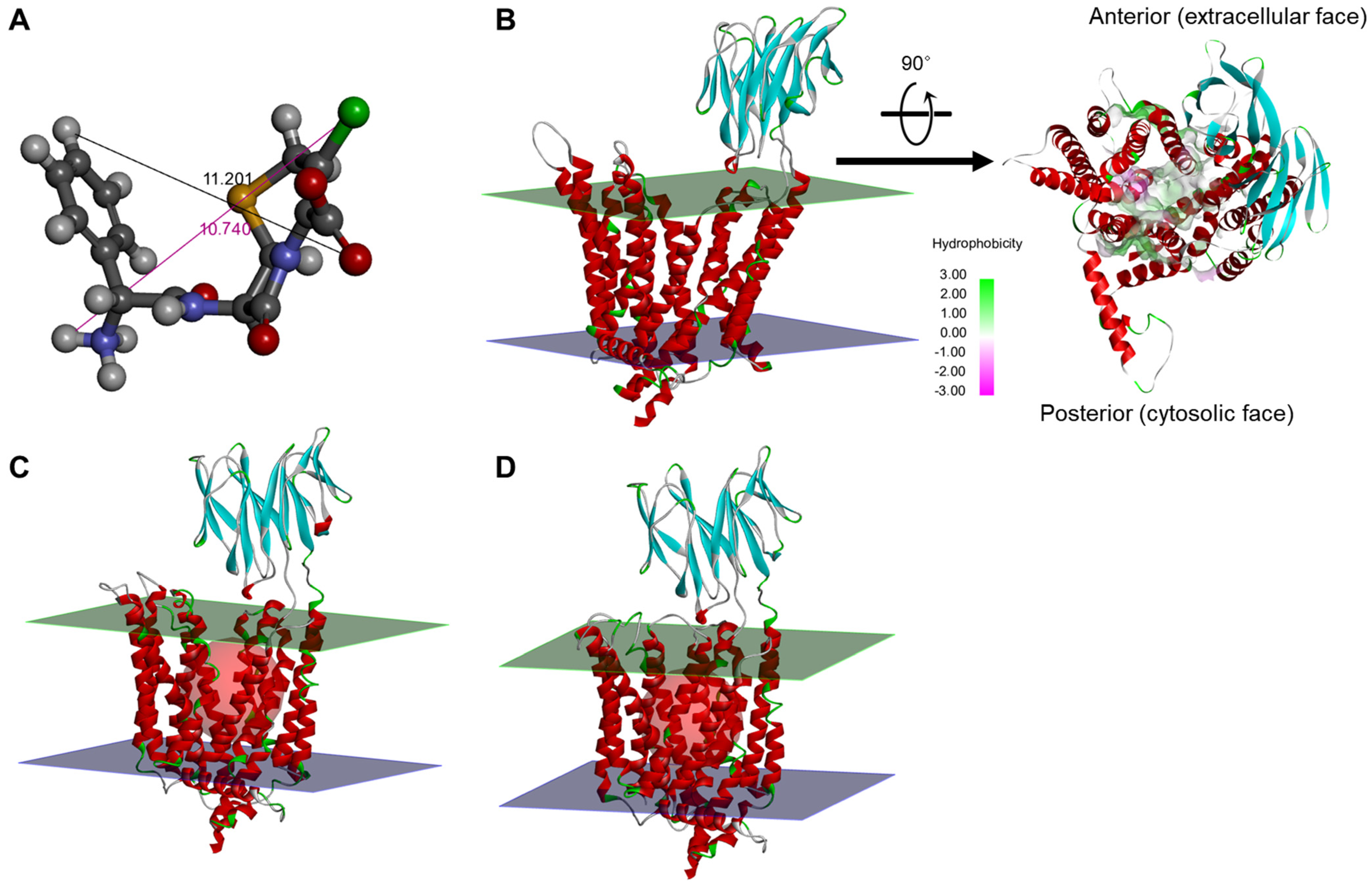
2.2. Molecular Modeling-Based Cefaclor-PEPT1 Interaction Analysis
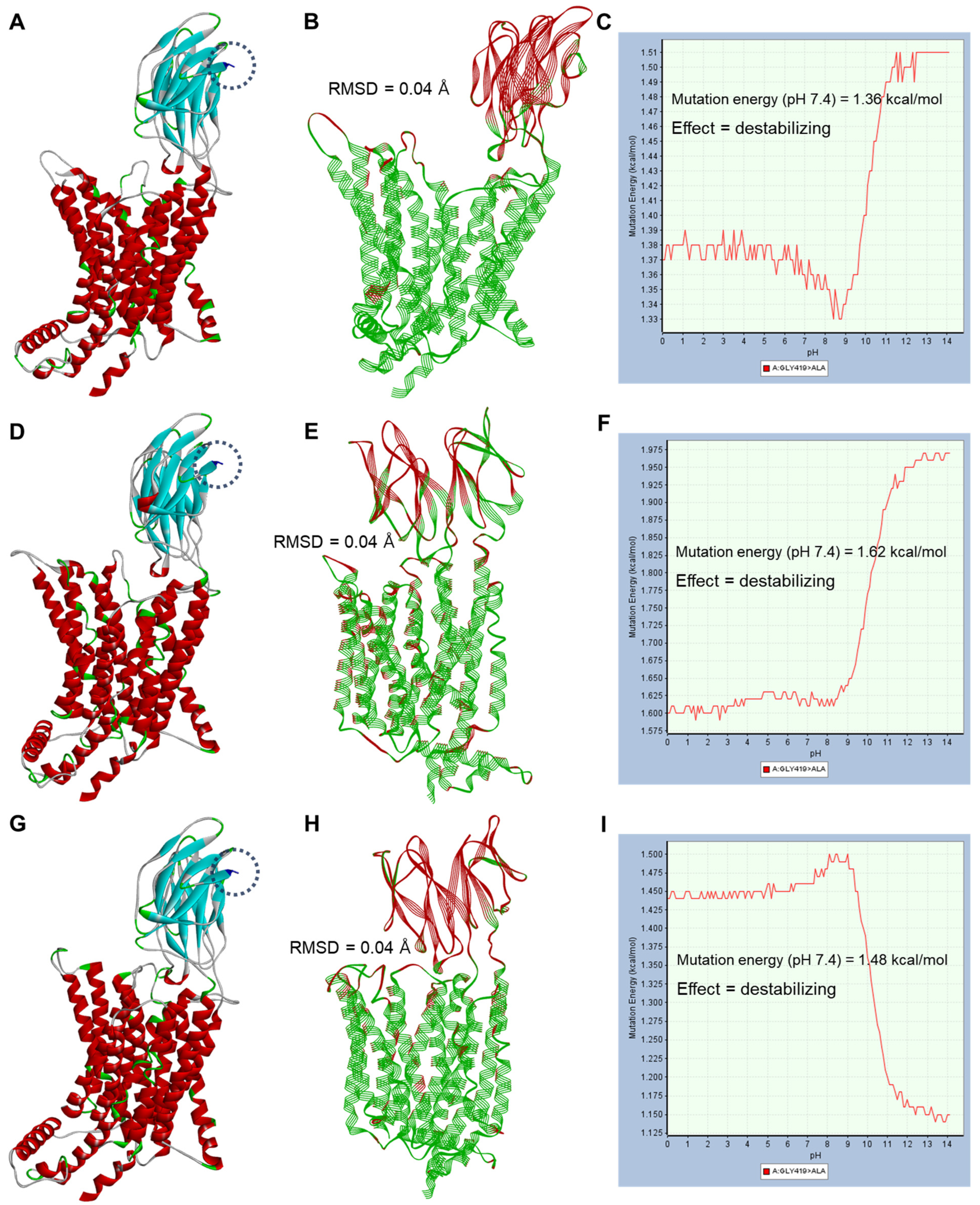
2.3. PEPT1 Structural Analysis According to Genetic Polymorphisms of SLC15A1 Exons 5 and 16
2.4. PEPT1 Structural Analysis through Application of Other SLC15A1 SNPs
2.5. Analysis of Genetic Polymorphism Effects Based on Molecular Dynamics Simulation
3. Materials and Methods
3.1. Research Approach
3.2. Pharmacokinetic Study
3.3. Pharmacokinetic Analysis
3.4. Covariate Analysis Using Population Pharmacokinetic Modeling
3.5. Molecular Modeling-Based Cefaclor-PEPT1 Interaction Analysis
3.6. Structural Analysis According to Genetic Polymorphisms of SLC15A1 Exons 5 and 16
3.7. Structural Analysis following Application of Other Additional SLC15A1 SNPs
3.8. Structural Analysis through Molecular Dynamics Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lima, L.M.; da Silva, B.N.M.; Barbosa, G.; Barreiro, E.J. β-lactam antibiotics: An overview from a medicinal chemistry perspective. Eur. J. Med. Chem. 2020, 208, 112829. [Google Scholar] [CrossRef] [PubMed]
- Nagai, K.; Davies, T.A.; Jacobs, M.R.; Appelbaum, P.C. Effects of amino acid alterations in penicillin-binding proteins (PBPs) 1a, 2b, and 2x on PBP affinities of penicillin, ampicillin, amoxicillin, cefditoren, cefuroxime, cefprozil, and cefaclor in 18 clinical isolates of penicillin-susceptible,-intermediate, and-resistant pneumococci. Antimicrob. Agents Chemother. 2002, 46, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Bach, V.T.; Khurana, M.M.; Thadepalli, H. In vitro activity of cefaclor against aerobic and anaerobic bacteria. Antimicrob. Agents Chemother. 1978, 13, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Bergman, H.D.; Jackson, E.A.; Cardoni, A.A. Cefaclor (Ceclor®-Eli Lilly and Company). Drug Intell. Clin. Pharm. 1980, 14, 11–16. [Google Scholar] [CrossRef]
- Shadomy, S.; Wagner, G.; Carver, M. In vitro activities of five oral cephalosporins against aerobic pathogenic bacteria. Antimicrob. Agents Chemother. 1977, 12, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Brumfitt, W.; Hamilton-Miller, J. Cefaclor into the millennium. J. Chemother. 1999, 11, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.-H.; Jang, J.-H.; Cho, H.-Y.; Lee, Y.-B. Population pharmacokinetic analysis of cefaclor in healthy Korean subjects. Pharmaceutics 2021, 13, 754. [Google Scholar] [CrossRef] [PubMed]
- Nix, D.E.; Symonds, W.T.; Hyatt, J.M.; Wilton, J.H.; Teal, M.A.; Reidenberg, P.; Affrime, M.B. Comparative pharmacokinetics of oral ceftibuten, cefixime, cefaclor, and cefuroxime axetil in healthy volunteers. Pharmacotherapy 1997, 17, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.-H.; Jeong, S.-H.; Cho, H.-Y.; Lee, Y.-B. Population pharmacokinetics of cis-, trans-, and total cefprozil in healthy male Koreans. Pharmaceutics 2019, 11, 531. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Anderson, G.D.; Phillips, B.R.; Kong, W.; Shen, D.D.; Wang, J. Interactions of amoxicillin and cefaclor with human renal organic anion and peptide transporters. Drug Metab. Dispos. 2006, 34, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Bretschneider, B.; Brandsch, M.; Neubert, R. Intestinal transport of β-lactam antibiotics: Analysis of the affinity at the H+/peptide symporter (PEPT1), the uptake into Caco-2 cell monolayers and the transepithelial flux. Pharm. Res. 1999, 16, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Dantzig, A.H.; Tabas, L.B.; Bergin, L. Cefaclor uptake by the proton-dependent dipeptide transport carrier of human intestinal Caco-2 cells and comparison to cephalexin uptake. Biochim. Biophys. Acta 1992, 1112, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Terada, T.; Inui, K.-i. Gene expression and regulation of drug transporters in the intestine and kidney. Biochem. Pharmacol. 2007, 73, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Luckner, P.; Brandsch, M. Interaction of 31 β-lactam antibiotics with the H+/peptide symporter PEPT2: Analysis of affinity constants and comparison with PEPT1. Eur. J. Pharm. Biopharm. 2005, 59, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Daniel, H.; Kottra, G. The proton oligopeptide cotransporter family SLC15 in physiology and pharmacology. Pflügers Archiv 2004, 447, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.E.; Clémençon, B.; Hediger, M.A. Proton-coupled oligopeptide transporter family SLC15: Physiological, pharmacological and pathological implications. Mol. Aspects Med. 2013, 34, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-M.; Lee, S.-N.; Kang, H.-A.; Cho, H.-Y.; Lee, I.-K.; Lee, Y.-B. Haplotype analysis and single nucleotide polymorphism frequency of PEPT1 gene (Exon 5 and 16) in Korean. J. Pharm. Investig. 2009, 39, 411–416. [Google Scholar] [CrossRef]
- Anderle, P.; Nielsen, C.U.; Pinsonneault, J.; Krog, P.L.; Brodin, B.; Sadée, W. Genetic variants of the human dipeptide transporter PEPT1. J. Pharmacol. Exp. Ther. 2006, 316, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Killer, M.; Wald, J.; Pieprzyk, J.; Marlovits, T.C.; Löw, C. Structural snapshots of human PepT1 and PepT2 reveal mechanistic insights into substrate and drug transport across epithelial membranes. Sci. Adv. 2021, 7, eabk3259. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Units | SLC15A1 Exon 5 | ||
|---|---|---|---|---|
| c.381GG (n = 7) | c.381GA (n = 12) | c.381AA (n = 5) | ||
| AUCall | h·μg/mL | 7.59 ± 1.52 | 7.97 ± 1.83 | 7.81 ± 2.22 |
| AUCinf | h·μg/mL | 7.74 ± 1.53 | 8.21 ± 1.86 | 7.97 ± 2.20 |
| CL/F | mL/hr | 33,503.74 ± 7249.84 | 32,189.36 ± 8599.64 | 33,277.68 ± 8681.99 |
| Cmax | μg/mL | 6.51 ± 1.90 | 7.20 ± 2.32 | 5.88 ± 2.66 |
| T1/2 | h | 0.60 ± 0.06 | 0.70 ± 0.20 | 0.62 ± 0.16 |
| MRT | h | 1.26 ± 0.19 | 1.43 ± 0.40 | 1.64 ± 0.34 |
| Tmax | h | 0.82 ± 0.35 | 0.77 ± 0.31 | 0.95 ± 0.11 |
| V/F | mL | 29,008.13 ± 5949.24 | 31,689.12 ± 9513.51 | 29,598.65 ± 10,516.69 |
| Parameters | Units | SLC15A1 Exon 16 | |
|---|---|---|---|
| c.1287GG (n = 22) | c.1287GC (n = 2) | ||
| AUCall | h·μg/mL | 7.92 ± 1.77 | 6.85 ± 1.69 |
| AUCinf | h·μg/mL | 8.11 ± 1.79 | 7.02 ± 1.69 |
| CL/F | mL/hr | 32,444.65 ± 7951.04 | 36,702.30 ± 8866.25 |
| Cmax | μg/mL | 6.77 ± 2.34 | 6.20 ± 0.23 |
| T1/2 | h | 0.67 ± 0.17 | 0.53 ± 0.11 |
| MRT | h | 1.44 ± 0.37 | 1.26 ± 0.09 |
| Tmax | h | 0.82 ± 0.30 | 0.88 ± 0.18 |
| V/F | mL | 30,769.65 ± 8896.89 | 27,193.69 ± 1112.81 |
| Exon | SNP No. | Nucleotide Change a | Amino Acid Change | Outward-Facing Apo-State (Stability Energy [kcal/mol], pH 7.4) | Outward-Facing Open Conformation (Stability Energy [kcal/mol], pH 7.4) | Outward-Facing Occluded Conformation (Stability Energy [kcal/mol], pH 7.4) | Outward-Facing Open Conformation (Binding Energy [kcal/mol], pH 7.4) | Outward-Facing Occluded Conformation (Binding Energy [kcal/mol], pH 7.4) |
|---|---|---|---|---|---|---|---|---|
| 3 | 2 | G>A | p.Val21Ile | −0.94 (stabilizing) a | −0.78 (stabilizing) b | −0.89 (stabilizing) b | 0.11 (neutral) b | −0.21 (neutral) b |
| 3 | 3 | T>A | p.Phe28Tyr | 0.08 (neutral) a | 0.63 (destabilizing) b | 0.25 (neutral) b | 0.04 (neutral) b | −0.33 (neutral) b |
| 5 | 4 | G>A | p.Ser117Asn | −0.23 (neutral) a | −0.64 (stabilizing) b | −0.45 (neutral) b | 0.00 (neutral) b | 0.01 (neutral) b |
| 5 | 5 | C>A | p.Ser117Arg | −1.74 (stabilizing) a | −2.07 (stabilizing) b | −1.03 (stabilizing) b | −0.01 (neutral) b | −0.09 (neutral) b |
| 5 | 6 | G>A | p.Val122Met | −0.62 (stabilizing) a | 0.18 (neutral) b | 0.33 (neutral) b | 0.11 (neutral) b | −0.10 (neutral) b |
| 16 | 3 | G>C | p.Gly419Ala | 1.36 (destabilizing) a | 1.62 (destabilizing) b | 1.48 (destabilizing) b | 0.01 (neutral) b | 0.01 (neutral) b |
| 17 | 2 | G>A | p.Val450Ile | −1.14 (stabilizing) a | −0.66 (stabilizing) b | −1.00 (stabilizing) b | 0.09 (neutral) b | 0.01 (neutral) b |
| 17 | 3 | C>A | p.Thr451Asn | 0.13 (neutral) a | 0.31 (neutral) b | −0.05 (neutral) b | −0.01 (neutral) b | 0.00 (neutral) b |
| 20 | 3 | C>T | p.Pro537Ser | −0.04 (neutral) a | 0.90 (destabilizing) b | 0.61 (destabilizing) b | 0.01 (neutral) b | −0.10 (neutral) b |
| Key Residues Interacting with Cefaclor in Outward-Facing Open Conformation a | Amino Acid Change b | Outward-Facing Open Conformation (Binding Energy [kcal/mol], pH 7.4) | Key Residues Interacting with Cefaclor in Outward-Facing Occluded Conformation a | Amino Acid Change b | Outward-Facing Occluded Conformation (Binding Energy [kcal/mol], pH 7.4) |
|---|---|---|---|---|---|
| 27 Arg | p.Arg27Glu | 2.22 (destabilizing) c | 27 Arg | p.Arg27Thr | 1.15 (destabilizing) c |
| 31 Tyr | p.Tyr31Gly | 1.95 (destabilizing) c | 31 Tyr | p.Tyr31Gly | 1.46 (destabilizing) c |
| 171 Asn | p.Asn171Phe | 2.48 (destabilizing) c | 171 Asn | p.Asn171Trp | 5.35 (destabilizing) c |
| 297 Phe | p.Phe297Gly | 0.75 (destabilizing) c | 297 Phe | p.Phe297Gly | 0.90 (destabilizing) c |
| 329 Asn | p.Asn329Gly | 0.69 (destabilizing) c | 329 Asn | p.Asn329Ile | 1.80 (destabilizing) c |
| 140 Lys | p.Lys140Glu | 1.50 (destabilizing) c | 64 Tyr | p.Tyr64Asp | 0.46 (neutral) c |
| 330 Ala | p.Ala330Tyr | 1.73 (destabilizing) c | 167 Tyr | p.Tyr167Thr | 0.72 (destabilizing) c |
| Amino Acid Change Site a | Outward-Facing Apo-State (Stability Energy [kcal/mol], pH 7.4) | Amino Acid Change Site a | Outward-Facing Open Conformation (Stability Energy [kcal/mol], pH 7.4) | Amino Acid Change Site a | Outward-Facing Occluded Conformation (Stability Energy [kcal/mol], pH 7.4) |
|---|---|---|---|---|---|
| 622 Trp | 4.90 (destabilizing) b | 264 Trp | 5.25 (destabilizing) b | 223 Tyr | 5.24 (destabilizing) b |
| 223 Tyr | 4.91 (destabilizing) b | 622 Trp | 5.31 (destabilizing) b | 622 Trp | 5.41 (destabilizing) b |
| 264 Trp | 5.29 (destabilizing) b | 294 Trp | 5.53 (destabilizing) b | 304 Trp | 6.02 (destabilizing) b |
| 465 Trp | 5.47 (destabilizing) b | 135 Gly | 6.21 (destabilizing) b | 313 Gly | 6.36 (destabilizing) b |
| 304 Trp | 7.02 (destabilizing) b | 304 Trp | 6.38 (destabilizing) b | 135 Gly | 7.21 (destabilizing) b |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, J.-H.; Jeong, S.-H. Structure-Based Analysis of Cefaclor Pharmacokinetic Diversity According to Human Peptide Transporter-1 Genetic Polymorphism. Int. J. Mol. Sci. 2024, 25, 6880. https://doi.org/10.3390/ijms25136880
Jang J-H, Jeong S-H. Structure-Based Analysis of Cefaclor Pharmacokinetic Diversity According to Human Peptide Transporter-1 Genetic Polymorphism. International Journal of Molecular Sciences. 2024; 25(13):6880. https://doi.org/10.3390/ijms25136880
Chicago/Turabian StyleJang, Ji-Hun, and Seung-Hyun Jeong. 2024. "Structure-Based Analysis of Cefaclor Pharmacokinetic Diversity According to Human Peptide Transporter-1 Genetic Polymorphism" International Journal of Molecular Sciences 25, no. 13: 6880. https://doi.org/10.3390/ijms25136880
APA StyleJang, J.-H., & Jeong, S.-H. (2024). Structure-Based Analysis of Cefaclor Pharmacokinetic Diversity According to Human Peptide Transporter-1 Genetic Polymorphism. International Journal of Molecular Sciences, 25(13), 6880. https://doi.org/10.3390/ijms25136880