
SAHA/5-AZA Enhances Acetylation and Degradation of mutp53, Upregulates p21 and Downregulates c-Myc and BRCA-1 in Pancreatic Cancer Cells
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
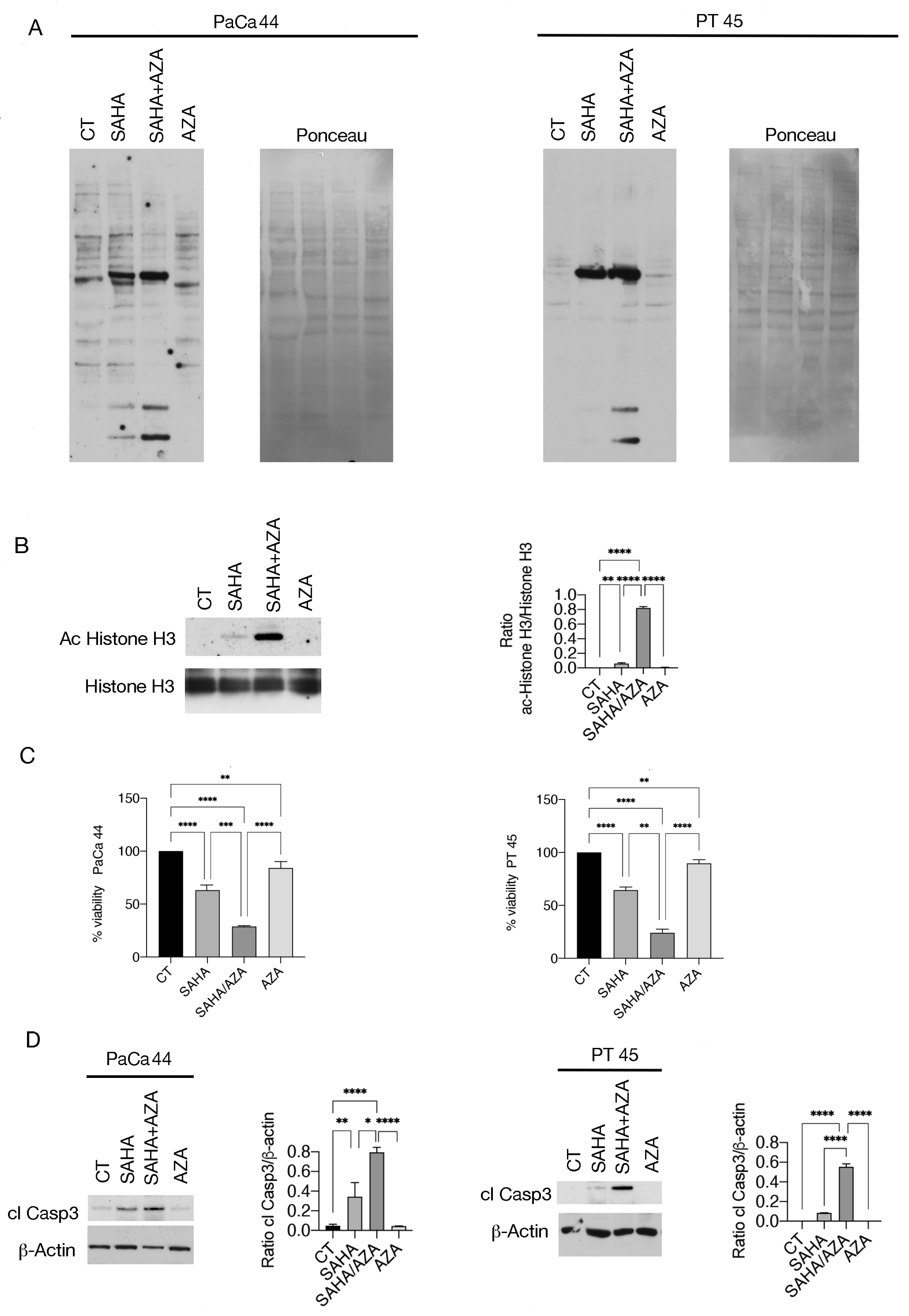
2.1. 5-AZA Enhances Pan-Lysine Acetylation and Cytotoxicity Induced by SAHA against Pancreatic Cancer Cells
2.2. SAHA/5-AZA Induces a Stronger Increase in MT2A Expression than SAHA
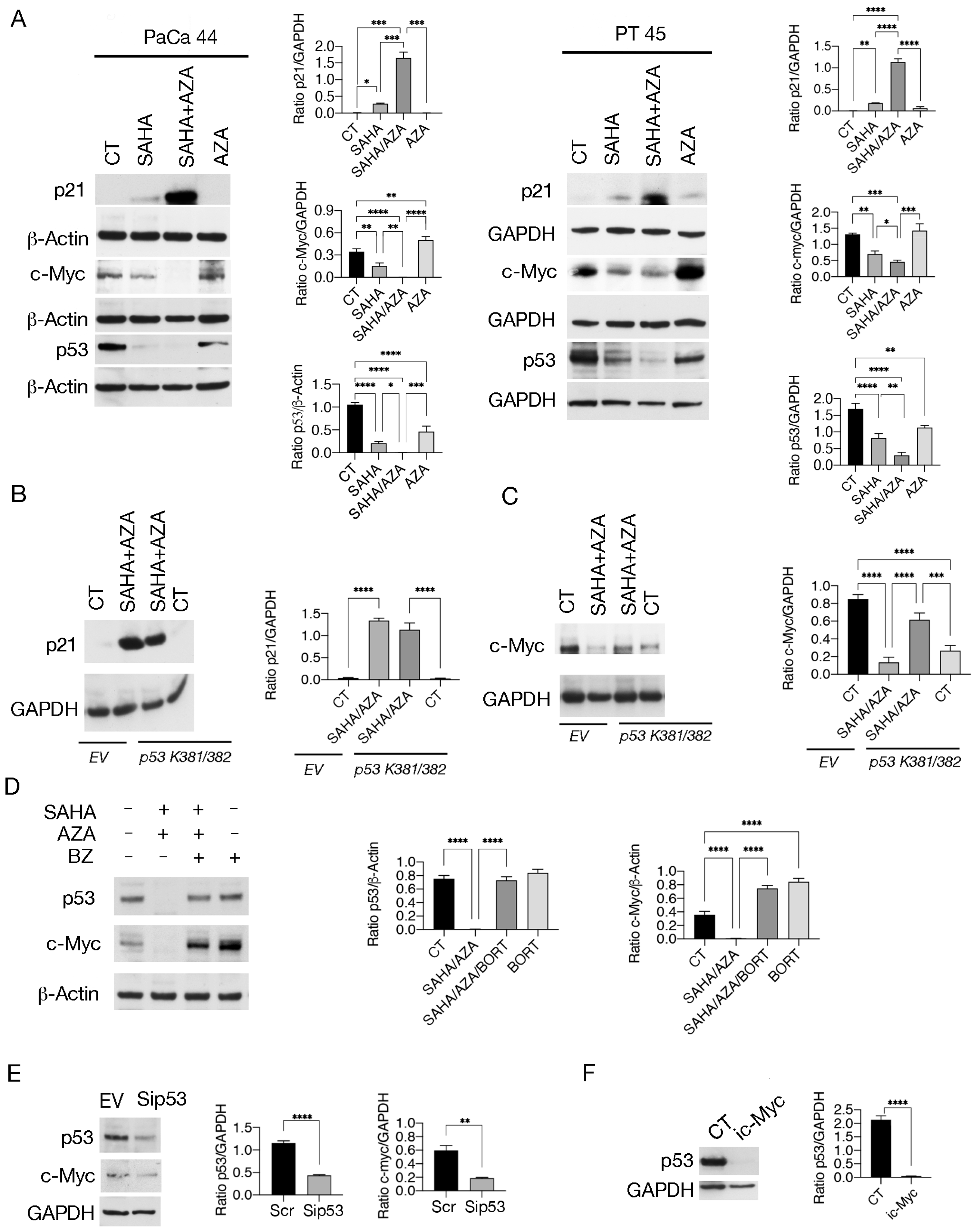
2.3. SAHA/5-AZA Increases mutp53 Acetylation Further Promoting Its Proteasomal Degradation
2.4. SAHA/5-AZA Interrupts the Positive Crosstalk between c-Myc and p53
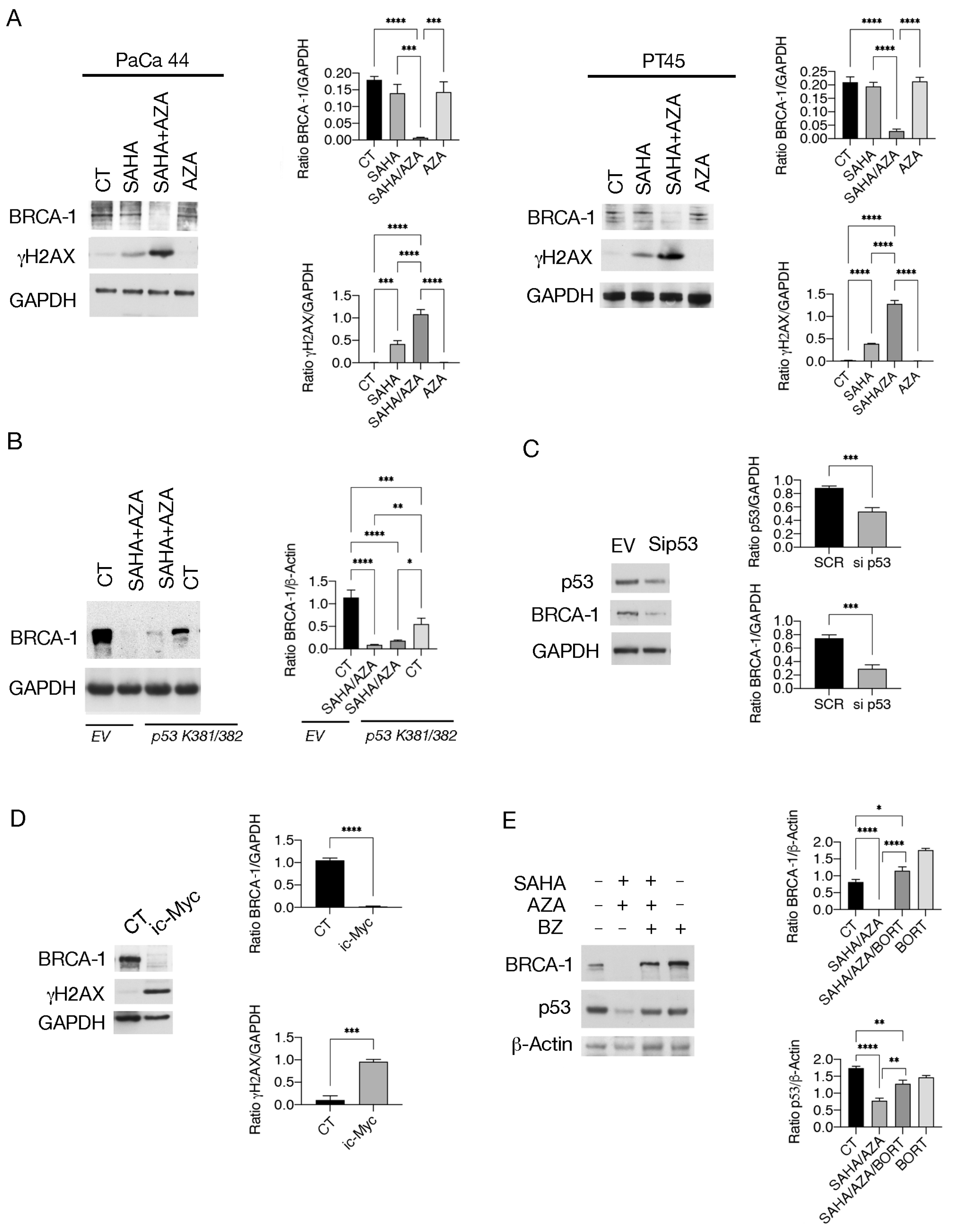
2.5. SAHA/5-AZA Downregulates BRCA-1 and Induces DNA Damage in Pancreatic Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Treatments
4.2. Cell Assay Viability
4.3. Western Blot Analysis
4.4. Antibodies
| Antibody | Isotype | Species | Diluition Ratio | Company | Catalog |
| anti-P53 (DO-1) | Monoclonal | Mouse | (1:500) | Santa Cruz Biotechnology | sc-126 |
| anti-Ac-lysine (AKL5C1) | Monoclonal | Mouse | (1:500) | Santa Cruz Biotechnology | sc-32268 |
| anti-γH2AX (Ser 139) | Monoclonal | Mouse | (1:100) | Santa Cruz Biotechnology | sc-517348 |
| anti-Histone H3 ac | Polyclonal | Rabbit | (1:500) | Invitrogen | PA5-114693 |
| anti-Histone H3 (D1H2) | Polyclonal | Rabbit | (1:4000) | Cell Signaling | 4499T |
| anti-c-myc | Polyclonal | Rabbit | (1:500) | Proteintech | 10828-AP |
| anti-p21(12D1) | Polyclonal | Rabbit | (1:500) | Cell Signaling | 2947T |
| anti-Acetyl-p53 (Lys373; Lys382) | Polyclonal | Rabbit | (1:500) | Merckmillipore | 06-758 |
| anti-BRCA-1 | Polyclonal | Rabbit | (1:2000) | Proteintech | 22362-1-AP |
| anti-βActin (AC-74) | Monoclonal | Mouse | (1:10,000) | Sigma-Aldrich | A5441 |
| anti-GAPDH (G-9) | Monoclonal | Mouse | (1:10,000) | Santa Cruz Biotechnology | A5316 |
| anti-caspase3 (31A1067) | Monoclonal | Mouse | (1:300) | Santa Cruz Biotechnology | sc-56053 |
4.5. RNA Isolation and Quantitative Real-Time PCR Analysis
- MT2A forw: 5′-CCGACTCTAGCCTCTT-3′
- MT2A rev: 5′-GTGGAAGTCGCGTTCTTTACA-3′
- MT1E forw:5′-GCTTGTTCGTCTCACTGGTG-3′
- MT1E rev: 5′-CAGGTTGTGCAGGTTCTA-3′
- ACT forw: 5′-TCACCCACACTGTGCCATCTACGA-3′
- ACT rev: 5′-CAGCGGAACCGCTCATTGCCAATGG-3′.
4.6. Knockdown of p53 and Transfection with p53 K381/382R Vector
4.7. Immunoprecipitation Assay
4.8. Densitometric Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, G.; Tian, Y.; Zhu, W.G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 576946. [Google Scholar] [CrossRef] [PubMed]
- Bieliauskas, A.V.; Weerasinghe, S.V.; Pflum, M.K. Structural requirements of HDAC inhibitors: SAHA analogs functionalized adjacent to the hydroxamic acid. Bioorg. Med. Chem. Lett. 2007, 17, 2216–2219. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.; Jiang, Q.; Zhang, L.; Song, W. Zinc binding groups for histone deacetylase inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Kai, L.; Samuel, S.K.; Levenson, A.S. Resveratrol enhances p53 acetylation and apoptosis in prostate cancer by inhibiting MTA1/NuRD complex. Int. J. Cancer 2010, 126, 1538–1548. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.W.; Shin, D.H.; Lee, D.H.; Choi, J.; Han, G.; Lee, K.Y.; Kwon, S.H. HDAC6 deacetylates p53 at lysines 381/382 and differentially coordinates p53-induced apoptosis. Cancer Lett. 2017, 391, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Scolnick, D.M.; Trievel, R.C.; Zhang, H.B.; Marmorstein, R.; Halazonetis, T.D.; Berger, S.L. p53 sites acetylated In Vitro by PCAF and p300 are acetylated In Vivo in response to DNA damage. Mol. Cell Biol. 1999, 19, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, K.; Herrera, J.E.; Saito, S.; Miki, T.; Bustin, M.; Vassilev, A.; Anderson, C.W.; Appella, E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes. Dev. 1998, 12, 2831–2841. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Mello, S.S.; Flowers, B.M.; Mazur, P.K.; Lee, J.J.; Muller, F.; Denny, S.K.; Ferreira, S.; Hanson, K.; Kim, S.K.; Greenleaf, W.J.; et al. Multifaceted role for p53 in pancreatic cancer suppression. Proc. Natl. Acad. Sci. USA 2023, 120, e2211937120. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Qian, W.; Yang, Z.; Zhang, Z.; Sun, P.; Wan, Q.; Yin, Y.; Hu, Y.; Gong, L.; Zhang, B.; et al. Acetylation halts missense mutant p53 aggregation and rescues tumor suppression in non-small cell lung cancers. iScience 2023, 26, 107003. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Xu, D.; Zhang, T.; Hu, W.; Feng, Z. Gain-of-function mutant p53 in cancer progression and therapy. J. Mol. Cell Biol. 2020, 12, 674–687. [Google Scholar] [CrossRef] [PubMed]
- D’Orazi, G.; Cordani, M.; Cirone, M. Oncogenic pathways activated by pro-inflammatory cytokines promote mutant p53 stability: Clue for novel anticancer therapies. Cell Mol. Life Sci. 2021, 78, 1853–1860. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Dowdy, S.C.; Podratz, K.C.; Jin, F.; Attewell, J.R.; Eberhardt, N.L.; Jiang, S.W. Histone deacetylase inhibitors decrease DNA methyltransferase-3B messenger RNA stability and down-regulate de novo DNA methyltransferase activity in human endometrial cells. Cancer Res. 2005, 65, 2684–2689. [Google Scholar] [CrossRef] [PubMed]
- Fuks, F.; Burgers, W.A.; Brehm, A.; Hughes-Davies, L.; Kouzarides, T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat. Genet. 2000, 24, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, K.; Datta, J.; Majumder, S.; Bai, S.; Dong, X.; Parthun, M.; Jacob, S.T. Inhibitors of histone deacetylase and DNA methyltransferase synergistically activate the methylated metallothionein I promoter by activating the transcription factor MTF-1 and forming an open chromatin structure. Mol. Cell Biol. 2002, 22, 8302–8319. [Google Scholar] [CrossRef] [PubMed]
- Stefanska, B.; Salame, P.; Bednarek, A.; Fabianowska-Majewska, K. Comparative effects of retinoic acid, vitamin D and resveratrol alone and in combination with adenosine analogues on methylation and expression of phosphatase and tensin homologue tumour suppressor gene in breast cancer cells. Br. J. Nutr. 2012, 107, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Kuo, C.W.; Tsai, S.L.; Cheng, S.M.; Chen, S.H.; Chan, H.H.; Lin, C.H.; Lin, K.Y.; Li, C.F.; Kanwar, J.R.; et al. Inhibition of HDAC3- and HDAC6-Promoted Survivin Expression Plays an Important Role in SAHA-Induced Autophagy and Viability Reduction in Breast Cancer Cells. Front. Pharmacol. 2016, 7, 81. [Google Scholar] [CrossRef] [PubMed]
- Ala, M. Target c-Myc to treat pancreatic cancer. Cancer Biol. Ther. 2022, 23, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Zeng, S.X.; Zhou, X.; Chen, T.; Zhou, F.; Cao, B.; Jung, J.H.; Del Sal, G.; Luo, S.; Lu, H. Mutant p53 Gains Its Function via c-Myc Activation upon CDK4 Phosphorylation at Serine 249 and Consequent PIN1 Binding. Mol. Cell 2017, 68, 1134–1146.e6. [Google Scholar] [CrossRef]
- Ganci, F.; Pulito, C.; Valsoni, S.; Sacconi, A.; Turco, C.; Vahabi, M.; Manciocco, V.; Mazza, E.M.C.; Meens, J.; Karamboulas, C.; et al. PI3K Inhibitors Curtail MYC-Dependent Mutant p53 Gain-of-Function in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2020, 26, 2956–2971. [Google Scholar] [CrossRef] [PubMed]
- Romeo, M.A.; Gilardini Montani, M.S.; Arena, A.; Benedetti, R.; D’Orazi, G.; Cirone, M. c-Myc Sustains Pancreatic Cancer Cell Survival and mutp53 Stability through the Mevalonate Pathway. Biomedicines 2022, 10, 2489. [Google Scholar] [CrossRef] [PubMed]
- Arena, A.; Gilardini Montani, M.S.; Romeo, M.A.; Benedetti, R.; Gaeta, A.; Cirone, M. DNA damage triggers an interplay between wtp53 and c-Myc affecting lymphoma cell proliferation and Kaposi sarcoma herpesvirus replication. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119168. [Google Scholar] [CrossRef]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.F.; Su, B.; Li, W. DNA demethylation is superior to histone acetylation for reactivating cancer-associated genes in ovarian cancer cells. Mol. Med. Rep. 2011, 4, 1273–1278. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Eden, S.; Hashimshony, T.; Keshet, I.; Cedar, H.; Thorne, A.W. DNA methylation models histone acetylation. Nature 1998, 394, 842. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Baltaci, A.K.; Yuce, K.; Mogulkoc, R. Zinc Metabolism and Metallothioneins. Biol. Trace Elem. Res. 2018, 183, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Madden, S.K.; de Araujo, A.D.; Gerhardt, M.; Fairlie, D.P.; Mason, J.M. Taking the Myc out of cancer: Toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 2021, 20, 3. [Google Scholar] [CrossRef] [PubMed]
- Garufi, A.; Ricci, A.; Trisciuoglio, D.; Iorio, E.; Carpinelli, G.; Pistritto, G.; Cirone, M.; D’Orazi, G. Glucose restriction induces cell death in parental but not in homeodomain-interacting protein kinase 2-depleted RKO colon cancer cells: Molecular mechanisms and implications for tumor therapy. Cell Death Dis. 2013, 4, e639. [Google Scholar] [CrossRef] [PubMed]
- Cecchinelli, B.; Lavra, L.; Rinaldo, C.; Iacovelli, S.; Gurtner, A.; Gasbarri, A.; Ulivieri, A.; Del Prete, F.; Trovato, M.; Piaggio, G.; et al. Repression of the antiapoptotic molecule galectin-3 by homeodomain-interacting protein kinase 2-activated p53 is required for p53-induced apoptosis. Mol. Cell Biol. 2006, 26, 4746–4757. [Google Scholar] [CrossRef] [PubMed]
- Puca, R.; Nardinocchi, L.; Sacchi, A.; Rechavi, G.; Givol, D.; D’Orazi, G. HIPK2 modulates p53 activity towards pro-apoptotic transcription. Mol. Cancer 2009, 8, 85. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Crosta, M.; Ragone, F.C.; Benedetti, R.; D’Orazi, G.; Gilardini Montani, M.S.; Cirone, M. SAHA/5-AZA Enhances Acetylation and Degradation of mutp53, Upregulates p21 and Downregulates c-Myc and BRCA-1 in Pancreatic Cancer Cells. Int. J. Mol. Sci. 2024, 25, 7020. https://doi.org/10.3390/ijms25137020
Di Crosta M, Ragone FC, Benedetti R, D’Orazi G, Gilardini Montani MS, Cirone M. SAHA/5-AZA Enhances Acetylation and Degradation of mutp53, Upregulates p21 and Downregulates c-Myc and BRCA-1 in Pancreatic Cancer Cells. International Journal of Molecular Sciences. 2024; 25(13):7020. https://doi.org/10.3390/ijms25137020
Chicago/Turabian StyleDi Crosta, Michele, Francesca Chiara Ragone, Rossella Benedetti, Gabriella D’Orazi, Maria Saveria Gilardini Montani, and Mara Cirone. 2024. "SAHA/5-AZA Enhances Acetylation and Degradation of mutp53, Upregulates p21 and Downregulates c-Myc and BRCA-1 in Pancreatic Cancer Cells" International Journal of Molecular Sciences 25, no. 13: 7020. https://doi.org/10.3390/ijms25137020