
From Inhalation to Neurodegeneration: Air Pollution as a Modifiable Risk Factor for Alzheimer’s Disease
 , , and
, , and
Abstract
:1. Introduction
2. Air Pollution: Composition and Sources
2.1. Gaseous Pollutants
2.2. Persistent Organic Pollutants
2.3. Heavy Metals
2.4. Particulate Matter
3. Neurotoxicity of Air Pollutants: Tracing the Path from Inhalation to the Brain
4. Pathophysiology of Alzheimer’s Disease: An Overview
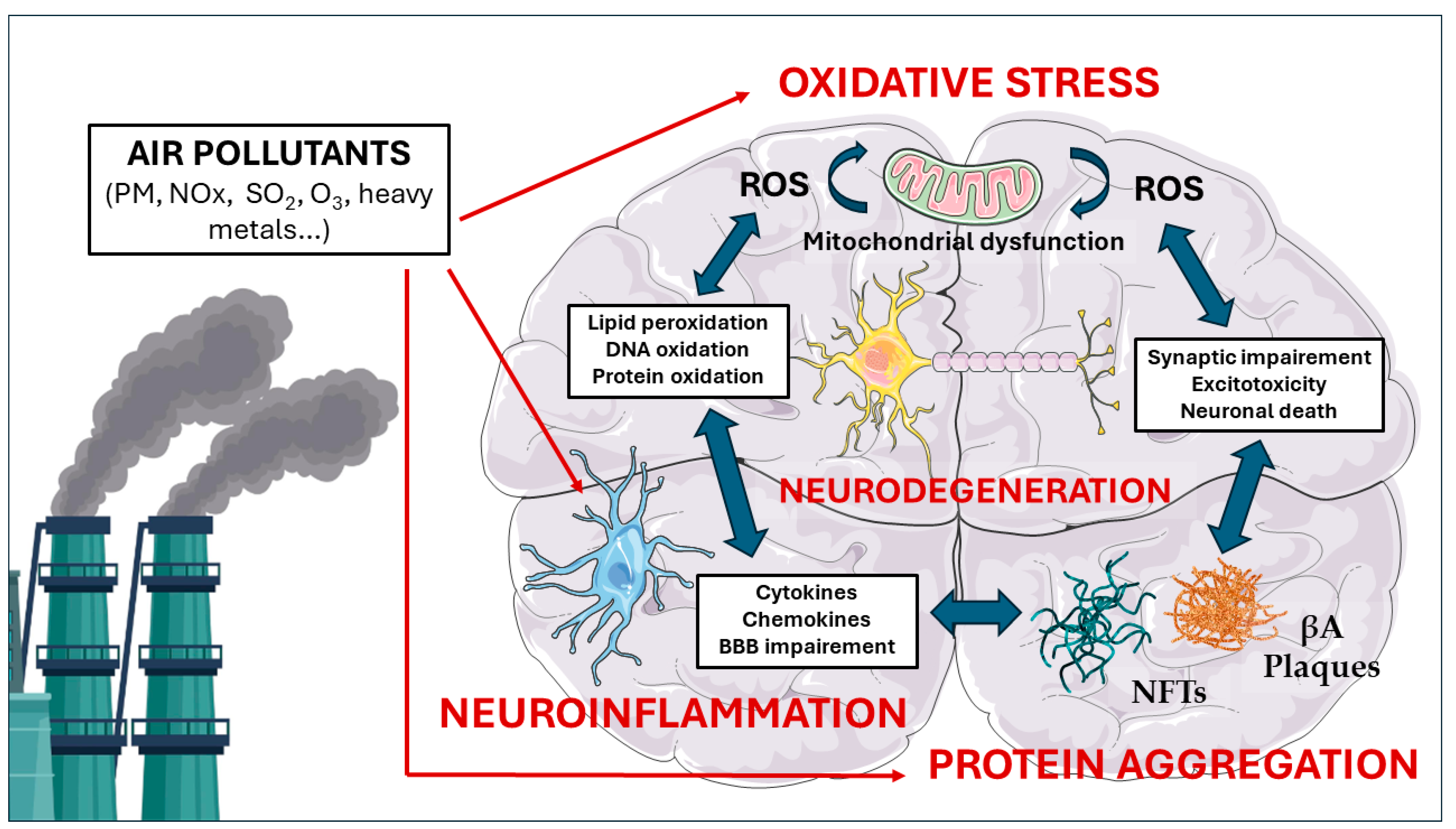
5. Molecular Pathways Linking Air Pollution to Neurodegeneration
5.1. Oxidative Stress
5.2. Neuroinflammation
5.3. Protein Aggregation
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Manisalidis, I.; Stavropoulou, E.; Stavropoulos, A.; Bezirtzoglou, E. Environmental and Health Impacts of Air Pollution: A Review. Front. Public Health 2020, 8, 14. [Google Scholar] [CrossRef]
- WHO. W.H.O. Air Pollution. Available online: https://www.who.int/health-topics/air-pollution#tab=tab_1 (accessed on 1 March 2024).
- Cohen, A.J.; Brauer, M.; Burnett, R.; Anderson, H.R.; Frostad, J.; Estep, K.; Balakrishnan, K.; Brunekreef, B.; Dandona, L.; Dandona, R.; et al. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: An analysis of data from the Global Burden of Diseases Study 2015. Lancet 2017, 389, 1907–1918. [Google Scholar] [CrossRef] [PubMed]
- Hunt, A.; Ferguson, J.; Hurley, F.; Searl, A. Social Costs of Morbidity Impacts of Air Pollution. In OECD Environment Working Papers; No. 99; OECD Publishing: Paris, France, 2016. [Google Scholar]
- Boogaard, H.; Walker, K.; Cohen, A.J. Air pollution: The emergence of a major global health risk factor. Int. Health 2019, 11, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model. Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Mensah-Kane, P.; Sumien, N. The potential of hyperbaric oxygen as a therapy for neurodegenerative diseases. Geroscience 2023, 45, 747–756. [Google Scholar] [CrossRef]
- Gammon, K. Neurodegenerative disease: Brain windfall. Nature 2014, 515, 299–300. [Google Scholar] [CrossRef] [PubMed]
- GBD 2019 Dementia Forecasting Collaborators. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef]
- 2023 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2023, 19, 1598–1695. [CrossRef]
- Khreis, H.; Bredell, C.; Wai Fung, K.; Hong, L.; Szybka, M.; Phillips, V.; Abbas, A.; Lim, Y.H.; Jovanovic Andersen, Z.; Woodcock, J.; et al. Impact of long-term air pollution exposure on incidence of neurodegenerative diseases: A protocol for a systematic review and exposure-response meta-analysis. Environ. Int. 2022, 170, 107596. [Google Scholar] [CrossRef] [PubMed]
- Pipal, A.S.; Dubey, S.; Taneja, A. Health Risk Assessment and Management of Air Pollutants. In Air Pollution and Environmental Health; Pallavi Saxena, A.S., Ed.; Springer: Singapore, 2020; Volume VII, pp. 209–232. [Google Scholar]
- Singh, N. Air Pollution Exposure Studies Related to Human Health. In Air Pollution and Environmental Health; Saxena, P., Srivastava, A., Eds.; Springer: Singapore, 2020; Volume VII, pp. 141–177. [Google Scholar]
- Taneja, A.; Saini, R.; Masih, A. Indoor air quality of houses located in the urban environment of Agra, India. Ann. N. Y. Acad. Sci. 2008, 1140, 228–245. [Google Scholar] [CrossRef] [PubMed]
- WHO. W.H.O. Household Air Pollution. Available online: https://www.who.int/en/news-room/fact-sheets/detail/household-air-pollution-and-health (accessed on 1 March 2024).
- Olloquequi, J.; Silva, O.R. Biomass smoke as a risk factor for chronic obstructive pulmonary disease: Effects on innate immunity. Innate Immun. 2016, 22, 373–381. [Google Scholar] [CrossRef]
- Leech, J.A.; Nelson, W.C.; Burnett, R.T.; Aaron, S.; Raizenne, M.E. It’s about time: A comparison of Canadian and American time-activity patterns. J. Expo. Anal. Environ. Epidemiol. 2002, 12, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Falkowska, L. Environmental characteristics of gaseous pollutants and related adverse health effects. In Synergic Influence of Gaseous Particulate, and Biological Pollutants on Human Health; Pastuszka, J.S., Ed.; CRC Press: Boca Raton, FL, USA, 2015; pp. 2–28. [Google Scholar]
- Mannucci, P.M.; Harari, S.; Martinelli, I.; Franchini, M. Effects on health of air pollution: A narrative review. Intern. Emerg. Med. 2015, 10, 657–662. [Google Scholar] [CrossRef]
- Jackson, A.V. Sources of Air Pollution. In Handbook of Atmospheric Science; Hewitt, C.N., Jackson, A.V., Eds.; Blackwell Science Ltd.: Hoboken, NJ, USA, 2003; pp. 124–155. [Google Scholar]
- Tang, Y. Sources of underground CO: Crushing and ambient temperature oxidation of coal. J. Loss Prev. Process Ind. 2015, 38, 50–57. [Google Scholar] [CrossRef]
- Chiang, P.-C.; Gao, X. SOx Control. In Air Pollution Control and Design; Chiang, P.-C., Gao, X., Eds.; Springer Nature: Singapore, 2022; pp. 7–47. [Google Scholar]
- Lin, C.K.; Lin, R.T.; Chen, P.C.; Wang, P.; De Marcellis-Warin, N.; Zigler, C.; Christiani, D.C. A Global Perspective on Sulfur Oxide Controls in Coal-Fired Power Plants and Cardiovascular Disease. Sci. Rep. 2018, 8, 2611. [Google Scholar] [CrossRef] [PubMed]
- Kleinman, L.I. The dependence of tropospheric ozone production rate on ozone precursors. Atmos. Environ. 2005, 39, 575–586. [Google Scholar] [CrossRef]
- Lee, D.S.; Köhler, I.; Grobler, E.; Rohrer, F.; Sausen, R.; Gallardo-Klenner, L.; Olivier, J.G.J.; Dentener, F.J.; Bouwman, A.F. Estimations of global no, emissions and their uncertainties. Atmos. Environ. 1997, 31, 1735–1749. [Google Scholar] [CrossRef]
- Saunois, M.; Stavert, A.R.; Poulter, B.; Bousquet, P.; Canadell, J.G.; Jackson, R.B.; Raymond, P.A.; Dlugokencky, E.J.; Houweling, S.; Patra, P.K. The global methane budget 2000–2017. Earth Syst. Sci. Data Discuss. 2019, 2019, 1–136. [Google Scholar] [CrossRef]
- Zhang, J.J.; Wei, Y.; Fang, Z. Ozone Pollution: A Major Health Hazard Worldwide. Front. Immunol 2019, 10, 2518. [Google Scholar] [CrossRef]
- Scheringer, M.; Strempel, S.; Hukari, S.; Ng, C.A.; Blepp, M.; Hungerbuhler, K. How many persistent organic pollutants should we expect? Atmos. Pollut. Res. 2012, 3, 383–391. [Google Scholar] [CrossRef]
- Liu, C.; Hou, H.S. Physical exercise and persistent organic pollutants. Heliyon 2023, 9, e19661. [Google Scholar] [CrossRef] [PubMed]
- Maring, T.; Kumar, S.; Jha, A.K.; Kumar, N.; Pandey, S.P. Airborne Particulate Matter and Associated Heavy Metals: A Review. Macromol. Symp. 2023, 407, 2100487. [Google Scholar] [CrossRef]
- Martín-Cruz, Y.; Gómez-Losada, Á. Risk Assessment and Source Apportionment of Metals on Atmospheric Particulate Matter in a Suburban Background Area of Gran Canaria (Spain). Int. J. Environ. Res. Public Health 2023, 20, 5763. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Mutlu, G.M. Particulate Matter Air Pollution: Effects on the Cardiovascular System. Front. Endocrinol. 2018, 9, 680. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.M.; Shi, J.P.; Shuhua, X.; Khan, A.; Mark, D.; Kinnersley, R.; Yin, J. Measurement of Number, Mass and Size Distribution of Particles in the Atmosphere. Philos. Trans. Math. Phys. Eng. Sci. 2000, 358, 2567–2580. [Google Scholar] [CrossRef]
- Harrison, R.M. Airborne particulate matter. Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering. Sciences 2020, 378, 20190319. [Google Scholar]
- Oberdörster, G.; Utell, M.J. Ultrafine particles in the urban air: To the respiratory tract--and beyond? Environ. Health Perspect. 2002, 110, A440–A441. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Garcidueñas, L.; Azzarelli, B.; Acuna, H.; Garcia, R.; Gambling, T.M.; Osnaya, N.; Monroy, S.; Tizapantzi, M.D.R.; Carson, J.L.; Villarreal-Calderon, A.; et al. Air pollution and brain damage. Toxicol. Pathol. 2002, 30, 373–389. [Google Scholar] [CrossRef]
- Calderón-Garcidueñas, L.; Maronpot, R.R.; Torres-Jardon, R.; Henríquez-Roldán, C.; Schoonhoven, R.; Acuña-Ayala, H.; Villarreal-Calderón, A.; Nakamura, J.; Fernando, R.; Reed, W.; et al. DNA damage in nasal and brain tissues of canines exposed to air pollutants is associated with evidence of chronic brain inflammation and neurodegeneration. Toxicol. Pathol. 2003, 31, 524–538. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.M.; Lu, D.; Liu, L.P.; Zhang, H.H.; Zhou, Y.Y. Olfactory dysfunction in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2016, 12, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Genc, S.; Zadeoglulari, Z.; Fuss, S.H.; Genc, K. The adverse effects of air pollution on the nervous system. J. Toxicol. 2012, 2012, 782462. [Google Scholar] [CrossRef] [PubMed]
- Oberdörster, G.; Elder, A.; Rinderknecht, A. Nanoparticles and the brain: Cause for concern? J. Nanosci. Nanotechnol. 2009, 9, 4996–5007. [Google Scholar] [CrossRef] [PubMed]
- Nemmar, A.; Hoet, P.H.; Vanquickenborne, B.; Dinsdale, D.; Thomeer, M.; Hoylaerts, M.F.; Vanbilloen, H.; Mortelmans, L.; Nemery, B. Passage of inhaled particles into the blood circulation in humans. Circulation 2002, 105, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Nemmar, A.; Zia, S.; Subramaniyan, D.; Al-Amri, I.; Al Kindi, M.A.; Ali, B.H. Interaction of diesel exhaust particles with human, rat and mouse erythrocytes in vitro. Cell. Physiol. Biochem. 2012, 29, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Geiser, M.; Rothen-Rutishauser, B.; Kapp, N.; Schürch, S.; Kreyling, W.; Schulz, H.; Semmler, M.; Im Hof, V.; Heyder, J.; Gehr, P. Ultrafine particles cross cellular membranes by nonphagocytic mechanisms in lungs and in cultured cells. Environ. Health Perspect. 2005, 113, 1555–1560. [Google Scholar] [CrossRef]
- Araujo, J.A.; Nel, A.E. Particulate matter and atherosclerosis: Role of particle size, composition and oxidative stress. Part. Fibre Toxicol. 2009, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Oberdörster, G.; Oberdörster, E.; Oberdörster, J. Nanotoxicology: An emerging discipline evolving from studies of ultrafine particles. Environ. Health Perspect. 2005, 113, 823–839. [Google Scholar] [CrossRef]
- Yarns, B.C.; Holiday, K.A.; Carlson, D.M.; Cosgrove, C.K.; Melrose, R.J. Pathophysiology of Alzheimer’s Disease. Psychiatr. Clin. N. Am. 2022, 45, 663–676. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.; Li, R.; Sterling, K.; Song, W. Amyloid β-based therapy for Alzheimer’s disease: Challenges, successes and future. Signal Transduct. Target. Ther. 2023, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Otero-Garcia, M.; Mahajani, S.U.; Wakhloo, D.; Tang, W.; Xue, Y.-Q.; Morabito, S.; Pan, J.; Oberhauser, J.; Madira, A.E.; Shakouri, T.; et al. Molecular signatures underlying neurofibrillary tangle susceptibility in Alzheimer’s disease. Neuron 2022, 110, 2929–2948.e8. [Google Scholar] [CrossRef]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef] [PubMed]
- Pfundstein, G.; Nikonenko, A.G.; Sytnyk, V. Amyloid precursor protein (APP) and amyloid β (Aβ) interact with cell adhesion molecules: Implications in Alzheimer’s disease and normal physiology. Front. Cell Dev. Biol. 2022, 10, 969547. [Google Scholar] [CrossRef] [PubMed]
- Roselli, S.; Satir, T.M.; Camacho, R.; Fruhwürth, S.; Bergström, P.; Zetterberg, H.; Agholme, L. APP-BACE1 Interaction and Intracellular Localization Regulate Aβ Production in iPSC-Derived Cortical Neurons. Cell. Mol. Neurobiol. 2023, 43, 3653–3668. [Google Scholar] [CrossRef] [PubMed]
- Rawat, P.; Sehar, U.; Bisht, J.; Selman, A.; Culberson, J.; Reddy, P.H. Phosphorylated Tau in Alzheimer’s Disease and Other Tauopathies. Int. J. Mol. Sci. 2022, 23, 12841. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Guerrero, J.; Santiago-Balmaseda, A.; Jeronimo-Aguilar, P.; Vargas-Rodríguez, I.; Cadena-Suárez, A.R.; Sánchez-Garibay, C.; Pozo-Molina, G.; Méndez-Catalá, C.F.; Cardenas-Aguayo, M.D.; Diaz-Cintra, S.; et al. Alzheimer’s Disease: An Updated Overview of Its Genetics. Int. J. Mol. Sci. 2023, 24, 3754. [Google Scholar] [CrossRef] [PubMed]
- Plachez, C.; Tsytsarev, V.; Zhao, S.; Erzurumlu, R.S. Amyloid Deposition and Dendritic Complexity of Corticocortical Projection Cells in Five Familial Alzheimer’s Disease Mouse. Neuroscience 2023, 512, 85–98. [Google Scholar] [CrossRef]
- Yang, Z.; Zou, Y.; Wang, L. Neurotransmitters in Prevention and Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 3841. [Google Scholar] [CrossRef]
- Govindpani, K.; McNamara, L.G.; Smith, N.R.; Vinnakota, C.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. Vascular Dysfunction in Alzheimer’s Disease: A Prelude to the Pathological Process or a Consequence of It? J. Clin. Med. 2019, 8, 651. [Google Scholar] [CrossRef]
- Ionescu-Tucker, A.; Cotman, C.W. Emerging roles of oxidative stress in brain aging and Alzheimer’s disease. Neurobiol. Aging 2021, 107, 86–95. [Google Scholar] [CrossRef]
- Thakur, S.; Dhapola, R.; Sarma, P.; Medhi, B.; Reddy, D.H. Neuroinflammation in Alzheimer’s Disease: Current Progress in Molecular Signaling and Therapeutics. Inflammation 2023, 46, 1–17. [Google Scholar] [CrossRef]
- Das, N.; Raymick, J.; Sarkar, S. Role of metals in Alzheimer’s disease. Metab. Brain Dis. 2021, 36, 1627–1639. [Google Scholar] [CrossRef]
- Kilian, J.; Kitazawa, M. The emerging risk of exposure to air pollution on cognitive decline and Alzheimer’s disease—Evidence from epidemiological and animal studies. Biomed. J. 2018, 41, 141–162. [Google Scholar] [CrossRef]
- Peters, R.; Ee, N.; Peters, J.; Booth, A.; Mudway, I.; Anstey, K.J. Air Pollution and Dementia: A Systematic Review. J. Alzheimer’s Dis. 2019, 70, S145–S163. [Google Scholar] [CrossRef] [PubMed]
- Clifford, A.; Lang, L.; Chen, R.; Anstey, K.J.; Seaton, A. Exposure to air pollution and cognitive functioning across the life course--A systematic literature review. Environ. Res. 2016, 147, 383–398. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.; Yung, K.K.L. Air Pollution and Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2020, 77, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Hahad, O.; Lelieveld, J.; Birklein, F.; Lieb, K.; Daiber, A.; Münzel, T. Ambient Air Pollution Increases the Risk of Cerebrovascular and Neuropsychiatric Disorders through Induction of Inflammation and Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 4306. [Google Scholar] [CrossRef]
- Lardelli, M. An Alternative View of Familial Alzheimer’s Disease Genetics. J. Alzheimer’s Dis. 2023, 96, 13–39. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Moulton, P.V.; Yang, W. Air Pollution, Oxidative Stress, and Alzheimer’s Disease. J. Environ. Public Health 2012, 2012, 472751. [Google Scholar] [CrossRef] [PubMed]
- Poon, H.F.; Calabrese, V.; Scapagnini, G.; Butterfield, D.A. Free radicals and brain aging. Clin. Geriatr. Med. 2004, 20, 329–359. [Google Scholar] [CrossRef] [PubMed]
- Migliore, L.; Coppedè, F. Environmental-induced oxidative stress in neurodegenerative disorders and aging. Mutat. Res. 2009, 674, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Kim, S.R. Linking Oxidative Stress and Proteinopathy in Alzheimer’s Disease. Antioxidants 2021, 10, 1231. [Google Scholar] [CrossRef] [PubMed]
- Ferré-González, L.; Peña-Bautista, C.; Baquero, M.; Cháfer-Pericás, C. Assessment of Lipid Peroxidation in Alzheimer’s Disease Differential Diagnosis and Prognosis. Antioxidants 2022, 11, 551. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Ashok, A.; Andrabi, S.S.; Mansoor, S.; Kuang, Y.; Kwon, B.K.; Labhasetwar, V. Antioxidant Therapy in Oxidative Stress-Induced Neurodegenerative Diseases: Role of Nanoparticle-Based Drug Delivery Systems in Clinical Translation. Antioxidants 2022, 11, 408. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Kalani, A.; Rai, S.; Swarnkar, S.; Tota, S.; Nath, C.; Tyagi, N. Mechanism of Oxidative Stress and Synapse Dysfunction in the Pathogenesis of Alzheimer’s Disease: Understanding the Therapeutics Strategies. Mol. Neurobiol. 2016, 53, 648–661. [Google Scholar] [CrossRef] [PubMed]
- Pardillo-Díaz, R.; Pérez-García, P.; Castro, C.; Nunez-Abades, P.; Carrascal, L. Oxidative Stress as a Potential Mechanism Underlying Membrane Hyperexcitability in Neurodegenerative Diseases. Antioxidants 2022, 11, 1511. [Google Scholar] [CrossRef]
- Cory-Slechta, D.A.; Merrill, A.; Sobolewski, M. Air Pollution-Related Neurotoxicity Across the Life Span. Annu. Rev. Pharmacol. Toxicol. 2023, 63, 143–163. [Google Scholar] [CrossRef]
- Mezzaroba, L.; Alfieri, D.F.; Colado Simão, A.N.; Vissoci Reiche, E.M. The role of zinc, copper, manganese and iron in neurodegenerative diseases. Neurotoxicology 2019, 74, 230–241. [Google Scholar] [CrossRef]
- Masaldan, S.; Bush, A.I.; Devos, D.; Rolland, A.S.; Moreau, C. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free. Radic. Biol. Med. 2019, 133, 221–233. [Google Scholar] [CrossRef]
- Numan, M.S.; Brown, J.P.; Michou, L. Impact of air pollutants on oxidative stress in common autophagy-mediated aging diseases. Int. J. Environ. Res. Public Health 2015, 12, 2289–2305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Staimer, N.; Gillen, D.L.; Tjoa, T.; Schauer, J.J.; Shafer, M.M.; Hasheminassab, S.; Pakbin, P.; Vaziri, N.D.; Sioutas, C.; et al. Associations of oxidative stress and inflammatory biomarkers with chemically-characterized air pollutant exposures in an elderly cohort. Environ. Res. 2016, 150, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Valdez, J.M.; Johnstone, A.F.M.; Richards, J.E.; Schmid, J.E.; Royland, J.E.; Kodavanti, P.R.S. Interaction of Diet and Ozone Exposure on Oxidative Stress Parameters within Specific Brain Regions of Male Brown Norway Rats. Int. J. Mol. Sci. 2018, 20, 11. [Google Scholar] [CrossRef]
- Calabró, V.; Garcés, M.; Cáceres, L.; Magnani, N.D.; Marchini, T.; Freire, A.; Vico, T.; Martinefski, M.; Vanasco, V.; Tripodi, V.; et al. Urban air pollution induces alterations in redox metabolism and mitochondrial dysfunction in mice brain cortex. Arch. Biochem. Biophys. 2021, 704, 108875. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, J.H.; Kim, Y.D.; Seo, J.H. High Vulnerability of Oligodendrocytes to Oxidative Stress Induced by Ultrafine Urban Particles. Antioxidants 2020, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Nicol, C.J.B.; Wan, C.; Chen, S.J.; Huang, R.N.; Chiang, M.C. Exposure to PM(2.5) induces neurotoxicity, mitochondrial dysfunction, oxidative stress and inflammation in human SH-SY5Y neuronal cells. Neurotoxicology 2022, 88, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Fagundes, L.S.; Fleck Ada, S.; Zanchi, A.C.; Saldiva, P.H.; Rhoden, C.R. Direct contact with particulate matter increases oxidative stress in different brain structures. Inhal. Toxicol. 2015, 27, 462–467. [Google Scholar] [CrossRef]
- Kim, J.Y.; Hong, S.; Bolormaa, O.; Seo, J.H.; Eom, S.Y.; Kim, Y.D.; Kim, H. Effects of diesel exhaust particles and urban particles on brain endothelial cells. Toxicol Res 2022, 38, 91–98. [Google Scholar] [CrossRef]
- Alkhalifa, A.E.; Al-Ghraiybah, N.F.; Odum, J.; Shunnarah, J.G.; Austin, N.; Kaddoumi, A. Blood-Brain Barrier Breakdown in Alzheimer’s Disease: Mechanisms and Targeted Strategies. Int. J. Mol. Sci. 2023, 24, 16288. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.B.; Zanchi, A.C.T.; Damaceno-Rodrigues, N.R.; Veras, M.M.; Saldiva, P.H.N.; Barros, H.M.T.; Rhoden, C.R. The impact of chronic exposure to air pollution over oxidative stress parameters and brain histology. Environ. Sci. Pollut. Res. Int. 2021, 28, 47407–47417. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Farina, F.; Botto, L.; Massimino, L.; Lonati, E.; Donzelli, E.; Ballarini, E.; Crippa, L.; Marmiroli, P.; Bulbarelli, A.; et al. Systemic Exposure to Air Pollution Induces Oxidative Stress and Inflammation in Mouse Brain, Contributing to Neurodegeneration Onset. Int. J. Mol. Sci. 2020, 21, 3699. [Google Scholar] [CrossRef] [PubMed]
- Kodavanti, P.R.S.; Valdez, M.; Richards, J.E.; Agina-Obu, D.I.; Phillips, P.M.; Jarema, K.A.; Kodavanti, U.P. Ozone-induced changes in oxidative stress parameters in brain regions of adult, middle-age, and senescent Brown Norway rats. Toxicol. Appl. Pharmacol. 2021, 410, 115351. [Google Scholar] [CrossRef] [PubMed]
- Velázquez-Pérez, R.; Rodríguez-Martínez, E.; Valdés-Fuentes, M.; Gelista-Herrera, N.; Gómez-Crisóstomo, N.; Rivas-Arancibia, S. Oxidative Stress Caused by Ozone Exposure Induces Changes in P2X7 Receptors, Neuroinflammation, and Neurodegeneration in the Rat Hippocampus. Oxid. Med. Cell. Longev. 2021, 2021, 3790477. [Google Scholar] [CrossRef] [PubMed]
- Rivas-Arancibia, S.; Guevara-Guzmán, R.; López-Vidal, Y.; Rodríguez-Martínez, E.; Zanardo-Gomes, M.; Angoa-Pérez, M.; Raisman-Vozari, R. Oxidative stress caused by ozone exposure induces loss of brain repair in the hippocampus of adult rats. Toxicol. Sci. 2010, 113, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Guxens, M.; Lubczynska, M.J.; Perez-Crespo, L.; Muetzel, R.L.; El Marroun, H.; Basagana, X.; Hoek, G.; Tiemeier, H. Associations of Air Pollution on the Brain in Children: A Brain Imaging Study. Res. Rep. Health Eff. Inst. 2022, 2022, 209. [Google Scholar] [PubMed]
- Shabani, K.; Hassan, B.A. The brain on time: Links between development and neurodegeneration. Development 2023, 150, dev200397. [Google Scholar] [CrossRef] [PubMed]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef]
- Norden, D.M.; Trojanowski, P.J.; Villanueva, E.; Navarro, E.; Godbout, J.P. Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia 2016, 64, 300–316. [Google Scholar] [CrossRef]
- Hensley, K. Neuroinflammation in Alzheimer’s disease: Mechanisms, pathologic consequences, and potential for therapeutic manipulation. J. Alzheimer’s Dis. 2010, 21, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Koyama, Y.; Shimada, S. Inflammation from Peripheral Organs to the Brain: How Does Systemic Inflammation Cause Neuroinflammation? Front. Aging Neurosci. 2022, 14, 903455. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Onyango, I.G.; Jauregui, G.V.; Čarná, M.; Bennett, J.P., Jr.; Stokin, G.B. Neuroinflammation in Alzheimer’s Disease. Biomedicines 2021, 9, 524. [Google Scholar] [CrossRef]
- Lyra e Silva, N.M.; Gonçalves, R.A.; Pascoal, T.A.; Lima-Filho, R.A.S.; Resende, E.d.P.F.; Vieira, E.L.M.; Teixeira, A.L.; de Souza, L.C.; Peny, J.A.; Fortuna, J.T.S.; et al. Pro-inflammatory interleukin-6 signaling links cognitive impairments and peripheral metabolic alterations in Alzheimer’s disease. Transl. Psychiatry 2021, 11, 251. [Google Scholar] [CrossRef]
- Plantone, D.; Pardini, M.; Righi, D.; Manco, C.; Colombo, B.M.; De Stefano, N. The Role of TNF-α in Alzheimer’s Disease: A Narrative Review. Cells 2023, 13, 54. [Google Scholar] [CrossRef]
- Saito, T.; Saido, T.C. Neuroinflammation in mouse models of Alzheimer’s disease. Clin. Exp. Neuroimmunol. 2018, 9, 211–218. [Google Scholar] [CrossRef]
- Kiraly, M.; Foss, J.F.; Giordano, T. Neuroinflammation, its Role in Alzheimer’s Disease and Therapeutic Strategie. J. Prev. Alzheimer’s Dis. 2023, 10, 686–698. [Google Scholar]
- Figueiredo-Pereira, M.E.; Rockwell, P.; Schmidt-Glenewinkel, T.; Serrano, P. Neuroinflammation and J2 prostaglandins: Linking impairment of the ubiquitin-proteasome pathway and mitochondria to neurodegeneration. Front. Mol. Neurosci. 2014, 7, 104. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Sama, P.; Long, T.C.; Hester, S.; Tajuba, J.; Parker, J.; Chen, L.C.; Veronesi, B. The cellular and genomic response of an immortalized microglia cell line (BV2) to concentrated ambient particulate matter. Inhal. Toxicol. 2007, 19, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.G.; Cole, T.B.; Coburn, J.; Chang, Y.C.; Dao, K.; Roque, P. Neurotoxicants are in the air: Convergence of human, animal, and in vitro studies on the effects of air pollution on the brain. Biomed Res. Int. 2014, 2014, 736385. [Google Scholar] [CrossRef] [PubMed]
- Hartz, A.M.; Bauer, B.; Block, M.L.; Hong, J.S.; Miller, D.S. Diesel exhaust particles induce oxidative stress, proinflammatory signaling, and P-glycoprotein up-regulation at the blood-brain barrier. FASEB J. 2008, 22, 2723–2733. [Google Scholar] [CrossRef]
- Seo, S.; Jang, M.; Kim, H.; Sung, J.H.; Choi, N.; Lee, K.; Kim, H.N. Neuro-Glia-Vascular-on-a-Chip System to Assess Aggravated Neurodegeneration via Brain Endothelial Cells upon Exposure to Diesel Exhaust Particles. Adv. Funct. Mater. 2023, 33, 2210123. [Google Scholar] [CrossRef]
- Liu, F.; Huang, Y.; Zhang, F.; Chen, Q.; Wu, B.; Rui, W.; Zheng, J.C.; Ding, W. Macrophages treated with particulate matter PM2.5 induce selective neurotoxicity through glutaminase-mediated glutamate generation. J. Neurochem. 2015, 134, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Li, X.; Ai, R.S.; Deng, S.Y.; Ye, Z.Q.; Deng, X.; Ma, W.; Xiao, S.; Wang, J.Z.; Wang, L.M.; et al. Atmospheric particulate matter aggravates cns demyelination through involvement of TLR-4/NF-kB signaling and microglial activation. eLife 2022, 11, e72247. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.; Oldham, M.; Becaria, A.; Bondy, S.C.; Meacher, D.; Sioutas, C.; Misra, C.; Mendez, L.B.; Kleinman, M. Particulate matter in polluted air may increase biomarkers of inflammation in mouse brain. Neurotoxicology 2005, 26, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Kleinman, M.T.; Araujo, J.A.; Nel, A.; Sioutas, C.; Campbell, A.; Cong, P.Q.; Li, H.; Bondy, S.C. Inhaled ultrafine particulate matter affects CNS inflammatory processes and may act via MAP kinase signaling pathways. Toxicol. Lett. 2008, 178, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Bos, I.; De Boever, P.; Emmerechts, J.; Buekers, J.; Vanoirbeek, J.; Meeusen, R.; Van Poppel, M.; Nemery, B.; Nawrot, T.; Panis, L.I. Changed gene expression in brains of mice exposed to traffic in a highway tunnel. Inhal. Toxicol. 2012, 24, 676–686. [Google Scholar] [CrossRef]
- Gerlofs-Nijland, M.E.; van Berlo, D.; Cassee, F.R.; Schins, R.P.; Wang, K.; Campbell, A. Effect of prolonged exposure to diesel engine exhaust on proinflammatory markers in different regions of the rat brain. Part. Fibre Toxicol. 2010, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- van Berlo, D.; Albrecht, C.; Knaapen, A.M.; Cassee, F.R.; Gerlofs-Nijland, M.E.; Kooter, I.M.; Palomero-Gallagher, N.; Bidmon, H.J.; van Schooten, F.J.; Krutmann, J.; et al. Comparative evaluation of the effects of short-term inhalation exposure to diesel engine exhaust on rat lung and brain. Arch. Toxicol. 2010, 84, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.; Yue, H.; Yun, Y.; Sang, N. Chronic SO2 inhalation above environmental standard impairs neuronal behavior and represses glutamate receptor gene expression and memory-related kinase activation via neuroinflammation in rats. Environ. Res. 2015, 137, 85–93. [Google Scholar] [CrossRef]
- Calderón-Garcidueñas, L.; Kavanaugh, M.; Block, M.; D’Angiulli, A.; Delgado-Chávez, R.; Torres-Jardón, R.; González-Maciel, A.; Reynoso-Robles, R.; Osnaya, N.; Villarreal-Calderon, R.; et al. Neuroinflammation, hyperphosphorylated tau, diffuse amyloid plaques, and down-regulation of the cellular prion protein in air pollution exposed children and young adults. J. Alzheimer’s Dis. 2012, 28, 93–107. [Google Scholar] [CrossRef]
- Teleanu, D.M.; Niculescu, A.G.; Lungu, I.I.; Radu, C.I.; Vladâcenco, O.; Roza, E.; Costăchescu, B.; Grumezescu, A.M.; Teleanu, R.I. An Overview of Oxidative Stress, Neuroinflammation, and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 5938. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Potapova, E.V.; Dremin, V.V.; Dunaev, A.V. Interaction of Oxidative Stress and Misfolded Proteins in the Mechanism of Neurodegeneration. Life 2020, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Nguyen, M.; Robert, A.; Meunier, B. Metal Ions in Alzheimer’s Disease: A Key Role or Not? Acc. Chem. Res. 2019, 52, 2026–2035. [Google Scholar] [CrossRef]
- Bush, A.I.; Pettingell, W.H.; Multhaup, G.; d Paradis, M.; Vonsattel, J.P.; Gusella, J.F.; Beyreuther, K.; Masters, C.L.; Tanzi, R.E. Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 1994, 265, 1464–1467. [Google Scholar] [CrossRef]
- Mantyh, P.W.; Ghilardi, J.R.; Rogers, S.; DeMaster, E.; Allen, C.J.; Stimson, E.R.; Maggio, J.E. Aluminum, iron, and zinc ions promote aggregation of physiological concentrations of beta-amyloid peptide. J. Neurochem. 1993, 61, 1171–1174. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Tamagno, E.; Guglielmotto, M.; Aragno, M.; Borghi, R.; Autelli, R.; Giliberto, L.; Muraca, G.; Danni, O.; Zhu, X.; Smith, M.A.; et al. Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J. Neurochem. 2008, 104, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Muche, A.; Arendt, T.; Schliebs, R. Oxidative stress affects processing of amyloid precursor protein in vascular endothelial cells. PLoS ONE 2017, 12, e0178127. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef] [PubMed]
- Devi, S.; Kim, J.J.; Singh, A.P.; Kumar, S.; Dubey, A.K.; Singh, S.K.; Singh, R.S.; Kumar, V. Proteotoxicity: A Fatal Consequence of Environmental Pollutants-Induced Impairments in Protein Clearance Machinery. J. Pers. Med. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Tamás, M.J.; Fauvet, B.; Christen, P.; Goloubinoff, P. Misfolding and aggregation of nascent proteins: A novel mode of toxic cadmium action in vivo. Curr. Genet. 2018, 64, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Haghani, A.; Johnson, R.G.; Dalton, H.; Feinberg, J.I.; Lewis, K.C.; Ladd-Acosta, C.; Woodward, N.C.; Safi, N.; Jaffe, A.E.; Allayee, H.; et al. Early developmental exposure to air pollution increases the risk of Alzheimers disease and amyloid production: Studies in mouse and Caenorhabditis elegans. Alzheimer’s Dement. 2020, 16, e043846. [Google Scholar] [CrossRef]
- Calderón-Garcidueñas, L.; Solt, A.C.; Henríquez-Roldán, C.; Torres-Jardón, R.; Nuse, B.; Herritt, L.; Villarreal-Calderón, R.; Osnaya, N.; Stone, I.; García, R.; et al. Long-term air pollution exposure is associated with neuroinflammation, an altered innate immune response, disruption of the blood-brain barrier, ultrafine particulate deposition, and accumulation of amyloid beta-42 and alpha-synuclein in children and young adults. Toxicol. Pathol. 2008, 36, 289–310. [Google Scholar] [PubMed]
- Kim, S.H.; Knight, E.M.; Saunders, E.L.; Cuevas, A.K.; Popovech, M.; Chen, L.C.; Gandy, S. Rapid doubling of Alzheimer’s amyloid-β40 and 42 levels in brains of mice exposed to a nickel nanoparticle model of air pollution. F1000Research 2012, 1, 70. [Google Scholar] [CrossRef] [PubMed]
- Sahu, B.; Mackos, A.R.; Floden, A.M.; Wold, L.E.; Combs, C.K. Particulate Matter Exposure Exacerbates Amyloid-β Plaque Deposition and Gliosis in APP/PS1 Mice. J. Alzheimer’s Dis. 2021, 80, 761–774. [Google Scholar] [CrossRef]
- Motesaddi Zarandi, S.; Shahsavani, A.; Khodagholi, F.; Fakhri, Y. Co-exposure to ambient PM2.5 plus gaseous pollutants increases amyloid β1–42 accumulation in the hippocampus of male and female rats. Toxin Rev. 2021, 40, 300–309. [Google Scholar] [CrossRef]
- Ku, T.; Chen, M.; Li, B.; Yun, Y.; Li, G.; Sang, N. Synergistic effects of particulate matter (PM(2.5)) and sulfur dioxide (SO(2)) on neurodegeneration via the microRNA-mediated regulation of tau phosphorylation. Toxicol. Res. 2017, 6, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Wenbin, K.; Xia, Y.; Wang, J.; Wang, Y. Sulfur Dioxide Promotes the Formation of Amyloid Fibrils through Enhanced Secondary Nucleation: A Molecular Dynamics Study. Acta Chim. Sin. 2016, 74, 694. [Google Scholar]
- Kaumbekova, S.; Torkmahalleh, M.A.; Shah, D. Ammonium Sulfate and Ultrafine Particles Affect Early Onset of Alzheimer’s Disease. Chem. Eng. Trans. 2021, 85, 187–192. [Google Scholar]
- Kaumbekova, S.; Torkmahalleh, M.A.; Shah, D. Ambient Benzo[a]pyrene’s Effect on Kinetic Modulation of Amyloid Beta Peptide Aggregation: A Tentative Association between Ultrafine Particulate Matter and Alzheimer’s Disease. Toxics 2022, 10, 786. [Google Scholar] [CrossRef] [PubMed]
- Cacciottolo, M.; Morgan, T.E.; Saffari, A.A.; Shirmohammadi, F.; Forman, H.J.; Sioutas, C.; Finch, C.E. Traffic-related air pollutants (TRAP-PM) promote neuronal amyloidogenesis through oxidative damage to lipid rafts. Free. Radic. Biol. Med. 2020, 147, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Greve, H.J.; Dunbar, A.L.; Lombo, C.G.; Ahmed, C.; Thang, M.; Messenger, E.J.; Mumaw, C.L.; Johnson, J.A., Jr.; Kodavanti, U.P.; Oblak, A.L.; et al. The bidirectional lung brain-axis of amyloid-β pathology: Ozone dysregulates the peri-plaque microenvironment. Brain 2022, 146, 991–1005. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Hajat, A.; Leary, C.S.; Ilango, S.; Semmens, E.O.; Adam, C.; Fitzpatrick, A.L.; Lopez, O.L.; Kaufman, J.D. Associations between long-term air pollution exposure and plasma amyloid beta in very old adults. Alzheimer’s Dement. 2021, 17, e054700. [Google Scholar] [CrossRef]
- Iaccarino, L.; La Joie, R.; Lesman-Segev, O.H.; Lee, E.; Hanna, L.; Allen, I.E.; Hillner, B.E.; Siegel, B.A.; Whitmer, R.A.; Carrillo, M.C.; et al. Association Between Ambient Air Pollution and Amyloid Positron Emission Tomography Positivity in Older Adults With Cognitive Impairment. JAMA Neurol. 2021, 78, 197–207. [Google Scholar] [CrossRef]
- Duchesne, J.; Gutierrez, L.A.; Chen, J.; Vienneau, D.; de Hoogh, K.; Jacquemin, B.; Ritchie, K.; Berr, C.; Mortamais, M. Association between ambient air pollution exposure and plasma β-amyloid levels in the French Three-City study: Preliminary results. ISEE Conf. Abstr. 2022, 32, 2022. [Google Scholar] [CrossRef]

{kind=link}
| Category | Pollutants | Main Sources |
|---|---|---|
| Gaseous Pollutants | Carbon monoxide (CO), sulfur oxides (SOx), nitrogen oxides (NOx), volatile organic compounds (VOCs), ozone (O3) | Incomplete combustion of carbon-containing materials, coal-burning power stations, industrial processes, vehicular traffic, biomass burning, photochemical reactions |
| Persistent Organic Pollutants (POPs) | Polychlorinated dibenzo-p-dioxins (PCDDs), polychlorinated dibenzofurans (PCDFs), hexachlorobenzene (HCB), organophosphates, dichlorodiphenyldichloroethylene (DDE), bisphenol A | Widespread use in controlling pests and diseases, enhancing crop production, and facilitating various industrial processes |
| Heavy Metals | Lead (Pb), mercury (Hg), cadmium (Cd), chromium (Cr), nickel (Ni), zinc (Zn), manganese (Mn), arsenic (As), and their inorganic salt compounds | Point sources: mines, foundries, smelters, coal-burning power plants; diffuse sources: combustion byproducts from industrial processes, residential heating, waste incineration, vehicle emissions |
| Particulate Matter (PM) | PM10 (coarse particles, ≤10 μm), PM2.5 (fine particles, ≤2.5 μm), ultrafine PM (UFPs, <0.1 μm); composed of sulfates, nitrates, endotoxins, polycyclic aromatic hydrocarbons (PAHs), heavy metals (iron, nickel, copper, zinc, vanadium) | Primary sources: road traffic exhaust, non-exhaust vehicular emissions (brake wear, tire abrasion, road dust resuspension), industrial processes (combustion, mining, construction), residential solid fuel burning (wood, coal), agricultural activities (crop residue burning, soil tillage); secondary particles: complex chemical reactions in the atmosphere |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olloquequi, J.; Díaz-Peña, R.; Verdaguer, E.; Ettcheto, M.; Auladell, C.; Camins, A. From Inhalation to Neurodegeneration: Air Pollution as a Modifiable Risk Factor for Alzheimer’s Disease. Int. J. Mol. Sci. 2024, 25, 6928. https://doi.org/10.3390/ijms25136928
Olloquequi J, Díaz-Peña R, Verdaguer E, Ettcheto M, Auladell C, Camins A. From Inhalation to Neurodegeneration: Air Pollution as a Modifiable Risk Factor for Alzheimer’s Disease. International Journal of Molecular Sciences. 2024; 25(13):6928. https://doi.org/10.3390/ijms25136928
Chicago/Turabian StyleOlloquequi, Jordi, Roberto Díaz-Peña, Ester Verdaguer, Miren Ettcheto, Carme Auladell, and Antoni Camins. 2024. "From Inhalation to Neurodegeneration: Air Pollution as a Modifiable Risk Factor for Alzheimer’s Disease" International Journal of Molecular Sciences 25, no. 13: 6928. https://doi.org/10.3390/ijms25136928