1. Introduction
Interest in structural studies of lectins has considerably increased due to the realization of their abilities to ‘read’ glycan-encoded information of cellular glycoconjugates and ‘translate’ it into cellular activities [
1]. In this field, the nature of modulatory events on activity and structure in terms of systematic profiling of ligand binding and protein folding has awaited clarification. An instructive example for the fundamental importance of such processes is provided by the alarmin high-mobility group box 1 protein (HMGB1) [
2,
3]. Intracellularly, its three cysteines are reduced allowing the protein to perform its nuclear function; upon non-classical secretion, HMGB1 associates with the chemokine CXCL12 to serve as a chemoattractant [
4]. The formation of the C23-C45 disulfide bridge is the structural reason for HMGB1 to induce proinflammatory cytokines, explaining its designation as alarmin [
5]. Adhesion/growth-regulatory ga(lactose-binding)lectins can also engage in multi-tasking (both intra- and extracellularly) following non-classical secretion and the ability to bind chemokines [
6,
7,
8,
9,
10,
11]. Intriguingly, this lectin group can undergo a redox-dependent change like HMGB1. Elucidating the nature of this change in human lectins is a prerequisite to defining biomedically relevant structure–activity relationships.
Several lines of evidence underscore the fundamental importance of cysteine residues in certain galectins: the need for a reducing agent such as dithiothreitol (DTT), the presence of a carboxamidomethylated derivative via treatment with iodoacetamide, and the strategic introduction of C-to-S mutations, which have been noted to maintain lectin activity. Most research has been conducted on mammalian galectin-1 (Gal-1) with its six cysteine residues [
12,
13,
14,
15,
16]. As a cellular protein, Gal-1 can be reacted with glutathione [
17] or hydrochloric acid and N-chloramines [
18] to detect the presence of its thiol groups in cell extracts. The six Cys residues in human Gal-1 can be entirely substituted with Ser residues without loss of lectin activity [
19,
20], indicating the potential for regulatory processes rather than involvement in ligand binding. On the chemical level, the oxidation of thiol groups and the formation of three disulfide bridges yields a mass reduction of 6 Da for bovine Gal-1 [
21], apparently converting reduced (intracellular) Gal-1 to the so-called oxidized Gal-1 that dissociates to its monomer state. This, in turn, leads to the loss of β-galactoside binding and acquisition of carbohydrate-independent growth regulation or axonal regeneration [
19,
22,
23,
24]. Obviously, a fundamental shift in quaternary structure and ligand binding properties occurs in a redox-dependent manner, as with alarmin (described above).
Structurally, the oxidation of Gal-1 causes a decrease in Trp fluorescence anisotropy [
25], a change in far-UV circular dichroism indicative of a reduction in β-strand content [
14,
26,
27], and a three-stage alteration in time-resolved Fourier transform infrared spectra [
28]. Mass spectrometric (MS) analysis of peptic/tryptic peptides has provided evidence of intramolecular disulfide bond formation via C2-C130/C16-C88/C42-C60 for bovine and human Gal-1 [
19,
21] and C2-C16/C42-C60 for rat Gal-1 [
29]. Chromatographically (via gel filtration), oxidized Gal-1 “adopts a number of different states” [
30] and shows structural heterogeneity. Oxidized Gal-1 can also appear as a covalently linked dimer (via an inter-molecular disulfide bond) under certain conditions [
15,
27]. Of conspicuous physiological relevance, the redox process appears to be reversible depending on environmental conditions [
14,
27,
30]. Unfortunately, crystallographic information as to how oxidized Gal-1 looks is not available. In contrast, this is the case for chicken galectin (CG)-1B, which was produced via duplication of the orthologous gene for avian Gal-1 to establish the paralogue pair CG1A/-1B [
31]. Oxidation of this galectin leads to intra-(C2-C7) or inter-molecular (C7-C7′) disulfide bridges, with resulting dimeric proteins being distinguished by their shape and type of association [
32]. In this case, establishment of this covalent bonding is favored by the local vicinity between cysteines. Given the prominent role of Gal-1 in acute and chronic inflammation, neurodegenerative diseases, and malignancy [
33,
34,
35,
36], this apparent gap in our structural knowledge on “oxidized Gal-1” has prompted us to follow the previous suggestion that “NMR studies in solution can help to clarify this problem” [
37]. By applying a strategic combination of methods, most importantly NMR spectroscopy, our present data shed light on the molecular details of the conversion of Gal-1 to its oxidized form(s).
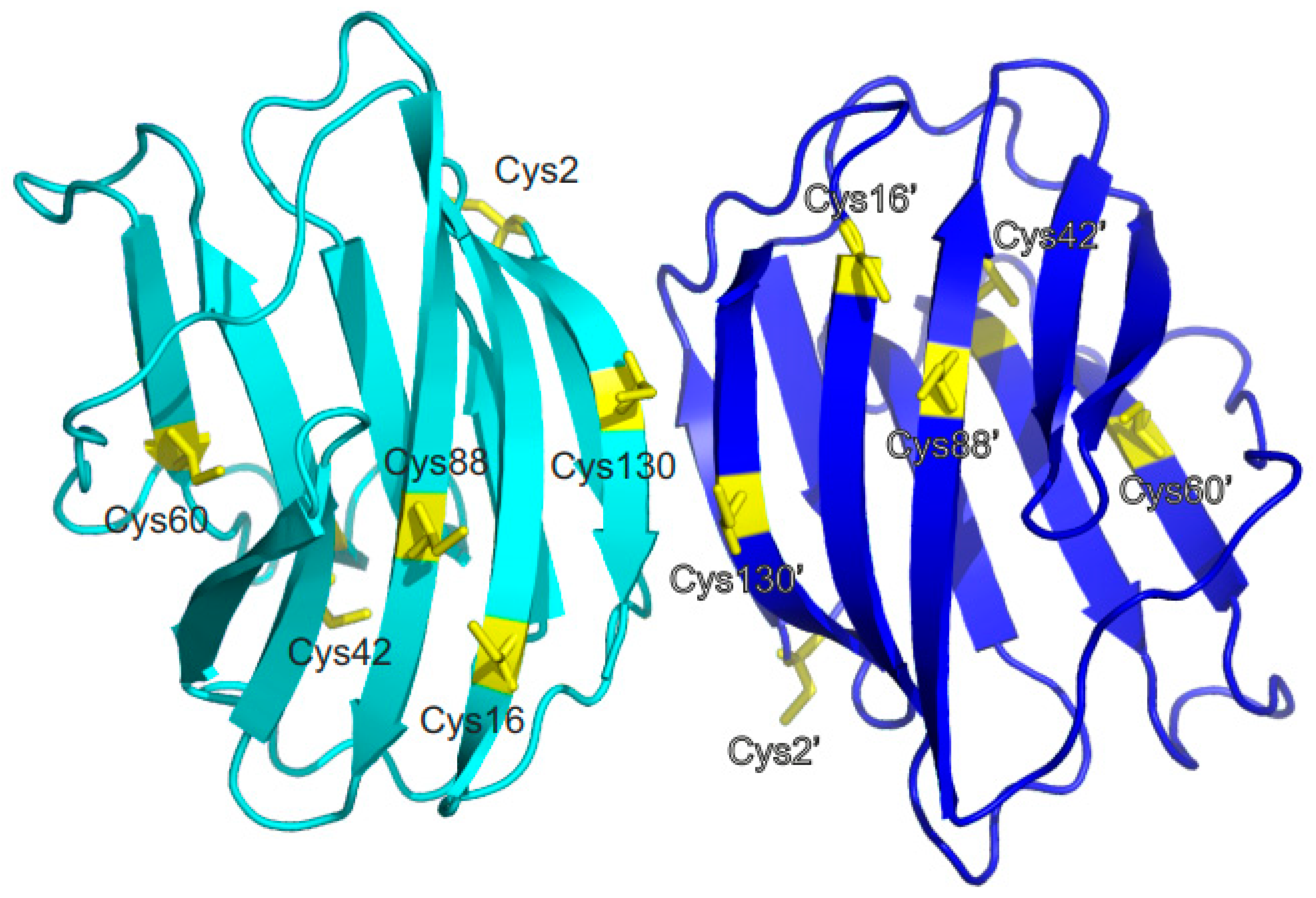
Figure 1 shows the distance profile of the six Cβ-SH atoms in human Gal-1. In order to pair cysteines, considering the MS data discussed above [
19,
21,
29], distances of 4.1 Å (for C16-C88), 10.3 Å (for C42-C60), and 20.2 Å (for C2-C130) between the paired moieties were measured in the Gal-1 crystal structure. A structural model must explain how cysteines more than 20 Å apart can connect. Internal motions and conformational fluctuations can somewhat reduce these distances. Surface accessibility decreases in the order of C2-C130 to C16-C88 and then to C42-C60, as reflected in reactivity to iodoacetamide, with C2 being most reactive, followed by C130, then C88/C16, and barely, if at all, C42/C88 cysteines [
14,
38]. When exposed to β-mercaptoethanol during crystallization, the SH groups of C88 and C130, as well as C16 (if not oxidized to sulfenate), are always found to be covalently modified [
39]. Within the dimer of Gal-1, the Cβ-SH atoms of the two C130 residues are only 4.1 Å apart (
Figure 1), presenting the possibility for covalently linked dimers [
15,
27]. Because the
1H,
13C,
15N backbone and side-chain assignments were available for reduced Gal-1 [
40], we performed heteronuclear single quantum coherence (HSQC) NMR spectroscopic experiments to monitor Gal-1 structural changes during the course of CuSO
4-mediated oxidation.
Based on experimental evidence, we report here that Gal-1 undergoes a relatively slow conversion of C16 and C88 thiols, as well as of C42 and C60, to disulfide bridges, followed lastly by C2/C130 pairing. The initial structural transitions occur via a significant oxidation-mediated increase in the lectin’s internal motional dynamics, thus promoting the proximity of more distant cysteines. Our model for this non-random order of oxidation steps is supported by spatial proximity between cysteines and by site-directed mutagenesis. Our results confirm concept-based predictions. Hemagglutination assays indicate that complete loss of Gal-1 ligand binding activity occurs in the fully oxidized state, whereas the partially oxidized intermediate functions in essentially the same as the reduced lectin. Gal-1 ligand binding activity can be regained via the addition of DTT to oxidized Gal-1, underscoring the potential for an underappreciated on/off mechanism of the extracellular functionality of Gal-1 that depends on the nature of the environment, e.g., changes in oxidative capacity during inflammation.
2. Results and Discussion
Gal-1 oxidation has been monitored via HSQC spectroscopy and MS. Disulfide bond formation in Gal-1 occurs extremely slowly (order of days) in the absence of reducing agents via air oxidation and much more quickly when catalyzed using CuSO
4 as an oxidizing agent (order of hours) [
17,
24].
Figure 2A shows time-dependent changes in the methyl/methylene region of
1H NMR spectra of Gal-1 (20 μM) in the presence of 0.5 μM CuSO
4 (30 °C). Several well-resolved methyl resonances were unambiguously assigned to I58, V76, V89, and I117, and their upfield positions indicate the proximity of their associated methyl groups to aromatic residues (ring current shift effects). Since these residues are located within the β-sandwich, chemical shifts in their resonances reflect the well-folded structure typical for this compact β-sandwich protein. As the incubation time under this oxidative condition proceeds, significant alterations arise in the NMR spectrum. These native-state, upfield-shifted methyl resonances, among others, are decreased in intensity, broadened, and/or shifted downfield, while new resonances appear and increase in intensity. The reduction in signal intensity is not the result of protein precipitation, because UV absorbance (220 nm) of the solution remains essentially constant at all stages, indicating that the protein stays in the solution with only its conformation being modified.
Figure 2B plots the change in net integral of the methyl/ methylene 1H region resonating between 2.52 and 0.07 ppm vs. incubation time. During the first 15 h of incubation, the change is relatively small; after that, it occurs more rapidly through a single exponential decay. A similar plot but now integrating the narrower region 0.41 to 0.08 ppm, that only contains native Gal-1 methyl peaks (
Figure 2C), show the distinct loss of folded structure reaching the 50% mark after ~12 h. Note that the resonances in these NMR spectra broaden over time, an effect that could result from the formation of molten globule states and/or reflect the presence of multiple oxidized states.
In order to figure out whether these time-dependent, NMR-based changes correlate directly with disulfide bond formation, corresponding evidence was obtained via MS for the occurrence of 2 Da losses due to (SH)
2 to S-S conversions. These results are shown in
Figure 2D. The MS trace of the native, fully reduced Gal-1 (0 h) shows a major
m/
z peak at 14,584 Da, as expected for the carbohydrate recognition domain (CRD) of Gal-1. As the period for oxidation increases, this
m/
z peak is decreased in value by three steps of 2 Da each; after 48 h, in the presence of 0.5 μM CuSO
4 (or by 6 h with 4 μM CuSO
4), the net reduction in the mass of Gal-1 is 6 amu (
m/
z = 14,578). This result is consistent with the generation of up to three S-S bonds from the six sulfhydryls in human Gal-1. In the absence of CuSO
4, air oxidation can take up to about 1 week to cause the same effect, as seen in respective NMR spectra and MS runs.
The observation of an initially slow phase in Gal-1 oxidation followed by a faster phase change indicates inequality in the rate and position of formation of individual disulfide bonds. This suggests that disulfide bonds are formed sequentially and not randomly. Since internuclear distances between C
β-SH atoms are conspicuously different in the range of 4.1 Å to 20.2 Å (
Figure 1), there is a grading for oxidative reactivity consistent with MS data and chemical reactivity to iodoacetamide. In this regard, the C16-C88 pair is the best choice to initiate the process and, thus, to initiate the slow step in the cascade. Nevertheless, it is also plausible to form an inter-molecular disulfide bridge, as detected via gel electrophoresis of Gal-1 in the absence of β-mercaptoethanol [
15,
27]. Because this is especially true between C130 residues of subunits within the Gal-1 dimer (C130-C130’s distance is about 4.1 Å), we gave special attention to this point when analyzing our MS data. Thus, we incubated Gal-1 at a higher concentration of 30 μM under oxidative conditions and found no significant build-up of such a dimer. Even though we did observe a very small
m/
z peak at 29,168 Da, consistent with a covalently linked Gal-1 dimer, we must note that this is usually observed with native Gal-1 and remains constant in intensity during the entire time course of oxidation. However, for the production of oxidized Gal-1 in published axonal regeneration assays [
19], different oxidation conditions were used (i.e., CuSO
4 at 0.7 μM, Tris-HCl pH 8, 4 °C). If we oxidize Gal-1 (25 μM) under these conditions, the normally small Gal-1 dimer peak at
m/
z 29,166 is increased in intensity by 10–15%, indicating that covalent S-S Gal-1 dimers are likely formed.
Following further inspection of our HSQC data, we extended our insight into oxidation-induced structural changes.
Figure 3 shows four
1H–
15N HSQC spectra for the control (
A) and three oxidation time points induced by 4 μM CuSO
4 (i.e., 3 h (
B), 12 h (
C), and 24 h (
D)). Compared to the HSQC spectrum of native Gal-1 (
Figure 3A), oxidation brings about significant spectral changes, in particular, the appearance of new resonances in the initial slow phase of oxidation, during which intramolecular disulfide bond formation starts to occur, as shown by our MS results (
Figure 2B). The appearance of multiple resonances is exemplified with F79 at 3 h incubation where two new resonances are observed (boxed in
Figure 3B), the most upfield of which is relatively minor. As oxidation proceeds, the major new F79 resonance (F79′) grows in intensity as the native F79 resonance decreases in intensity. At the 12 h time point (
Figure 3C), most HSQC resonances are broadened and only the new F79′ state is apparent, whereas the native state F79 resonance (black circle in
Figure 3C) is not observed at this HSQC contour level. By 24 h (
Figure 3D), when MS data indicate complete (or nearly complete) oxidation (i.e., all six cysteines become part of three disulfide bonds), most backbone resonances are too broad to be observed and those that arise from Asn and Gln side chains are still apparent. Extensive broadening of backbone NH resonances is likely explained by dynamic interconversion among the various Gal-1 conformers, a scenario indicative of a more open and/or unfolded protein structure than the native compact β-sandwich fold. What seems counterintuitive is that the presence of an intracellular disulfide bridge promotes backbone flexibility to Gal-1. Nevertheless, we have observed a similar phenomenon upon ligand binding to Gal-1 in which internal motions are enhanced upon lactose binding, thus increasing conformational entropy to lower the free energy of ligand binding [
41]. As the oxidation process proceeds, intramolecular inter-cysteine distances above 10 Å or more were no longer an impediment to disulfide bond formation.
For a clearer view of F79 and F79′ resonance intensity changes,
Figure 3E shows the time course of their spectral changes with slices through the
1H dimension of HSQC spectra. At the 3 h time point, the relative intensities of F79′ compared to native-state F79 are ~10% and ~90%, respectively. As oxidation proceeds, the intensity of F79′ increases, whereas that for the native state decreases. At about 12 h, the intensities of the F79 and F79′ peaks are nearly equal and a second new resonance, albeit minor, becomes apparent (see
Figure 3B). The kinetics are consistent with what we observed in
Figure 2A with a 50% change in net
1H resonance intensity occurring as well at ~12 h. At this 50% point, MS data indicate an average formation of two disulfide bonds (
Figure 2B). These new resonances are most likely associated with Gal-1 in a partially oxidized state, where one or two intramolecular disulfide bonds reside in the CRD. If that were the case, then monitoring cysteine NH cross peaks should report valuable confirmatory information.
Supplemental Figure S1 shows expansions of
1H–
15N HSQC spectra, where cysteine NH resonances for C16, C42, C60, C88, and C130 are observed. Indeed, these resolvable cysteine NH resonances are affected during the oxidation process, and this occurs in a rather similar manner even though the order of Cys pairing cannot be deduced unambiguously. NH cross peaks for C42, and possibly for C60, appear to be altered first, but this could simply be the consequence of resonance broadening due to changes in protein dynamics at those sites. Looking at chemical shifts, the C16/C88 pair appears in a prominent position, and resonances from C16 and C88 are the most shifted during oxidation. This suggests that a disulfide bond between these most spatially close residues is formed first during the initial oxidation phase. In that case, a C16S mutant should be rather insusceptible to oxidative stress (see below).
Whereas these data are not yet entirely predictive as to which disulfide bond is formed first,
1H–
13C HSQC data indicate that C2 is the last cysteine to be oxidized. This important conclusion is based on the analysis of cysteine
13C
βH
2 resonances shown in
Figure 4A–G.
13C
βH
2 resonances are observed for all cysteines, and most of these resonances are relatively broad, even in native Gal-1, most likely due to internal motions and/or conformational exchange. In this respect, the C
βH
2 resonances of C2 are exceptional, owing to their position at the relatively mobile N-terminus of Gal-1. The point to be made here is that no new C2 C
βH
2 resonances appear during the formation of the partially oxidized state(s). The only change measured is the decrease in their intensity during the oxidation process. Moreover, a plot of the fractional change in this intensity (plotted as ln [1-fraction intensity]) vs. time is linear over the first 28 h of oxidation (
Figure 4H). This plot deviates from linearity at time points greater than ~30 h, something that could be dependent on other processes (see below). The apparent linearity over most of the oxidation process further supports our reasoning that the transition involving C2 is primarily a second-state process. In this case, the intermediate has a reduced C2 thiol group that finally becomes oxidized to generate the fully oxidized Gal-1. Overall, this data set strongly suggests that the C2-C130 bond is the final one of the three disulfide bridges to be formed. In that case, the C2S mutant of Gal-1 should maintain one free sulfhydryl at C130 (see below). Also,
1H–
13C HSQC data provide further relevant information.
Supplemental Figure S2 shows expansions of the methyl region from these same
1H–
13C HSQC spectra, acquired over the duration of oxidation. Resonances for I89, I117, M120, and I128 are boxed and labeled, and complete
1H–
13C resonance assignments are provided in
Supplemental Figure S3 for reference. As observed with our
1H–
15N HSQC data, new resonances appear and increase in intensity during oxidation, whereas those for native Gal-1 decrease in intensity. Again, considering our MS data (
Figure 2D), these new resonances must be attributed to various oxidized states of Gal-1, as intramolecular disulfide bonds are being formed.
Figure 5A shows the
1H–
13C HSQC methyl region for the 18 h time point, expanded with more resonances labeled. At this time point, four cross peaks for M120 are observed. The most upfield one at 1.87 ppm
1H is associated with the native state (labeled “N”), and the others (M120′) originate from different species of oxidized Gal-1 (labeled P1 and P2). The fourth M120′ peak is minor and not labeled. Other new resonances are associated with Ala, Thr, Ile, Leu, and Val residues (e.g., CH
3 resonances from I58, I89, I117, L9, L17, L32, L96, V76, V131, V131, A94, A116, A132, and T90), representing sites throughout the protein.
Figure 5C plots the change in resonance intensities for these M120 signals vs. oxidation time. Although we can discern four peaks (or states) for M120 at 18 h oxidation, this number varies with the time or stage of oxidation. The most upfield resonance arises from native (N) Gal-1. Time-dependent intensity changes for N, P1, and P2 resonances are plotted in
Figure 5B. However, because line widths for P1 and P2 are about 2-fold greater than those for N, the populations of P1 and P2 should be increased by about 2-fold relative to that of N. Thus, these data reveal that the population for the native state N decreases over time, as expected, whereas the populations for P1 and P2 first increase and then decrease in intensity during the oxidation process. P1 rises in intensity in the first oxidation phase, during which P2 displays an initial lag phase. These trends for P1 and P2 indicate a sequential (or serial) order of formation of Gal-1 oxidation intermediates, with the overall scheme being from N to P1, P2, and finally into O, the fully oxidized state. Although other intermediates aside from P1 and P2 are possible, they would be either very short-lived, highly broadened, and/or have very small populations below the level of HSQC detection limit.
For the N state, the decay curve is non-linear with respect to longer time points (
Figure 2C) and requires the use of a double exponential function to be fitted optimally. This trend parallels that observed in
1H–
13C HSQC spectra with cysteine C2 C
βH
2 resonances (
Figure 4) and indicates that at least two events occur during oxidation of the N state, one event that is initially slow and the other(s) that subsequently proceed faster. This observation supports the sequential model introduced above. Although the initial part of the curve up to ~25 h can be fitted with a linear function, the remainder of the curve deviates substantially from linearity. Simple linear fits to initial and final parts of the curve show that the rates are only about a factor or two apart, i.e., ~0.7 × 10
−3 min
−1 and ~1.3 × 10
−3 min
−1, respectively. The initial slow rate of signal reduction for the N state is estimated to be the same as the initial rate of formation of P1, not indicating a lag in P2 formation. The conversion rate from P1 to P2 appears to be faster than that from N to P1. The final, fast oxidation phase starts at about 25 h after P1 has formed sufficiently and P2 production is underway. At this point, about two-thirds of N has already been converted, equivalent to forming of up to two disulfide bonds per Gal-1 monomer.
At the ~7 h time point, when the intensity of P1 is maximal and very little P2 has formed, our MS data (
Figure 2D, which also shows slow and fast oxidation phases) indicate that the net Gal-1 mass has been reduced by only ~1–2 amu. Although there is, in principle, more than one way to explain the average, the easiest one is to consider 50% of Gal-1 molecules in the fully reduced state and 50% of them having only one disulfide bond formed. As mentioned above, our data indicate that Gal-1 is modified during the oxidation process (initial and later stages). At the initial stages of oxidation (slow kinetics phase), in which the native structure of Gal-1 is mostly preserved, albeit with increased dynamics, one possibility is that the dimer symmetry is broken by the formation of a single disulfide bond in one of the subunits. This is consistent with the appearance of P1, which reaches an equal (or near-equal) population with the N state and could indicate the formation of a Gal-1 heterodimer in which one subunit still is fully reduced and the other has one disulfide bond. An alternative explanation is that P1 represents an oxidized Gal-1 monomer species, i.e., the dimer is dissociated upon generating a single intramolecular disulfide bond. In that case, the resulting monomer would no longer function in the hemagglutinin assay (see below).
Regarding dynamics, our results serve to address the pertinent question as to the distribution of this parameter. In order to present the observed NMR spectroscopic data graphically, an overview of backbone NH groups that are most perturbed in
1H–
15N HSQC spectra during the oxidation process is presented in
Figure 6. Residues associated with resonances that are most perturbed (i.e., chemically shifted, formation of double/multiple cross peaks, and/or strongly reduced peak intensity) during the initial hours of oxidation are highlighted in red, and those that are only slightly perturbed are in blue. Grey dots indicate no change in peak position or relative intensity.
Figure 6A has the F-face (the face of the β-sandwich opposite to the lactose-binding S-face) of the dimer in front.
Figure 6B rotates the structure by 90°, and
Figure 6C rotates the structure in
Figure 6A by 180° to show a frontal view of the S-face. During the initial stage of the oxidation process, perturbations at backbone NH positions are not localized. Instead, they occur throughout the folded structure of Gal-1, as suggested by the
13C HSQC data on methyl groups (
Figure 5). This suggests that the extent of internal motions within oxidized Gal-1 is generally increased, possibly promoting further oxidation by allowing more distant cysteines to come closer together.
An analysis of methyl resonances in
1H–
13C HSQC spectra was added to further refine this picture. Most methyl groups of Ala, Thr, Ile, Leu, and Val side chains are located within the β-sheet sandwich. These include the M120 case discussed above, as well as, e.g., I58, I89, I117, L9, L17, L32, L96, V76, V131, V131, A94, A116, A132, and T90.
Figure 6 highlights methyl groups (green spheres) whose resonances are most perturbed during oxidation. Even though some residues like I58, V76, and L96 near the lactose-binding site and relatively distant from any cysteine are perturbed at the initial stages of oxidation, the most perturbed methyls are close to C16 and C88, consistent with our Cys oxidation model. Many of these aliphatic side chains interact with each other through the β-sandwich interface. For example, side chains of V87 and I89, which flank C88 on β-strand 9, come into contact with those of L32 and L34 on the opposing β-strand 3. Also, the side chain of L17, flanking C16 on β-strand 2, interacts with that of M120 on the opposing β-strand 10. Moreover, intra-β-sandwich residues from all four strands contact each other. Since M120 is central to these interactions, its environment would be very sensitive (conformationally and/or dynamically) to the formation of the C16-C88 disulfide bond. Having detected only a few perturbations around C42 and C60 during the initial oxidation stage, it is most likely that the C16-C88 disulfide bond is formed first during Gal-1 oxidation, followed by C42-C60.
So far, we have shown that the formation of disulfide bonds in Gal-1 occurs in a sequential fashion, with the formation of the first one (probably C16-C88) having minimal, if any, effect on Gal-1 folding. Based on structural grounds, in order for the free sulfhydryls of the remaining cysteines C42-C60 and C2-C130 to come into proximity to facilitate Cys oxidation, there should, however, be a significant increase in conformational flexibility following the formation of the C16-C88 disulfide bond. Accordingly, resonances of intermediate oxidation states (e.g., see M120 P1 and P2 peaks in
Figure 5) formed during the initial stages of Gal-1 oxidation exhibit broader line widths than in the native state, supporting the proposed increase in conformational exchange. Regrettably, our NMR data cannot inform on conformational changes that occur at later stages of oxidation, because the resonances are so highly broadened and, thus, unobservable. What is possible is to look at ligand binding during the early stages of oxidation.
Carbohydrate binding to partially oxidized Gal-1. Because our HSQC data indicate that the native structure of Gal-1 is essentially preserved upon partial oxidation of Gal-1, we raised the question as to whether this state can still bind carbohydrates. To assess this, we performed a lactose titration with a sample of partially oxidized Gal-1, where resonances from both N and P1 states could be followed simultaneously. Overlays of
1H–
15N HSQC spectral expansions are shown as a function of lactose concentration for F79, R73, V76, and A116 for the 12 h oxidation time point where N and P1 states are readily observed (
Figure 7A). The contour in black is in the absence of lactose, and the contour in red is the endpoint at 45 mM lactose, with other colors for intermediate lactose concentrations, as stated in the legend of
Figure 7. Because only F79 and V76 show clear changes during the lactose titration,
Figure 7B plots
1H–
15N-weighted chemical shift changes for these resonances vs. the [lactose]/[protein] concentration ratio. Based on these plots, it is apparent that the affinity for lactose is essentially the same for native (N) and the P1 partially oxidized state of Gal-1. We also found that the HSQC spectra of Gal-1 acquired in the presence or absence of 10 mM lactose look the same during the CuSO
4-dependent oxidation process. The only difference is that the oxidation rate is reduced by about half in the presence of this saturating amount of lactose. Thus, the lactose-loaded state of Gal-1 attenuates the oxidation process.
Independently, the effect of oxidation on carbohydrate binding by Gal-1 was monitored using hemagglutination studies, which are traditionally used to provide insight into galectin-mediated cell–cell adhesion. Indeed, (ga)lectin-dependent bridging of glycoconjugates in cis/trans states underlies most aspects of lectin functionality [
42,
43]. Of note, this aspect of galectin activity will also report on the status of the Gal-1 dimer state, because our MS data (
Figure 2D) exclude the generation of a covalently linked dimer under our oxidation conditions.
Figure 8 plots IC
50 values for agglutination vs. oxidation time, and the insert exemplifies the percent agglutination as a function of Gal-1 concentration, with data collected at three time points during the oxidation time course. Agglutination was quantified by plotting the absorbance at 650 nm (A
650) as a function of Gal-1 concentration at each time point. Note that as the time of oxidation increases, the mid-point of each curve (IC
50) shifts to higher Gal-1 concentrations. During the initial phase of oxidation, when mostly native and partially oxidized states of Gal-1 are present, IC
50 values increase minimally. In contrast, IC
50 values increase dramatically at later times as more Gal-1 is converted to the fully oxidized state. The time course for changes in agglutination IC
50 values correlates well (regression coefficient R = 0.9) with that of the appearance of broad-resonance components in our NMR spectra (insert to
Figure 2). The reduction in IC
50 values during oxidation can be explained by increasing the amount of fully oxidized, and thus inactive, Gal-1, with partially oxidized Gal-1 remaining active. This observation is fully consistent with our NMR data in that partially oxidized Gal-1 maintains a native or native-like structure that can still bind lactose, whereas fully oxidized Gal-1 has lost its native structure and cannot bind lactose. In order to examine what happens to the quaternary structure, we added further NMR-spectroscopy-based data.
Gal-1 dimers dissociate upon complete oxidation. Our agglutination data presented in
Figure 8 suggest that partially oxidized Gal-1 maintains bridging capacity. Otherwise, aggregation of red blood cells would not occur. At the quaternary structure level, the Gal-1 dimer state should, thus, be maintained. We confirmed this by performing pulsed field gradient (PFG) NMR diffusion experiments during oxidation, where mostly native and partially oxidized Gal-1 are present. The resulting diffusion coefficient (
D) for native Gal-1 dimer (N) and for partially oxidized Gal-1 (P1) is essentially the same as ~1.05 × 10
−6 cm
2/s. For fully oxidized Gal-1, on the other hand, the
D value is increased to 1.13 × 10
−6 cm
2/s. Although this
D value is far from that of a Gal-1 monomer (
D = 1.35 × 10
−6 cm
2/s), it does move in that direction. The adoption of an elongated shape, as suggested for Gal-1 based on dynamic light scattering [
30], for CG-1B based on gel filtration and ultracentrifugation [
32], and for dimeric CG-2 [
44,
45], may underlie this phenomenon. In any event, fully oxidized Gal-1 is not a well-folded CRD but rather a variant with its own hydrodynamic properties that can interact with new function-promoting partners but not with β-galactosides. Considering the physiological significance of fully oxidized Gal-1 (e.g., in late-stage inflammation where it can inhibit the proliferation of activated T lymphocytes and trigger interferon-γ-mediated apoptosis [
46] thereby limiting the duration of the effector activity of this aspect of immune defense), the question arises as to whether the Gal-1 oxidation process is reversible or not.
Native Gal-1 structure recovered from oxidized Gal-1. The structure and activity of native Gal-1 can indeed be recovered from fully (or partially) oxidized Gal-1 via removal of the oxidizing agent CuSO
4, and subsequent incubation with a reducing agent (i.e., DTT).
Supplemental Figure S2 illustrates HSQC spectra acquired sequentially overnight, starting with native Gal-1 (S2
q) in buffered solution with 0.5 mM CuSO
4 and ending with fully oxidized Gal-1 ~41 h later (S2q
Q). When the Cu
2SO
4 is neutralized with EDTA and 8 mM DTT is added to the solution, the sample of fully oxidized Gal-1 reverts to its native state. Basically, the HSQC spectrum of reduced oxidized-Gal-1 (S2
Rr) looks no different from that of native Gal-1 (
A). In addition, lactose binding and glycan-dependent cross-linking activity are also fully regained.
The recovery kinetics can be followed by analyzing
1H NMR spectra acquired over time starting with adding 8 mM DTT to the sample of oxidized Gal-1 (
Figure 9A). Within the first 10–15 min upon the addition of DTT, most of the spectrum of oxidized Gal-1 has been converted to that of native or native-like Gal-1, with the remainder of the conversion taking considerably longer. An initial fast recovery phase is followed by a slower one, as the oxidation route is reversed. We quantified these kinetic phases by integrating resonance intensities during the time course of recovery. Two spectral regions were considered: case 1 with integration performed from 2.52 ppm to 0.07 ppm, where methyl groups and some methylene groups from all states (native, partially oxidized, and fully oxidized) resonate (
Figure 9B); and in case with integration performed from 0.41 ppm to 0.08 ppm, where methyl resonances of I58, V76, V98, and I117 alone are found (
Figure 9C). The methyl groups of I58, V76, V98, and I117 are located within the β-sandwich of native Gal-1 and, therefore, are suitable sensors for the well-folded structure of native Gal-1. For both cases, inserts are provided to show greater detail as to what happens over the first 3 h of recovery, during which >95% of native (or native-like) structure is recovered. These curves were fitted with single and double exponential functions. The formula of the double exponential function is shown in Equation (1):
where p and k stand for population and rate constant, respectively. Fits to these data with the double exponential function are shown in
Figure 9B,C. While a single exponential fit to the data for case 2 was acceptable, with a regression coefficient of R = 0.98, the fit was less than optimal for case 1, with R = 0.87. Nevertheless, the double exponential fit gave satisfying data in both cases, with R = 0.998 (
Table 1).
This analysis indicates that 83% of the native (or native-like) structure is rapidly recovered in both cases. The partitioning of 83% (fast recovery) and 17% (slow recovery) may be explained by once again considering the total of six disulfide bonds in the native Gal-1 dimers and reversing the order in the sequential model discussed above. In this regard, the rapid reduction of five disulfide bonds per dimer would account for the population of 83%. Our data show that, within the rapid recovery phase, certain disulfide bonds are very quickly reduced, promoting the formation of native-like dimers. One disulfide bond (presumably one of the two C16-C88 bonds in the dimer) remains longer intact and is more slowly reduced. The 12-fold difference in the slow recovery rate for native-state methyl groups I58, V76, V89, and I117 compared to other methyl and some methylene groups could be attributed to the presence of both rapid and slow conformational re-shuffling, respectively, as the native-state structure returns. It is possible that I58, V76, V89, and I117 are involved in building the native folding core more rapidly than other aliphatic residues. Recovery of the native structure from oxidized Gal-1 is supported by MS data in which we observe a corresponding mass increase following DTT treatment of oxidized Gal-1. With rat Gal-1, the shift “in the trough in the circular dichroism spectrum going from 216 nm for native lectin to 207 nm for oxidized lectin” was indicative of substantial secondary structure alteration; “the shift was reversed by reduction with DTT (unpublished results)” [
14]. However, reversibility can depend on the “circumstances” [
26] and, thus, may only be partial [
47] or involve protein aggregation that limits the extent of recovery [
27], or even fail to occur for certain species [
30].
Testing our sequential oxidation model using mutagenesis. Based on our data, we propose that oxidation is a stepwise, non-random process. The C16-C88 disulfide bridge likely forms first, causing conformational alterations that bring C42 and C60 into proximity, allowing the formation of the second disulfide bond. This partially oxidized state promotes further conformational shifting and dynamic changes to bring C2 into proximity with C130 to form the third S-S bond. At this stage, oxidized Gal-1 becomes “unfolded”. This process is reversible upon reduction of the oxidized state. To test this hypothesis, we produced two Gal-1 Cys-to-Ser mutants, i.e., C2S and C16S. The C2S mutant would be unable to form the final disulfide bridge, yet it should pass through the first phase of oxidation before becoming arrested prior to the final oxidation step. In contrast, the C16S mutant should be insensitive to oxidation if the C16 sulfhydryl indeed ignites the oxidation pathway. As with native Gal-1, 1H–15N HSQC data with each of these 15N-enriched mutants were determined as a function of oxidation time.
With the C2S mutant, HSQC spectra changed, as seen before in the initial phase of Gal-1 oxidation. However, as predicted, the broadened component characteristic of phase 2 was not observed, and the NMR signal was not degraded over time. The final “unfolded” state need for native Gal-1 to be attained could not be reached using the C2S mutant. As a sensor for detecting the remaining free sulfhydryls, application of Ellman’s reagent indicated that, as expected, one free SH group (i.e., C130) was present at the end of the oxidation process. The marked stability of the C2S mutant, in terms of binding activity seen by Hirabayashi and Kasai [
3], is fully in line with the possibility of phase 1 transitions that do not impair lectin activity. Due to the stability of the C2S mutant, we could readily follow intensity changes for F79 N and P1 states, without concern for further oxidation of the P1 state.
Supplemental Figure S4A shows the oxidation time course for N and P1 states from 2 to 40 h. During the oxidation process, the N state decreases in intensity, while the P1 state increases in intensity until it is the only observable state remaining.
Because the Gal-1 C2S and C16S mutants appeared to be relatively stable in solution, we tried to elucidate their P1 3D structures by using NMR. We first made a C2S sample and started running an
15N-edited NOESY experiment. However, the following morning, we observed that the signal had degraded significantly, and upon looking at the NMR tube itself, it was apparent that the sample had mostly precipitated overnight, making structural elucidation impossible. However, the broadened, aggregated spectrum can be rescued partially by increasing the temperature from 30 to 40 °C (
Figure S6). Because of this, we ran a CD spectrum of C2S and observed that its CD trace was essentially the same as that of native, reduced Gal-1 (Nesmelova et al., 2010). Therefore, it appears that C2S maintains the secondary structural characteristics of Gal-1, with its structure being more dynamic and possibly like that of a molten globule state. This is consistent with our hemagglutination data (
Figure 9), which show that at least some activity remains up to the formation of this partially oxidized state. The same can be said for the C16S mutant.
The C16S mutant displays even more delayed kinetics of its P1 state (
Figure S4B) even at 4 μM CuSO
4, having a short ca. 10 h time window where the metastable oxidized state becomes conformationally pure to be structurally studied by NMR (
Figure S5).
This evidence argues strongly in favor of C16-C88 bond oxidation, initiating structural/dynamical conversions. Both of these cysteines had been previously identified as critical via mutational analysis [
27]. Because no oxidation-dependent conformational alteration then appears to occur, the C16S mutant could substitute for a chemically stabilized wild-type Gal-1 in functional assays, unlike the C2S mutant, which can still be converted to an intermediate. However, the C-to-S substitution at position 2 changes the orientation of the side chain of D123 by 180° and also the thermodynamics of lactose binding [
39]. The role of C16 as a molecular switch is underscored by its high level of conservation in mammalian galectins, and cases like CG-1A (without C2 and C130, but still susceptible to oxidative damage to lectin activity) give incentive to further studying the role of individual constellations of cysteines in galectin activity in vertebrates.



 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}