
Protein Tyrosine Phosphatase 1B (PTP1B): A Comprehensive Review of Its Role in Pathogenesis of Human Diseases



Abstract
:1. Introduction
2. Role of PTP1B in Pathogenesis of Human Diseases and Prospects of Therapy with Inhibitors of the Enzyme
2.1. Diabetes Mellitus and Obesity
2.2. Alzheimer’s Disease
2.3. Major Depressive Disorder
2.4. Fatty Liver Disease
2.5. Cancers
2.6. Other Diseases
3. Fundamental Origins of PTP1B Overexpression
4. Overview of Strategies Providing Adequate Enzyme Selectivity of PTP1B Inhibitors
5. Future Perspectives for Therapeutic Use of PTP1B Inhibitors
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tonks, N.K.; Diltz, C.D.; Fischer, E.H. Purification of the major protein-tyrosine-phosphatases of human placenta. J. Biol. Chem. 1988, 263, 6722–6730. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K.; Diltz, C.D.; Fischer, E.H. Characterization of the major protein-tyrosine-phosphatases of human placenta. J. Biol. Chem. 1988, 263, 6731–6737. [Google Scholar] [CrossRef] [PubMed]
- Barford, D.; Flint, A.J.; Tonks, N.K. Crystal structure of human protein tyrosine phosphatase 1B. Science 1994, 263, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Wiener, J.R.; Hurteau, J.A.; Kerns, B.J.; Whitaker, R.S.; Conaway, M.R.; Berchuck, A.; Bast, R.C., Jr. Overexpression of the tyrosine phosphatase PTP1B is associated with human ovarian carcinomas. Am. J. Obstet. Gynecol. 1994, 170, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Wiener, J.R.; Kerns, B.J.; Harvey, E.L.; Conaway, M.R.; Iglehart, J.D.; Berchuck, A.; Bast, R.C., Jr. Overexpression of the protein tyrosine phosphatase PTP1B in human breast cancer: Association with p185c-erbB-2 protein expression. J. Natl. Cancer Inst. 1994, 86, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Barr, A.J. Protein tyrosine phosphatases as drug targets: Strategies and challenges of inhibitor development. Future Med. Chem. 2010, 2, 1563–1576. [Google Scholar] [CrossRef] [PubMed]
- Iversen, L.F.; Moller, K.B.; Pedersen, A.K.; Peters, G.H.; Petersen, A.S.; Andersen, H.S.; Branner, S.; Mortensen, S.B.; Moller, N.P. Structure determination of T cell protein-tyrosine phosphatase. J. Biol. Chem. 2002, 277, 19982–19990. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.P.; Li, Y.; Chen, Y.Y.; Hsu, S.D.; Page, R.; Peti, W.; Meng, T.C. The catalytic activity of TCPTP is auto-regulated by its intrinsically disordered tail and activated by Integrin alpha-1. Nat. Commun. 2022, 13, 94. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Lan, J.; Tang, J.; Luo, N. PTPN2 in the Immunity and Tumor Immunotherapy: A Concise Review. Int. J. Mol. Sci. 2022, 23, 10025. [Google Scholar] [CrossRef]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef]
- Baumgartner, C.K.; Ebrahimi-Nik, H.; Iracheta-Vellve, A.; Hamel, K.M.; Olander, K.E.; Davis, T.G.R.; McGuire, K.A.; Halvorsen, G.T.; Avila, O.I.; Patel, C.H.; et al. The PTPN2/PTPN1 inhibitor ABBV-CLS-484 unleashes potent anti-tumour immunity. Nature 2023, 622, 850–862. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Dodd, G.T.; Tiganis, T. Protein Tyrosine Phosphatases in Hypothalamic Insulin and Leptin Signaling. Trends Pharmacol. Sci. 2015, 36, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Tiganis, T. PTP1B and TCPTP--nonredundant phosphatases in insulin signaling and glucose homeostasis. FEBS J. 2013, 280, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Sun, R.; Yagimuma, H.; Taki, K.; Mizoguchi, A.; Kobayashi, T.; Sugiyama, M.; Onoue, T.; Tsunekawa, T.; Takagi, H.; et al. Protein Tyrosine Phosphatase 1B Deficiency Improves Glucose Homeostasis in Type 1 Diabetes Treated With Leptin. Diabetes 2022, 71, 1902–1914. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Andersen, J.N.; Salmeen, A.; Barford, D.; Tonks, N.K. Conformation-sensing antibodies stabilize the oxidized form of PTP1B and inhibit its phosphatase activity. Cell 2011, 147, 185–198. [Google Scholar] [CrossRef]
- Melander, S.A.; Larsen, A.T.; Karsdal, M.A.; Henriksen, K. Are insulin sensitizers the new strategy to treat Type 1 diabetes? A long-acting dual amylin and calcitonin receptor agonist improves insulin-mediated glycaemic control and controls body weight. Br. J. Pharmacol. 2024, 181, 1829–1842. [Google Scholar] [CrossRef] [PubMed]
- You-Ten, K.E.; Muise, E.S.; Itié, A.; Michaliszyn, E.; Wagner, J.; Jothy, S.; Lapp, W.S.; Tremblay, M.L. Impaired bone marrow microenvironment and immune function in T cell protein tyrosine phosphatase-deficient mice. J. Exp. Med. 1997, 186, 683–693. [Google Scholar] [CrossRef]
- Lee, G.B.; Etherton-Beer, C.; Hosking, S.M.; Pasco, J.A.; Page, A.T. The patterns and implications of potentially suboptimal medicine regimens among older adults: A narrative review. Ther. Adv. Drug Saf. 2022, 13, 20420986221100117. [Google Scholar] [CrossRef]
- International Diabetes Federation. IDF Diabetes Atlas 2021—10th Edition. 2022. Available online: https://diabetesatlas.org/atlas/tenth-edition/ (accessed on 11 May 2024).
- National Institute for Health and Care Excellence. Type 2 Diabetes in Adults: Management. 29 June 2022. Available online: https://www.nice.org.uk/guidance/ng28/chapter/Recommendations#drug-treatment (accessed on 11 May 2024).
- Wang, M.Y.; Chen, L.; Clark, G.O.; Lee, Y.; Stevens, R.D.; Ilkayeva, O.R.; Wenner, B.R.; Bain, J.R.; Charron, M.J.; Newgard, C.B.; et al. Leptin therapy in insulin-deficient type I diabetes. Proc. Natl. Acad. Sci. USA 2010, 107, 4813–4819. [Google Scholar] [CrossRef]
- Shulman, G.I. Cellular mechanisms of insulin resistance. J. Clin. Investig. 2000, 106, 171–176. [Google Scholar] [CrossRef]
- Larsen, J.; Brekke, M.; Sandvik, L.; Arnesen, H.; Hanssen, K.F.; Dahl-Jorgensen, K. Silent coronary atheromatosis in type 1 diabetic patients and its relation to long-term glycemic control. Diabetes 2002, 51, 2637–2641. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, K.; Rana, M.B.M.; Sultan, S. Oral Hypoglycemic Medications. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- World Health Organization. The Selection and Use of Essential Medicines 2023: Web Annex A: World Health Organization Model List of Essential Medicines: 23rd List (2023). 2023. Available online: https://iris.who.int/handle/10665/371090 (accessed on 11 May 2024).
- Kane, S.P. The Top 300 of 2021, ClinCalc DrugStats Database, Version 2024.01. 1 January 2024. Available online: https://clincalc.com/DrugStats/Top300Drugs.aspx (accessed on 11 May 2024).
- Fujita, Y.; Inagaki, N. Metformin: New Preparations and Nonglycemic Benefits. Curr. Diabetes Rep. 2017, 17, 5. [Google Scholar] [CrossRef]
- Buntz, B. Best-Selling Pharmaceuticals of 2023 Reveal a Shift in Pharma Landscape. 27 March 2024. Available online: https://www.drugdiscoverytrends.com/best-selling-pharmaceuticals-2023/ (accessed on 11 May 2024).
- US Food and Drug Administration. FDA Drug Safety Communication: FDA Warns that DPP-4 Inhibitors for Type 2 Diabetes May Cause Severe Joint Pain. 23 June 2016. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-warns-dpp-4-inhibitors-type-2-diabetes-may-cause-severe-joint-pain (accessed on 11 May 2024).
- Hsia, D.S.; Grove, O.; Cefalu, W.T. An update on sodium-glucose co-transporter-2 inhibitors for the treatment of diabetes mellitus. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, J.; Ferrannini, E. Euglycemic Diabetic Ketoacidosis: A Predictable, Detectable, and Preventable Safety Concern With SGLT2 Inhibitors. Diabetes Care 2015, 38, 1638–1642. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. FDA Drug Safety Communication: Updated FDA Review Concludes that Use of Type 2 Diabetes Medicine Pioglitazone May Be Linked to an Increased Risk of Bladder Cancer. 11 December 2017. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-updated-fda-review-concludes-use-type-2-diabetes-medicine-pioglitazone (accessed on 11 May 2024).
- Fortune Business Insights. Anti-Obesity Drugs Market Size, Share & Industry Analysis, By Type (Prescription Drugs and OTC Drugs), By Distribution Channel (Hospital Pharmacy and Retail and Online Pharmacy), and Regional Forecast, 2024–2032. April 2024.. Available online: https://www.fortunebusinessinsights.com/anti-obesity-drugs-market-104783 (accessed on 11 May 2024).
- Davies, M.; Pieber, T.R.; Hartoft-Nielsen, M.L.; Hansen, O.K.H.; Jabbour, S.; Rosenstock, J. Effect of Oral Semaglutide Compared With Placebo and Subcutaneous Semaglutide on Glycemic Control in Patients With Type 2 Diabetes: A Randomized Clinical Trial. JAMA 2017, 318, 1460–1470. [Google Scholar] [CrossRef] [PubMed]
- Weghuber, D.; Barrett, T.; Barrientos-Pérez, M.; Gies, I.; Hesse, D.; Jeppesen, O.K.; Kelly, A.S.; Mastrandrea, L.D.; Sørrig, R.; Arslanian, S.; et al. Once-Weekly Semaglutide in Adolescents with Obesity. N. Engl. J. Med. 2022, 387, 2245–2257. [Google Scholar] [CrossRef]
- Nuffer, W.A.; Trujillo, J.M. Liraglutide: A New Option for the Treatment of Obesity. Pharmacotherapy 2015, 35, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.G.; Ruan, J.Q.; Lai, C.; Sun, Z.; Yang, X. Efficacy and Safety of Phentermine/Topiramate in Adults with Overweight or Obesity: A Systematic Review and Meta-Analysis. Obesity 2021, 29, 985–994. [Google Scholar] [CrossRef]
- Liu, Z.; Gao, H.; Zhao, Z.; Huang, M.; Wang, S.; Zhan, J. Status of research on natural protein tyrosine phosphatase 1B inhibitors as potential antidiabetic agents: Update. Biomed. Pharmacother. 2023, 157, 113990. [Google Scholar] [CrossRef]
- Zhao, X.; An, X.; Yang, C.; Sun, W.; Ji, H.; Lian, F. The crucial role and mechanism of insulin resistance in metabolic disease. Front. Endocrinol. 2023, 14, 1149239. [Google Scholar] [CrossRef]
- Teimouri, M.; Hosseini, H.; ArabSadeghabadi, Z.; Babaei-Khorzoughi, R.; Gorgani-Firuzjaee, S.; Meshkani, R. The role of protein tyrosine phosphatase 1B (PTP1B) in the pathogenesis of type 2 diabetes mellitus and its complications. J. Physiol. Biochem. 2022, 78, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wertheimer, S.J.; Lin, C.H.; Katz, S.L.; Amrein, K.E.; Burn, P.; Quon, M.J. Protein-tyrosine phosphatases PTP1B and syp are modulators of insulin-stimulated translocation of GLUT4 in transfected rat adipose cells. J. Biol. Chem. 1997, 272, 8026–8031. [Google Scholar] [CrossRef] [PubMed]
- Meek, T.H.; Morton, G.J. The role of leptin in diabetes: Metabolic effects. Diabetologia 2016, 59, 928–932. [Google Scholar] [CrossRef]
- D’souza, A.M.; Neumann, U.H.; Glavas, M.M.; Kieffer, T.J. The glucoregulatory actions of leptin. Mol. Metab. 2017, 6, 1052–1065. [Google Scholar] [CrossRef] [PubMed]
- Flores-Cordero, J.A.; Pérez-Pérez, A.; Jiménez-Cortegana, C.; Alba, G.; Flores-Barragán, A.; Sánchez-Margalet, V. Obesity as a Risk Factor for Dementia and Alzheimer’s Disease: The Role of Leptin. Int. J. Mol. Sci. 2022, 23, 5202. [Google Scholar] [CrossRef] [PubMed]
- Tsou, R.C.; Bence, K.K. The Genetics of PTPN1 and Obesity: Insights from Mouse Models of Tissue-Specific PTP1B Deficiency. J. Obes. 2012, 2012, 926857. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, S.; Bhattarai, B.R.; Cho, H.; Choi, J.K.; Cho, H. PTP1B inhibitor Ertiprotafib is also a potent inhibitor of IkappaB kinase beta (IKK-beta). Bioorg. Med. Chem. Lett. 2007, 17, 2728–2730. [Google Scholar] [CrossRef] [PubMed]
- Lantz, K.A.; Hart, S.G.; Planey, S.L.; Roitman, M.F.; Ruiz-White, I.A.; Wolfe, H.R.; McLane, M.P. Inhibition of PTP1B by trodusquemine (MSI-1436) causes fat-specific weight loss in diet-induced obese mice. Obesity 2010, 18, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Ohta, T.; Sakata, S.; Morinaga, H.; Ito, M.; Nakagawa, Y.; Tanaka, M.; Matsushita, M. Pharmacological profiles of a novel protein tyrosine phosphatase 1B inhibitor, JTT-551. Diabetes Obes. Metab. 2010, 12, 299–306. [Google Scholar] [CrossRef]
- Swarbrick, M.M.; Havel, P.J.; Levin, A.A.; Bremer, A.A.; Stanhope, K.L.; Butler, M.; Booten, S.L.; Graham, J.L.; McKay, R.A.; Murray, S.F.; et al. Inhibition of protein tyrosine phosphatase-1B with antisense oligonucleotides improves insulin sensitivity and increases adiponectin concentrations in monkeys. Endocrinology 2009, 150, 1670–1679. [Google Scholar] [CrossRef]
- Liu, R.; Mathieu, C.; Berthelet, J.; Zhang, W.; Dupret, J.M.; Rodrigues Lima, F. Human Protein Tyrosine Phosphatase 1B (PTP1B): From Structure to Clinical Inhibitor Perspectives. Int. J. Mol. Sci. 2022, 23, 7027. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh Grewal, A.; Grover, R.; Sharma, N.; Chopra, B.; Kumar Dhingra, A.; Arora, S.; Redhu, S.; Lather, V. Recent updates on development of protein-tyrosine phosphatase 1B inhibitors for treatment of diabetes, obesity and related disorders. Bioorg. Chem. 2022, 121, 105626. [Google Scholar] [CrossRef] [PubMed]
- Coronell-Tovar, A.; Pardo, J.P.; Rodríguez-Romero, A.; Sosa-Peinado, A.; Vásquez-Bochm, L.; Cano-Sánchez, P.; Álvarez-Añorve, L.I.; González-Andrade, M. Protein tyrosine phosphatase 1B (PTP1B) function, structure, and inhibition strategies to develop antidiabetic drugs. FEBS Lett. 2024. [Google Scholar] [CrossRef] [PubMed]
- Delibegović, M.; Dall’Angelo, S.; Dekeryte, R. Protein tyrosine phosphatase 1B in metabolic diseases and drug development. Nat. Rev. Endocrinol. 2024, 20, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, H.; Min, X.; Sun, J.; Liang, B.; Xu, L.; Li, J.; Wang, S.H.; Xu, X. Identification of 1,3,4-Thiadiazolyl-Containing Thiazolidine-2,4-dione Derivatives as Novel PTP1B Inhibitors with Antidiabetic Activity. J. Med. Chem. 2024, 67, 8406–8419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yang, X.; Wu, L.; Liu, F.; Dong, K.; Guo, C.; Gong, L.; Dong, G.; Shi, Y.; Gu, Z.; et al. Site-Specifically Modified Peptide Inhibitors of Protein Tyrosine Phosphatase 1B and T-Cell Protein Tyrosine Phosphatase with Enhanced Stability and Improved In Vivo Long-Acting Activity. ACS Pharmacol. Transl. Sci. 2024, 7, 1426–1437. [Google Scholar] [CrossRef] [PubMed]
- Kyriakou, E.; Schmidt, S.; Dodd, G.T.; Pfuhlmann, K.; Simonds, S.E.; Lenhart, D.; Geerlof, A.; Schriever, S.C.; De Angelis, M.; Schramm, K.W.; et al. Celastrol Promotes Weight Loss in Diet-Induced Obesity by Inhibiting the Protein Tyrosine Phosphatases PTP1B and TCPTP in the Hypothalamus. J. Med. Chem. 2018, 61, 11144–11157. [Google Scholar] [CrossRef] [PubMed]
- Wainaina, M.N.; Chen, Z.; Zhong, C. Environmental factors in the development and progression of late-onset Alzheimer’s disease. Neurosci. Bull. 2014, 30, 253–270. [Google Scholar] [CrossRef]
- Kamboh, M.I. Molecular genetics of late-onset Alzheimer’s disease. Ann. Hum. Genet. 2004, 68 Pt 4, 381–404. [Google Scholar] [CrossRef]
- Koedam, E.L.; Lauffer, V.; van der Vlies, A.E.; van der Flier, W.M.; Scheltens, P.; Pijnenburg, Y.A. Early-versus late-onset Alzheimer’s disease: More than age alone. J. Alzheimers Dis. 2010, 19, 1401–1408. [Google Scholar] [CrossRef]
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K.; et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 1995, 269, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Solomon, A.; Turunen, H.; Ngandu, T.; Peltonen, M.; Levälahti, E.; Helisalmi, S.; Antikainen, R.; Bäckman, L.; Hänninen, T.; Jula, A.; et al. Effect of the Apolipoprotein E Genotype on Cognitive Change During a Multidomain Lifestyle Intervention: A Subgroup Analysis of a Randomized Clinical Trial. JAMA Neurol. 2018, 75, 462–470. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 2020, 23, 194–208, Erratum in Nat. Neurosci. 2020, 23, 294; Erratum in Nat. Neurosci. 2020, 23, 1308. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, L.K.; Dufresne, M.; Joppé, S.E.; Petryszyn, S.; Aumont, A.; Calon, F.; Barnabé-Heider, F.; Furtos, A.; Parent, M.; Chaurand, P.; et al. Aberrant Lipid Metabolism in the Forebrain Niche Suppresses Adult Neural Stem Cell Proliferation in an Animal Model of Alzheimer’s Disease. Cell Stem Cell 2015, 17, 397–411. [Google Scholar] [CrossRef]
- Farmer, B.C.; Kluemper, J.; Johnson, L.A. Apolipoprotein E4 Alters Astrocyte Fatty Acid Metabolism and Lipid Droplet Formation. Cells 2019, 8, 182. [Google Scholar] [CrossRef]
- Czech, C.; Tremp, G.; Pradier, L. Presenilins and Alzheimer’s disease: Biological functions and pathogenic mechanisms. Prog. Neurobiol. 2000, 60, 363–384. [Google Scholar] [CrossRef]
- van der Flier, W.M.; Scheltens, P. Epidemiology and risk factors of dementia. J. Neurol. Neurosurg. Psychiatry 2005, 76 (Suppl. S5), v2–v7. [Google Scholar] [CrossRef]
- Mason, L.H.; Harp, J.P.; Han, D.Y. Pb neurotoxicity: Neuropsychological effects of lead toxicity. Biomed. Res. Int. 2014, 2014, 840547. [Google Scholar] [CrossRef] [PubMed]
- Basha, M.R.; Murali, M.; Siddiqi, H.K.; Ghosal, K.; Siddiqi, O.K.; Lashuel, H.A.; Ge, Y.W.; Lahiri, D.K.; Zawia, N.H. Lead (Pb) exposure and its effect on APP proteolysis and Abeta aggregation. FASEB J. 2005, 19, 2083–2084. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, B.S.; Stewart, W.F.; Bolla, K.I.; Simon, P.D.; Bandeen-Roche, K.; Gordon, P.B.; Links, J.M.; Todd, A.C. Past adult lead exposure is associated with longitudinal decline in cognitive function. Neurology 2000, 55, 1144–1150, Erratum in Neurology 2001, 56, 283. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Mazurek, M.F.; Ellison, D.W.; Kowall, N.W.; Solomon, P.R.; Pendlebury, W.W. Neurochemical characteristics of aluminum-induced neurofibrillary degeneration in rabbits. Neuroscience 1989, 29, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.B.; Wenzel, H.J.; Kafer, K.E.; Schwartzkroin, P.A.; Palmiter, R.D. Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc. Natl. Acad. Sci. USA 1999, 96, 1716–1721. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I. The metallobiology of Alzheimer’s disease. Trends Neurosci. 2003, 26, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guenette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef] [PubMed]
- Ricke, K.M.; Cruz, S.A.; Qin, Z.; Farrokhi, K.; Sharmin, F.; Zhang, L.; Zasloff, M.A.; Stewart, A.F.R.; Chen, H.H. Neuronal Protein Tyrosine Phosphatase 1B Hastens Amyloid β-Associated Alzheimer’s Disease in Mice. J. Neurosci. 2020, 40, 1581–1593. [Google Scholar] [CrossRef] [PubMed]
- Pannacciulli, N.; Del Parigi, A.; Chen, K.; Le, D.S.; Reiman, E.M.; Tataranni, P.A. Brain abnormalities in human obesity: A voxel-based morphometric study. Neuroimage 2006, 31, 1419–1425. [Google Scholar] [CrossRef]
- Yokum, S.; Ng, J.; Stice, E. Relation of regional gray and white matter volumes to current BMI and future increases in BMI: A prospective MRI study. Int. J. Obes. 2012, 36, 656–664. [Google Scholar] [CrossRef]
- Esteban-Cornejo, I.; Mora-Gonzalez, J.; Cadenas-Sanchez, C.; Contreras-Rodriguez, O.; Verdejo-Román, J.; Henriksson, P.; Migueles, J.H.; Rodriguez-Ayllon, M.; Molina-García, P.; Suo, C.; et al. Fitness, cortical thickness and surface area in overweight/obese children: The mediating role of body composition and relationship with intelligence. Neuroimage 2019, 186, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Veit, R.; Kullmann, S.; Heni, M.; Machann, J.; Häring, H.U.; Fritsche, A.; Preissl, H. Reduced cortical thickness associated with visceral fat and BMI. Neuroimage Clin. 2014, 6, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Ronan, L.; Alexander-Bloch, A.; Fletcher, P.C. Childhood Obesity, Cortical Structure, and Executive Function in Healthy Children. Cereb. Cortex 2020, 30, 2519–2528. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, E.J.; Bongard, V.; Beiser, A.S.; Lamon-Fava, S.; Robins, S.J.; Au, R.; Tucker, K.L.; Kyle, D.J.; Wilson, P.W.; Wolf, P.A. Plasma phosphatidylcholine docosahexaenoic acid content and risk of dementia and Alzheimer disease: The Framingham Heart Study. Arch. Neurol. 2006, 63, 1545–1550. [Google Scholar] [CrossRef] [PubMed]
- Olloquequi, J.; Ettcheto, M.; Cano, A.; Fortuna, A.; Bicker, J.; Sánchez-Lopez, E.; Paz, C.; Ureña, J.; Verdaguer, E.; Auladell, C.; et al. Licochalcone A: A Potential Multitarget Drug for Alzheimer’s Disease Treatment. Int. J. Mol. Sci. 2023, 24, 14177. [Google Scholar] [CrossRef] [PubMed]
- Kuban-Jankowska, A.; Kostrzewa, T.; Musial, C.; Barone, G.; Lo Bosco, G.; Lo Celso, F.; Górska-Ponikowska, M. Green Tea Catechins Induce Inhibition of PTP1B Phosphatase in Breast Cancer Cells with Potent Anti-Cancer Properties: In Vitro Assay, Molecular Docking, and Dynamics Studies. Antioxidants 2020, 9, 1208. [Google Scholar] [CrossRef] [PubMed]
- Ettcheto, M.; Cano, A.; Manzine, P.R.; Busquets, O.; Verdaguer, E.; Castro-Torres, R.D.; García, M.L.; Beas-Zarate, C.; Olloquequi, J.; Auladell, C.; et al. Epigallocatechin-3-Gallate (EGCG) Improves Cognitive Deficits Aggravated by an Obesogenic Diet Through Modulation of Unfolded Protein Response in APPswe/PS1dE9 Mice. Mol. Neurobiol. 2020, 57, 1814–1827. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.F.; Hu, L.H.; Lou, F.C.; Li, J.; Shen, Q. PTP1B inhibitors from Ardisia japonica. J. Asian Nat. Prod. Res. 2005, 7, 13–18. [Google Scholar] [CrossRef]
- Barai, P.; Raval, N.; Acharya, S.; Borisa, A.; Bhatt, H.; Acharya, N. Neuroprotective effects of bergenin in Alzheimer’s disease: Investigation through molecular docking, in vitro and in vivo studies. Behav. Brain Res. 2019, 356, 18–40. [Google Scholar] [CrossRef]
- Wang, H.; Sun, X.; Zhang, N.; Ji, Z.; Ma, Z.; Fu, Q.; Qu, R.; Ma, S. Ferulic acid attenuates diabetes-induced cognitive impairment in rats via regulation of PTP1B and insulin signaling pathway. Physiol. Behav. 2017, 182, 93–100. [Google Scholar] [CrossRef]
- Bai, X.; Zhao, X.; Liu, K.; Yang, X.; He, Q.; Gao, Y.; Li, W.; Han, W. Mulberry Leaf Compounds and Gut Microbiota in Alzheimer’s Disease and Diabetes: A Study Using Network Pharmacology, Molecular Dynamics Simulation, and Cellular Assays. Int. J. Mol. Sci. 2024, 25, 4062. [Google Scholar] [CrossRef]
- Hasin, D.S.; Sarvet, A.L.; Meyers, J.L.; Saha, T.D.; Ruan, W.J.; Stohl, M.; Grant, B.F. Epidemiology of Adult DSM-5 Major Depressive Disorder and Its Specifiers in the United States. JAMA Psychiatry 2018, 75, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Belmaker, R.H. Bipolar disorder. N. Engl. J. Med. 2004, 351, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Kendler, K.S.; Bulik, C.M.; Silberg, J.; Hettema, J.M.; Myers, J.; Prescott, C.A. Childhood sexual abuse and adult psychiatric and substance use disorders in women: An epidemiological and cotwin control analysis. Arch. Gen. Psychiatry 2000, 57, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Caspi, A.; Sugden, K.; Moffitt, T.E.; Taylor, A.; Craig, I.W.; Harrington, H.; McClay, J.; Mill, J.; Martin, J.; Braithwaite, A.; et al. Influence of life stress on depression: Moderation by a polymorphism in the 5-HTT gene. Science 2003, 301, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Buckholtz, J.W.; Meyer-Lindenberg, A. MAOA and the neurogenetic architecture of human aggression. Trends Neurosci. 2008, 31, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Nolen, W.A.; van de Putte, J.J.; Dijken, W.A.; Kamp, J.S.; Blansjaar, B.A.; Kramer, H.J.; Haffmans, J. Treatment strategy in depression. II. MAO inhibitors in depression resistant to cyclic antidepressants: Two controlled crossover studies with tranylcypromine versus L-5-hydroxytryptophan and nomifensine. Acta Psychiatr. Scand. 1988, 78, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ming, Q.; Zhong, X.; Dong, D.; Li, C.; Xiong, G.; Cheng, C.; Cao, W.; He, J.; Wang, X.; et al. The MAOA Gene Influences the Neural Response to Psychosocial Stress in the Human Brain. Front. Behav. Neurosci. 2020, 14, 65. [Google Scholar] [CrossRef] [PubMed]
- Varghese, F.P.; Brown, E.S. The Hypothalamic-Pituitary-Adrenal Axis in Major Depressive Disorder: A Brief Primer for Primary Care Physicians. Prim. Care Companion J. Clin. Psychiatry 2001, 3, 151–155. [Google Scholar] [CrossRef]
- Smith, M.A.; Makino, S.; Kvetnansky, R.; Post, R.M. Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J. Neurosci. 1995, 15 Pt 1, 1768–1777. [Google Scholar] [CrossRef]
- Qin, Z.; Zhou, X.; Pandey, N.R.; Vecchiarelli, H.A.; Stewart, C.A.; Zhang, X.; Lagace, D.C.; Brunel, J.M.; Béïque, J.C.; Stewart, A.F.; et al. Chronic stress induces anxiety via an amygdalar intracellular cascade that impairs endocannabinoid signaling. Neuron 2015, 85, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Notaras, M.; van den Buuse, M. Neurobiology of BDNF in fear memory, sensitivity to stress, and stress-related disorders. Mol. Psychiatry 2020, 25, 2251–2274. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, F.; Carpinteiro, A.; Edwards, M.J.; Wilson, G.C.; Keitsch, S.; Soddemann, M.; Wilker, B.; Kleuser, B.; Becker, K.A.; Müller, C.P.; et al. Stress induces major depressive disorder by a neutral sphingomyelinase 2-mediated accumulation of ceramide-enriched exosomes in the blood plasma. J. Mol. Med. 2022, 100, 1493–1508. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Pandey, N.R.; Zhou, X.; Stewart, C.A.; Hari, A.; Huang, H.; Stewart, A.F.; Brunel, J.M.; Chen, H.H. Functional properties of Claramine: A novel PTP1B inhibitor and insulin-mimetic compound. Biochem. Biophys. Res. Commun. 2015, 458, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Pandey, N.R.; Zhou, X.; Qin, Z.; Zaman, T.; Gomez-Smith, M.; Keyhanian, K.; Anisman, H.; Brunel, J.M.; Stewart, A.F.; Chen, H.H. The LIM domain only 4 protein is a metabolic responsive inhibitor of protein tyrosine phosphatase 1B that controls hypothalamic leptin signaling. J. Neurosci. 2013, 33, 12647–12655. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.J.; Freedland, K.E.; Clouse, R.E.; Lustman, P.J. The prevalence of comorbid depression in adults with diabetes: A meta-analysis. Diabetes Care 2001, 24, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Knol, M.J.; Twisk, J.W.; Beekman, A.T.; Heine, R.J.; Snoek, F.J.; Pouwer, F. Depression as a risk factor for the onset of type 2 diabetes mellitus. A meta-analysis. Diabetologia 2006, 49, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Zarouna, S.; Wozniak, G.; Papachristou, A.I. Mood disorders: A potential link between ghrelin and leptin on human body? World J. Exp. Med. 2015, 5, 103–109. [Google Scholar] [CrossRef]
- Li, Y.; Wang, N.; Pan, J.; Wang, X.; Zhao, Y.; Guo, Z. Hippocampal miRNA-144 Modulates Depressive-Like Behaviors in Rats by Targeting PTP1B. Neuropsychiatr. Dis. Treat. 2021, 17, 389–399. [Google Scholar] [CrossRef]
- Diks, S.H.; Richel, D.J.; Peppelenbosch, M.P. LPS signal transduction: The picture is becoming more complex. Curr. Top. Med. Chem. 2004, 4, 1115–1126. [Google Scholar] [CrossRef]
- Fessler, M.B.; Malcolm, K.C.; Duncan, M.W.; Worthen, G.S. A genomic and proteomic analysis of activation of the human neutrophil by lipopolysaccharide and its mediation by p38 mitogen-activated protein kinase. J. Biol. Chem. 2002, 277, 31291–31302. [Google Scholar] [CrossRef]
- Zhao, B.; Bowden, R.A.; Stavchansky, S.A.; Bowman, P.D. Human endothelial cell response to gram-negative lipopolysaccharide assessed with cDNA microarrays. Am. J. Physiol. Cell Physiol. 2001, 281, C1587–C1595. [Google Scholar] [CrossRef]
- Alexander, C.; Rietschel, E.T. Bacterial lipopolysaccharides and innate immunity. J. Endotoxin Res. 2001, 7, 167–202. [Google Scholar] [CrossRef]
- Parlesak, A.; Schäfer, C.; Schütz, T.; Bode, J.C.; Bode, C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J. Hepatol. 2000, 32, 742–747. [Google Scholar] [CrossRef]
- Khoruts, A.; Stahnke, L.; McClain, C.J.; Logan, G.; Allen, J.I. Circulating tumor necrosis factor, interleukin-1 and interleukin-6 concentrations in chronic alcoholic patients. Hepatology 1991, 13, 267–276. [Google Scholar] [CrossRef]
- He, G.; Karin, M. NF-κB and STAT3—Key players in liver inflammation and cancer. Cell Res. 2011, 21, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Larrea, E.; Aldabe, R.; Molano, E.; Fernandez-Rodriguez, C.M.; Ametzazurra, A.; Civeira, M.P.; Prieto, J. Altered expression and activation of signal transducers and activators of transcription (STATs) in hepatitis C virus infection: In vivo and in vitro studies. Gut 2006, 55, 1188–1196. [Google Scholar] [CrossRef]
- Yang, L.; Sun, Y.Y.; Liu, Y.R.; Yin, N.N.; Bu, F.T.; Yu, H.X.; Du, X.S.; Li, J.; Huang, C. PTP1B promotes macrophage activation by regulating the NF-κB pathway in alcoholic liver injury. Toxicol. Lett. 2020, 319, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Pike, K.A.; Tremblay, M.L. TC-PTP and PTP1B: Regulating JAK-STAT signaling, controlling lymphoid malignancies. Cytokine 2016, 82, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.F.; Koike, S.; Mello, A.; Nagy, L.E.; Haj, F.G. Hepatic protein-tyrosine phosphatase 1B disruption and pharmacological inhibition attenuate ethanol-induced oxidative stress and ameliorate alcoholic liver disease in mice. Redox Biol. 2020, 36, 101658. [Google Scholar] [CrossRef]
- Dietrich, P.; Hellerbrand, C. Non-alcoholic fatty liver disease, obesity and the metabolic syndrome. Best. Pract. Res. Clin. Gastroenterol. 2014, 28, 637–653. [Google Scholar] [CrossRef] [PubMed]
- Zabolotny, J.M.; Kim, Y.B.; Welsh, L.A.; Kershaw, E.E.; Neel, B.G.; Kahn, B.B. Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. J. Biol. Chem. 2008, 283, 14230–14241. [Google Scholar] [CrossRef]
- Asrih, M.; Jornayvaz, F.R. Metabolic syndrome and nonalcoholic fatty liver disease: Is insulin resistance the link? Mol. Cell Endocrinol. 2015, 418 Pt 1, 55–65. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Falck-Ytter, Y.; Younossi, Z.M.; Marchesini, G.; McCullough, A.J. Clinical features and natural history of nonalcoholic steatosis syndromes. Semin. Liver Dis. 2001, 21, 17–26. [Google Scholar] [CrossRef]
- Ferlay, J.; Ervik, M.; Lam, F.; Laversanne, M.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today; International Agency for Research on Cancer: Lyon, France, 2024; Available online: https://gco.iarc.who.int/today (accessed on 11 May 2024).
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Delibegovic, M.; Zimmer, D.; Kauffman, C.; Rak, K.; Hong, E.G.; Cho, Y.R.; Kim, J.K.; Kahn, B.B.; Neel, B.G.; Bence, K.K. Liver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced endoplasmic reticulum stress. Diabetes 2009, 58, 590–599. [Google Scholar] [CrossRef]
- Pagliassotti, M.J. Endoplasmic reticulum stress in nonalcoholic fatty liver disease. Annu. Rev. Nutr. 2012, 32, 17–33. [Google Scholar] [CrossRef] [PubMed]
- González-Rodríguez, Á.; Valdecantos, M.P.; Rada, P.; Addante, A.; Barahona, I.; Rey, E.; Pardo, V.; Ruiz, L.; Laiglesia, L.M.; Moreno-Aliaga, M.J.; et al. Dual role of protein tyrosine phosphatase 1B in the progression and reversion of non-alcoholic steatohepatitis. Mol. Metab. 2018, 7, 132–146. [Google Scholar] [CrossRef]
- Xu, H.; An, H.; Hou, J.; Han, C.; Wang, P.; Yu, Y.; Cao, X. Phosphatase PTP1B negatively regulates MyD88- and TRIF-dependent proinflammatory cytokine and type I interferon production in TLR-triggered macrophages. Mol. Immunol. 2008, 45, 3545–3552. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, X.L.; Yi, Q.; Zhang, X.L.; Tian, J.Y.; Wang, D.M.; Wu, S.; Ye, F. Investigating the effects of compound WS090152 on non-alcoholic fatty liver in mice. Yao Xue Xue Bao 2016, 51, 919–925. [Google Scholar]
- Li, J.; Zhang, X.; Tian, J.; Li, J.; Li, X.; Wu, S.; Liu, Y.; Han, J.; Ye, F. CX08005, a Protein Tyrosine Phosphatase 1B Inhibitor, Attenuated Hepatic Lipid Accumulation and Microcirculation Dysfunction Associated with Nonalcoholic Fatty Liver Disease. Pharmaceuticals 2023, 16, 106. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, L.L.; Tang, W.J.; Tang, B. Astragaloside IV inhibits protein tyrosine phosphatase 1B and improves insulin resistance in insulin-resistant HepG2 cells and triglyceride accumulation in oleic acid (OA)-treated HepG2 cells. J. Ethnopharmacol. 2021, 268, 113556. [Google Scholar] [CrossRef] [PubMed]
- Bourebaba, L.; Łyczko, J.; Alicka, M.; Bourebaba, N.; Szumny, A.; Fal, A.M.; Marycz, K. Inhibition of Protein-tyrosine Phosphatase PTP1B and LMPTP Promotes Palmitate/Oleate-challenged HepG2 Cell Survival by Reducing Lipoapoptosis, Improving Mitochondrial Dynamics and Mitigating Oxidative and Endoplasmic Reticulum Stress. J. Clin. Med. 2020, 9, 1294. [Google Scholar] [CrossRef] [PubMed]
- Bartolomé, R.A.; Martín-Regalado, Á.; Jaén, M.; Zannikou, M.; Zhang, P.; de Los Ríos, V.; Balyasnikova, I.V.; Casal, J.I. Protein Tyrosine Phosphatase-1B Inhibition Disrupts IL13Rα2-Promoted Invasion and Metastasis in Cancer Cells. Cancers 2020, 12, 500. [Google Scholar] [CrossRef]
- Fujisawa, T.; Joshi, B.; Nakajima, A.; Puri, R.K. A novel role of interleukin-13 receptor alpha2 in pancreatic cancer invasion and metastasis. Cancer Res. 2009, 69, 8678–8685. [Google Scholar] [CrossRef]
- Bartolomé, R.A.; Casal, J.I. IL13Rα2 signaling in colorectal cancer. Oncoscience 2015, 2, 787–788. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Aguilar, B.; Starr, R.; Yang, X.; Chang, W.C.; Weng, L.; Chang, B.; Sarkissian, A.; Brito, A.; Sanchez, J.F.; et al. Optimization of IL13Rα2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Mol. Ther. 2018, 26, 31–44. [Google Scholar] [CrossRef]
- Bernard, J.; Treton, D.; Vermot-Desroches, C.; Boden, C.; Horellou, P.; Angevin, E.; Galanaud, P.; Wijdenes, J.; Richard, Y. Expression of interleukin 13 receptor in glioma and renal cell carcinoma: IL13Ralpha2 as a decoy receptor for IL13. Lab. Investig. 2001, 81, 1223–1231. [Google Scholar] [CrossRef]
- Takenouchi, M.; Hirai, S.; Sakuragi, N.; Yagita, H.; Hamada, H.; Kato, K. Epigenetic modulation enhances the therapeutic effect of anti-IL-13R(alpha)2 antibody in human mesothelioma xenografts. Clin. Cancer Res. 2011, 17, 2819–2829. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Yoshimatsu, Y.; Tomizawa, T.; Kunita, A.; Takayama, R.; Morikawa, T.; Komura, D.; Takahashi, K.; Oshima, T.; Sato, M.; et al. Interleukin-13 receptor α2 is a novel marker and potential therapeutic target for human melanoma. Sci. Rep. 2019, 9, 1281. [Google Scholar] [CrossRef] [PubMed]
- Márquez-Ortiz, R.A.; Contreras-Zárate, M.J.; Tesic, V.; Alvarez-Eraso, K.L.F.; Kwak, G.; Littrell, Z.; Costello, J.C.; Sreekanth, V.; Ormond, D.R.; Karam, S.D.; et al. IL13Rα2 Promotes Proliferation and Outgrowth of Breast Cancer Brain Metastases. Clin. Cancer Res. 2021, 27, 6209–6221. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wu, Y.; Zhu, S.; Liang, W.; Wang, Z.; Wang, Y.; Lv, T.; Yao, Y.; Yuan, D.; Song, Y. PTP1B promotes cell proliferation and metastasis through activating src and ERK1/2 in non-small cell lung cancer. Cancer Lett. 2015, 359, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Barderas, R.; Bartolomé, R.A.; Fernandez-Aceñero, M.J.; Torres, S.; Casal, J.I. High expression of IL-13 receptor α2 in colorectal cancer is associated with invasion, liver metastasis, and poor prognosis. Cancer Res. 2012, 72, 2780–2790. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.G.; Dubé, N.; Read, M.; Penney, J.; Paquet, M.; Han, Y.; Kennedy, B.P.; Muller, W.J.; Tremblay, M.L. Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat. Genet. 2007, 39, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Tanner, M.M.; Tirkkonen, M.; Kallioniemi, A.; Isola, J.; Kuukasjärvi, T.; Collins, C.; Kowbel, D.; Guan, X.Y.; Trent, J.; Gray, J.W.; et al. Independent amplification and frequent co-amplification of three nonsyntenic regions on the long arm of chromosome 20 in human breast cancer. Cancer Res. 1996, 56, 3441–3445. [Google Scholar] [PubMed]
- Wang, N.; She, J.; Liu, W.; Shi, J.; Yang, Q.; Shi, B.; Hou, P. Frequent amplification of PTP1B is associated with poor survival of gastric cancer patients. Cell Cycle 2015, 14, 732–743. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, E.; Das, A.M.; Swets, M.; Cao, W.; van der Woude, C.J.; Bruno, M.J.; Peppelenbosch, M.P.; Kuppen, P.J.; Ten Hagen, T.L.; Fuhler, G.M. Increased PTP1B expression and phosphatase activity in colorectal cancer results in a more invasive phenotype and worse patient outcome. Oncotarget 2016, 7, 21922–21938. [Google Scholar] [CrossRef]
- Lessard, L.; Labbé, D.P.; Deblois, G.; Bégin, L.R.; Hardy, S.; Mes-Masson, A.M.; Saad, F.; Trotman, L.C.; Giguère, V.; Tremblay, M.L. PTP1B is an androgen receptor-regulated phosphatase that promotes the progression of prostate cancer. Cancer Res. 2012, 72, 1529–1537. [Google Scholar] [CrossRef]
- Dubé, N.; Bourdeau, A.; Heinonen, K.M.; Cheng, A.; Loy, A.L.; Tremblay, M.L. Genetic ablation of protein tyrosine phosphatase 1B accelerates lymphomagenesis of p53-null mice through the regulation of B-cell development. Cancer Res. 2005, 65, 10088–10095. [Google Scholar] [CrossRef] [PubMed]
- Beales, I.L.P.; Garcia-Morales, C.; Ogunwobi, O.O.; Mutungi, G. Adiponectin inhibits leptin-induced oncogenic signalling in oesophageal cancer cells by activation of PTP1B. Mol. Cell Endocrinol. 2014, 382, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Warabi, M.; Nemoto, T.; Ohashi, K.; Kitagawa, M.; Hirokawa, K. Expression of protein tyrosine phosphatases and its significance in esophageal cancer. Exp. Mol. Pathol. 2000, 68, 187–195, Erratum in Exp. Mol. Pathol. 2000, 69, 165. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Radisky, D.C.; Nelson, C.M.; Zhang, H.; Fata, J.E.; Roth, R.A.; Bissell, M.J. Mechanism of Akt1 inhibition of breast cancer cell invasion reveals a protumorigenic role for TSC2. Proc. Natl. Acad. Sci. USA 2006, 103, 4134–4139. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, N.; Koveal, D.; Miller, D.H.; Xue, B.; Akshinthala, S.D.; Kragelj, J.; Jensen, M.R.; Gauss, C.M.; Page, R.; Blackledge, M.; et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 2014, 10, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Tran, E.; Du, X.; Dong, J.; Sudholz, H.; Chen, H.; Qu, Z.; Huntington, N.D.; Babon, J.J.; Kershaw, N.J.; et al. A small molecule inhibitor of PTP1B and PTPN2 enhances T cell anti-tumor immunity. Nat. Commun. 2023, 14, 4524. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhang, W.; Gou, P.; Berthelet, J.; Nian, Q.; Chevreux, G.; Legros, V.; Moroy, G.; Bui, L.C.; Wang, L.; et al. Cisplatin causes covalent inhibition of protein-tyrosine phosphatase 1B (PTP1B) through reaction with its active site cysteine: Molecular, cellular and in vivo mice studies. Biomed. Pharmacother. 2022, 153, 113372. [Google Scholar] [CrossRef]
- Garbe, C.; Eigentler, T.K. Vemurafenib. In Small Molecules in Oncology; Recent Results in Cancer Research; Springer: Cham, Swizterland, 2018; Volume 211, pp. 77–89. [Google Scholar] [CrossRef]
- Cordeiro, H.G.; de Sousa Faria, A.V.; Ferreira-Halder, C.V. Vemurafenib downmodulates aggressiveness mediators of colorectal cancer (CRC): Low Molecular Weight Protein Tyrosine Phosphatase (LMWPTP), Protein Tyrosine Phosphatase 1B (PTP1B) and Transforming Growth Factor β (TGFβ). Biol. Chem. 2020, 401, 1063–1069. [Google Scholar] [CrossRef]
- Lee, Y.J.; Song, H.; Yoon, Y.J.; Park, S.J.; Kim, S.Y.; Cho Han, D.; Kwon, B.M. Ethacrynic acid inhibits STAT3 activity through the modulation of SHP2 and PTP1B tyrosine phosphatases in DU145 prostate carcinoma cells. Biochem. Pharmacol. 2020, 175, 113920. [Google Scholar] [CrossRef]
- Li, D.W.; Zhang, M.; Feng, L.; Huang, S.S.; Zhang, B.J.; Liu, S.S.; Deng, S.; Wang, C.; Ma, X.C.; Leng, A.J. Alkaloids from the nearly ripe fruits of Evodia rutaecarpa and their bioactivities. Fitoterapia 2020, 146, 104668. [Google Scholar] [CrossRef]
- Kostrzewa, T.; Przychodzen, P.; Górska-Ponikowska, M.; Kuban-Jankowska, A. Curcumin and Cinnamaldehyde as PTP1B Inhibitors With Antidiabetic and Anticancer Potential. Anticancer Res. 2019, 39, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Kostrzewa, T.; Wołosewicz, K.; Jamrozik, M.; Drzeżdżon, J.; Siemińska, J.; Jacewicz, D.; Górska-Ponikowska, M.; Kołaczkowski, M.; Łaźny, R.; Kuban-Jankowska, A. Curcumin and Its New Derivatives: Correlation between Cytotoxicity against Breast Cancer Cell Lines, Degradation of PTP1B Phosphatase and ROS Generation. Int. J. Mol. Sci. 2021, 22, 10368. [Google Scholar] [CrossRef] [PubMed]
- Kuban-Jankowska, A.; Górska-Ponikowska, M.; Sahu, K.K.; Kostrzewa, T.; Wozniak, M.; Tuszynski, J. Docosahexaenoic Acid Inhibits PTP1B Phosphatase and the Viability of MCF-7 Breast Cancer Cells. Nutrients 2019, 11, 2554. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Yao, S.; Cai, S.; Li, J.; He, L.; Zou, J.; Zhang, Q.; Fan, H.; Zhou, L.; Yu, S. miR-34c inhibits proliferation of glioma by targeting PTP1B. Acta Biochim. Biophys. Sin. 2021, 53, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Zhang, L.; Cruz, S.A.; Stewart, A.F.R.; Chen, H.H. Activation of tyrosine phosphatase PTP1B in pyramidal neurons impairs endocannabinoid signaling by tyrosine receptor kinase trkB and causes schizophrenia-like behaviors in mice. Neuropsychopharmacology 2020, 45, 1884–1895. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.W.; Chen, N.F.; Chan, T.F.; Chen, W.F. Therapeutic Role of Protein Tyrosine Phosphatase 1B in Parkinson’s Disease via Antineuroinflammation and Neuroprotection In Vitro and In Vivo. Park. Dis. 2020, 2020, 8814236. [Google Scholar] [CrossRef] [PubMed]
- De-la-Cruz-Martínez, L.; Duran-Becerra, C.; González-Andrade, M.; Páez-Franco, J.C.; Germán-Acacio, J.M.; Espinosa-Chávez, J.; Torres-Valencia, J.M.; Pérez-Villanueva, J.; Palacios-Espinosa, J.F.; Soria-Arteche, O.; et al. Indole- and Pyrazole-Glycyrrhetinic Acid Derivatives as PTP1B Inhibitors: Synthesis, In Vitro and In Silico Studies. Molecules 2021, 26, 4375. [Google Scholar] [CrossRef] [PubMed]
- Farid, H.A.; Sayed, R.H.; El-Shamarka, M.E.; Abdel-Salam, O.M.E.; El Sayed, N.S. PI3K/AKT signaling activation by roflumilast ameliorates rotenone-induced Parkinson’s disease in rats. Inflammopharmacology 2024, 32, 1421–1437. [Google Scholar] [CrossRef] [PubMed]
- Kołodziej, D.; Sobczak, Ł.; Łączkowski, K.Z. New opportunities for treatment and prevention of neurodegenerative diseases with PTP1B inhibitors. Future Med. Chem. 2023, 15, 1443–1447. [Google Scholar] [CrossRef]
- Liu, F.; Chen, J.; Hu, W.; Gao, C.; Zeng, Z.; Cheng, S.; Yu, K.; Qian, Y.; Xu, D.; Zhu, G.; et al. PTP1B Inhibition Improves Mitochondrial Dynamics to Alleviate Calcific Aortic Valve Disease Via Regulating OPA1 Homeostasis. JACC Basic. Transl. Sci. 2022, 7, 697–712. [Google Scholar] [CrossRef]
- Greenberg, H.Z.E.; Zhao, G.; Shah, A.M.; Zhang, M. Role of oxidative stress in calcific aortic valve disease and its therapeutic implications. Cardiovasc. Res. 2022, 118, 1433–1451. [Google Scholar] [CrossRef] [PubMed]
- Gomez, E.; Vercauteren, M.; Kurtz, B.; Ouvrard-Pascaud, A.; Mulder, P.; Henry, J.P.; Besnier, M.; Waget, A.; Hooft Van Huijsduijnen, R.; Tremblay, M.L.; et al. Reduction of heart failure by pharmacological inhibition or gene deletion of protein tyrosine phosphatase 1B. J. Mol. Cell Cardiol. 2012, 52, 1257–1264. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Sarkar, A.; Banerjee, T.; Maji, A.; Sarkar, S.; Paul, S.; Karmakar, S.; Ghosh, N.; Maity, T.K. Structural and molecular insights of Protein Tyrosine Phosphatase 1B (PTP1B) and its inhibitors as anti-diabetic agents. J. Mol. Struct. 2023, 1293, 136258. [Google Scholar] [CrossRef]
- Agrawal, N.; Dhakrey, P.; Pathak, S. A comprehensive review on the research progress of PTP1B inhibitors as antidiabetics. Chem. Biol. Drug Des. 2023, 102, 921–938. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Depetris, R.S.; Barford, D.; Chernoff, J.; Hubbard, S.R. Crystal structure of a complex between protein tyrosine phosphatase 1B and the insulin receptor tyrosine kinase. Structure 2005, 13, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Choy, M.S.; Li, Y.; Machado, L.E.S.F.; Kunze, M.B.A.; Connors, C.R.; Wei, X.; Lindorff-Larsen, K.; Page, R.; Peti, W. Conformational Rigidity and Protein Dynamics at Distinct Timescales Regulate PTP1B Activity and Allostery. Mol. Cell 2017, 65, 644–658.e5. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, L.; Shi, D. The design strategy of selective PTP1B inhibitors over TCPTP. Bioorg. Med. Chem. 2016, 24, 3343–3352. [Google Scholar] [CrossRef]
- Xu, Y.; Gong, M.; Wang, Y.; Yang, Y.; Liu, S.; Zeng, Q. Global trends and forecasts of breast cancer incidence and deaths. Sci. Data 2023, 10, 334. [Google Scholar] [CrossRef]
- Johnson, K.J.; Peck, A.R.; Liu, C.; Tran, T.H.; Utama, F.E.; Sjolund, A.B.; Schaber, J.D.; Witkiewicz, A.K.; Rui, H. PTP1B suppresses prolactin activation of Stat5 in breast cancer cells. Am. J. Pathol. 2010, 177, 2971–2983. [Google Scholar] [CrossRef]
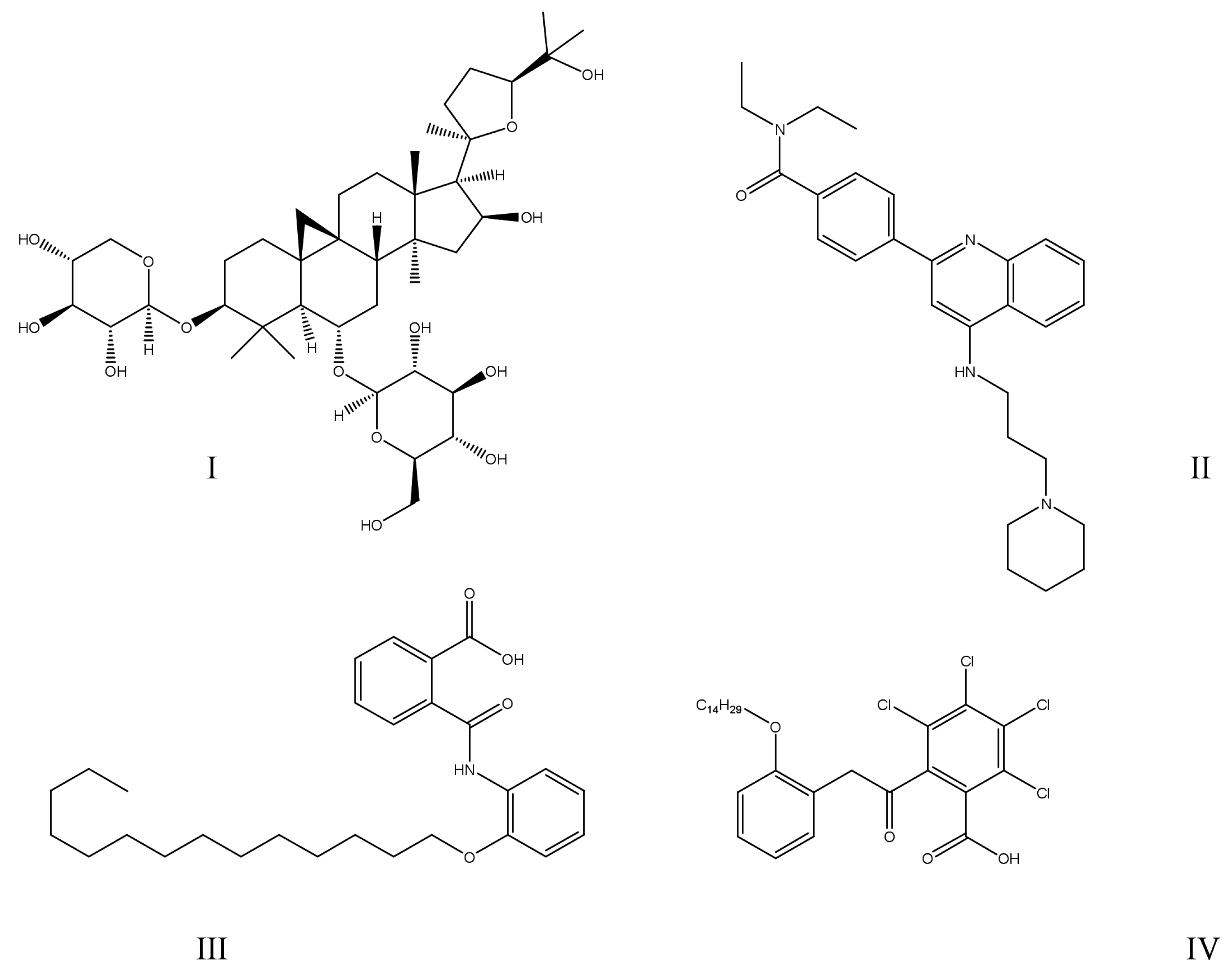

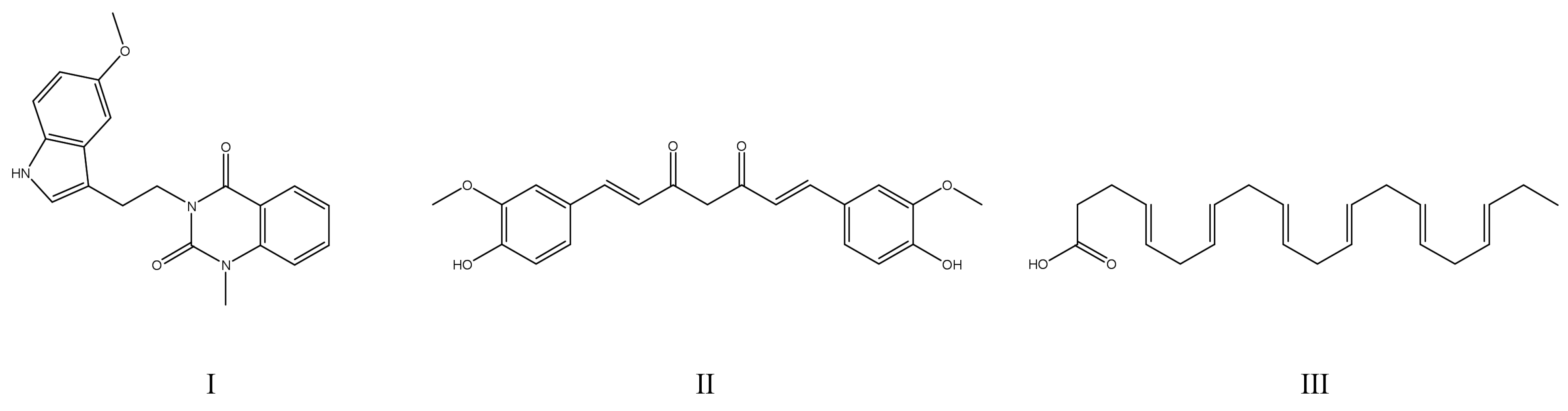


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
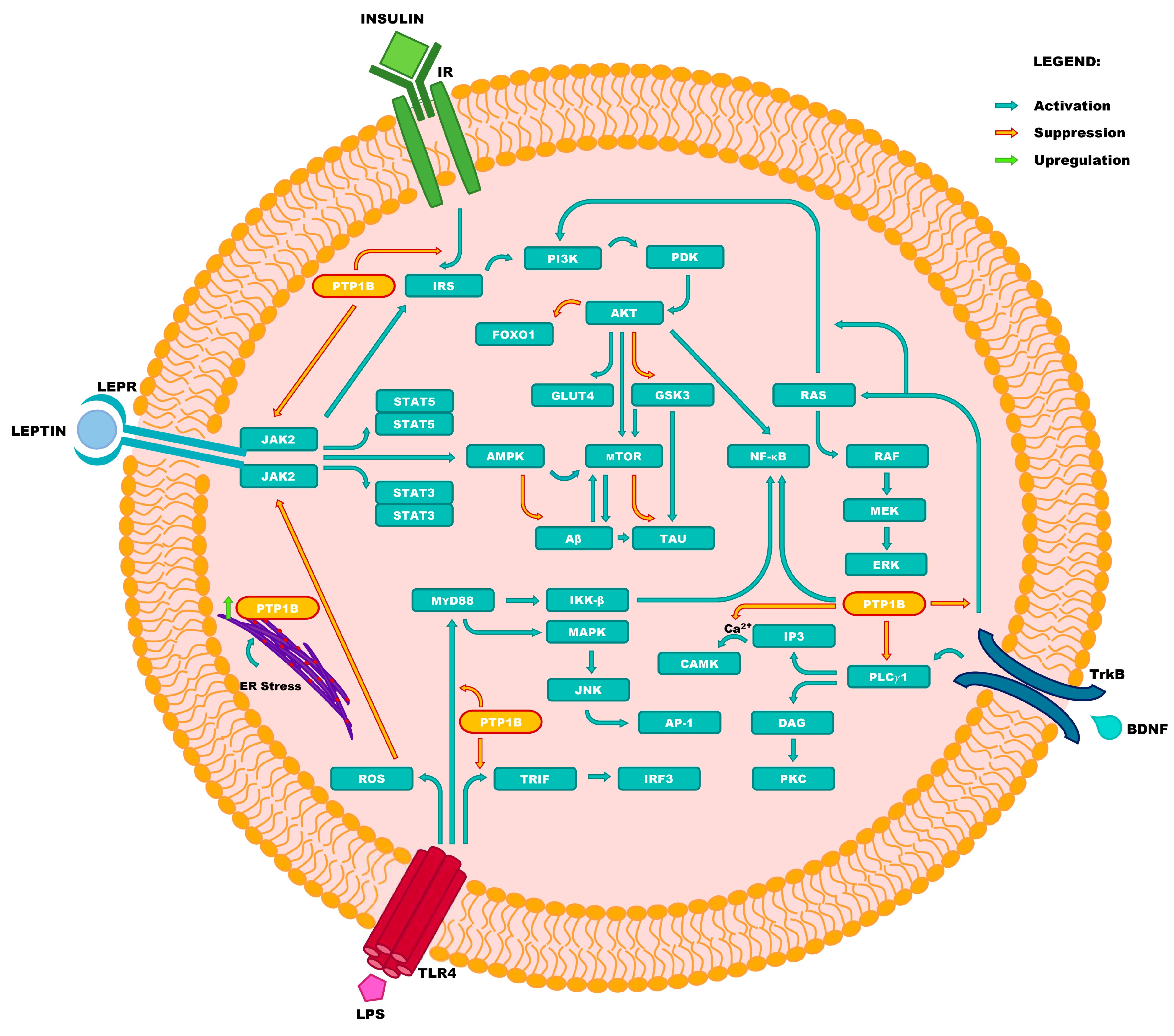
| Abbreviation | Full Name |
|---|---|
| AKT | protein kinase B |
| AMPK | 5′-AMP-activated protein kinase |
| AP-1 | activator protein 1 |
| Aβ | amyloid beta |
| BDNF | brain-derived neurotrophic factor |
| CAMK | Ca2+/calmodulin-dependent protein kinase(s) |
| DAG | diacylglycerol |
| ER | endoplasmic reticulum |
| ERK | extracellular signal-regulated kinase |
| FOXO1 | forkhead box protein O1 |
| GLUT4 | glucose transporter type 4 |
| GSK3 | glycogen synthase kinase 3 |
| IKK-β | inhibitor of nuclear factor kappa-B kinase subunithhv Xlh Thfbeta |
| IP3 | inositol triphosphate |
| IR | insulin receptor |
| IRF3 | interferon regulatory factor 3 |
| IRS | insulin receptor substrate |
| JAK2 | Janus kinase 2 |
| JNK | c-Jun N-terminal kinase(s) |
| LEPR | leptin receptor |
| LPS | lipopolysaccharide |
| MAPK | mitogen-activated protein kinase |
| MEK | mitogen-activated protein kinase kinase |
| mTOR | mammalian target of rapamycin kinase |
| MyD88 | myeloid differentiation primary response 88 protein |
| NF-κB | nuclear factor kappa-light-chain- enhancer of activated B cells |
| PDK | phosphoinositide-dependent kinase |
| PI3K | phosphoinositide 3-kinase |
| PKC | protein kinase C |
| PLCγ1 | phospholipase C gamma 1 |
| PTP1B | protein tyrosine phosphatase 1B |
| RAF | rapidly accelerated fibrosarcoma kinase(s) |
| RAS | rat sarcoma virus (G protein) |
| ROS | reactive oxygen species |
| STAT3 | signal transducer and activator of transcription 3 |
| STAT5 | signal transducer and activator of transcription 5 |
| TAU | tau protein |
| TLR4 | toll-like receptor 4 |
| TRIF | TIR-domain-containing adapter-inducing interferon-beta |
| TrkB | tropomyosin receptor kinase B |
| Name | Sequence |
|---|---|
| IONIS (ISIS) 113715 | 5′-GCUCCTTCCACTGATCCUGC-3′ |
| IONIS (ISIS) PTP1BRx | 5′-AATGGTTTATTCCATGGCCA-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kołodziej-Sobczak, D.; Sobczak, Ł.; Łączkowski, K.Z. Protein Tyrosine Phosphatase 1B (PTP1B): A Comprehensive Review of Its Role in Pathogenesis of Human Diseases. Int. J. Mol. Sci. 2024, 25, 7033. https://doi.org/10.3390/ijms25137033
Kołodziej-Sobczak D, Sobczak Ł, Łączkowski KZ. Protein Tyrosine Phosphatase 1B (PTP1B): A Comprehensive Review of Its Role in Pathogenesis of Human Diseases. International Journal of Molecular Sciences. 2024; 25(13):7033. https://doi.org/10.3390/ijms25137033
Chicago/Turabian StyleKołodziej-Sobczak, Dominika, Łukasz Sobczak, and Krzysztof Z. Łączkowski. 2024. "Protein Tyrosine Phosphatase 1B (PTP1B): A Comprehensive Review of Its Role in Pathogenesis of Human Diseases" International Journal of Molecular Sciences 25, no. 13: 7033. https://doi.org/10.3390/ijms25137033
APA StyleKołodziej-Sobczak, D., Sobczak, Ł., & Łączkowski, K. Z. (2024). Protein Tyrosine Phosphatase 1B (PTP1B): A Comprehensive Review of Its Role in Pathogenesis of Human Diseases. International Journal of Molecular Sciences, 25(13), 7033. https://doi.org/10.3390/ijms25137033