
Mitigating the Functional Deficit after Neurotoxic Motoneuronal Loss by an Inhibitor of Mitochondrial Fission
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
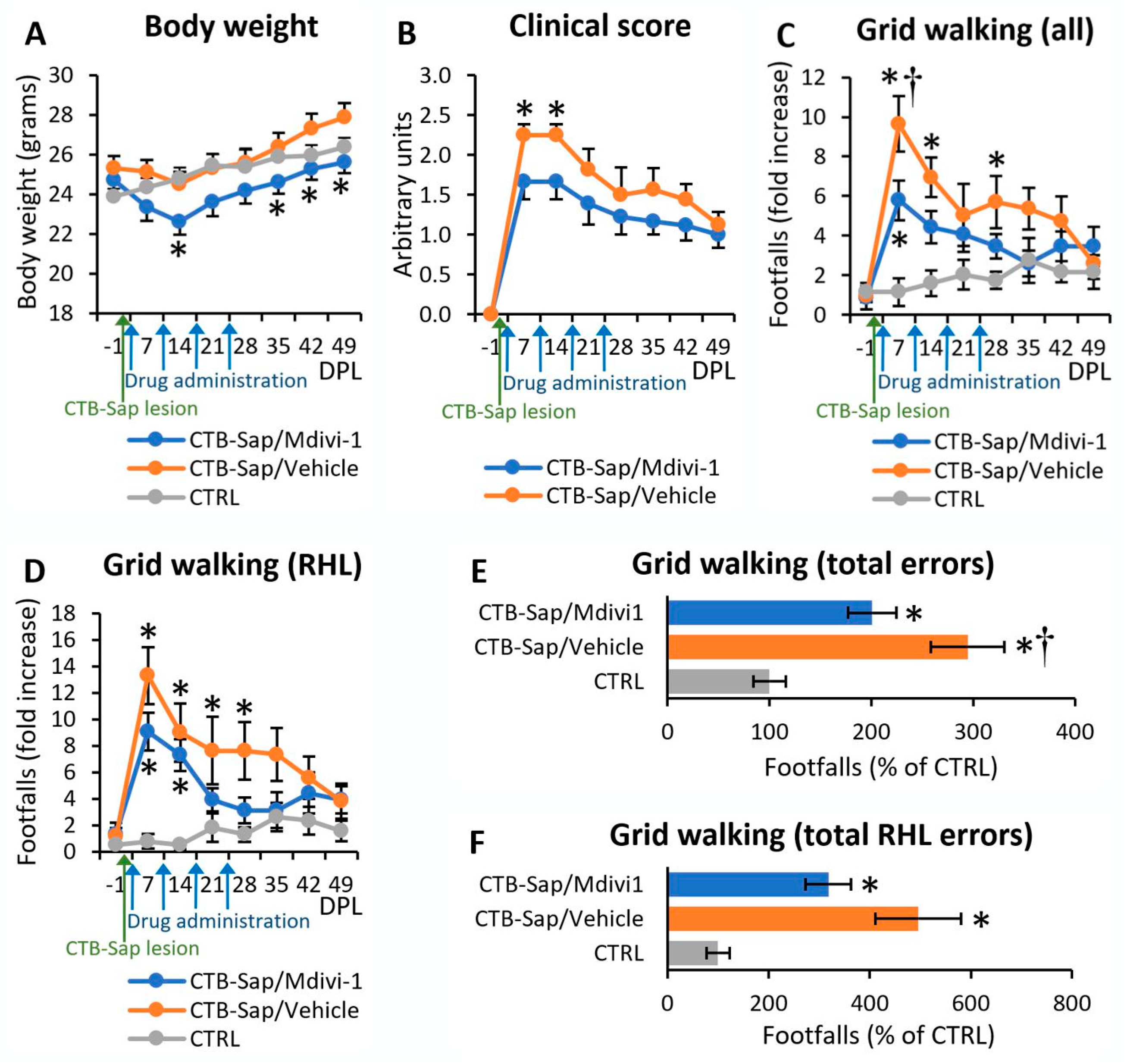
2.1. Mdivi-1 Reduced Motor Deficits after CTB-Sap Lesion
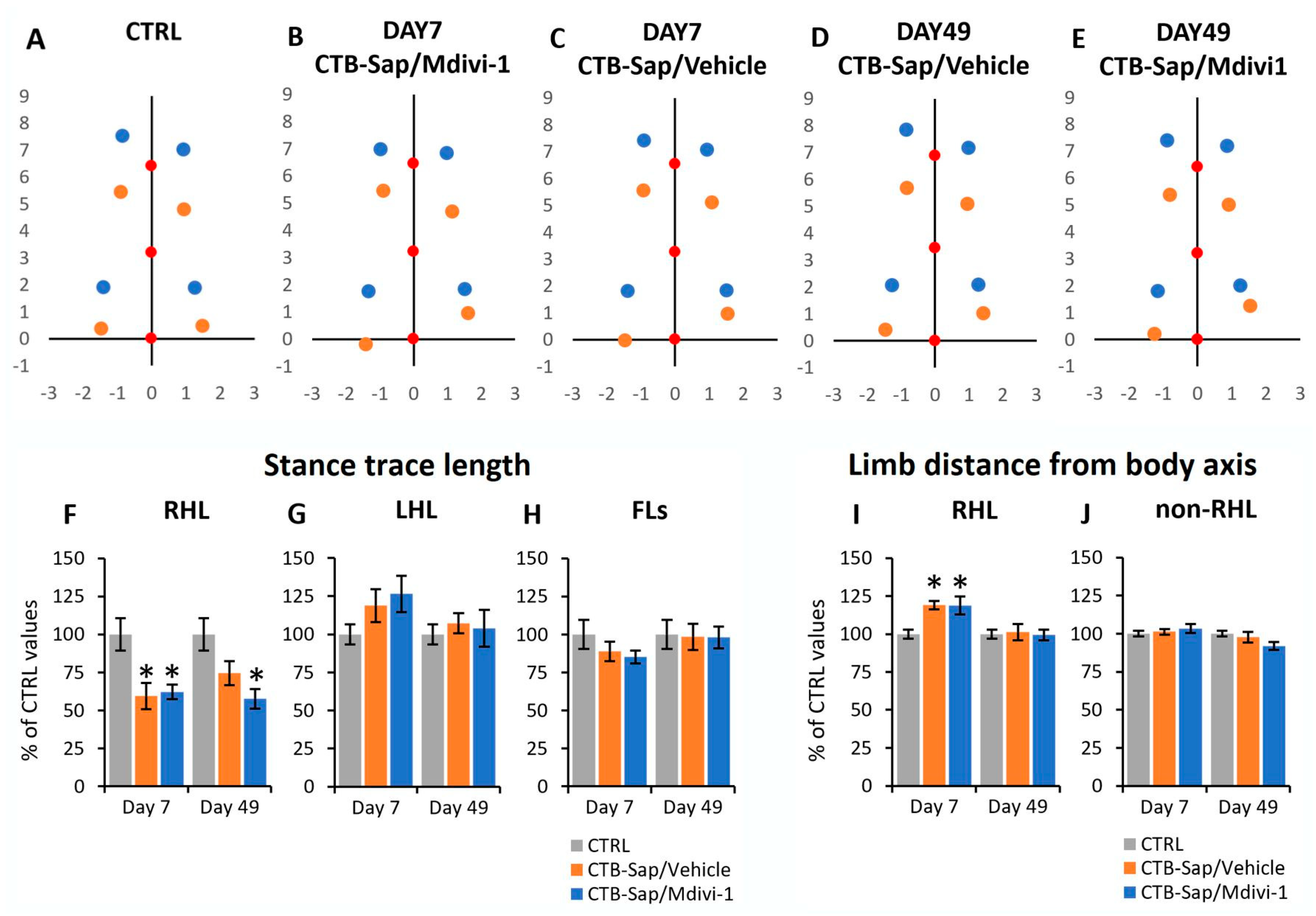
2.2. Effects of CTB-Sap Lesion and Drug Treatment on the Gait Parameters
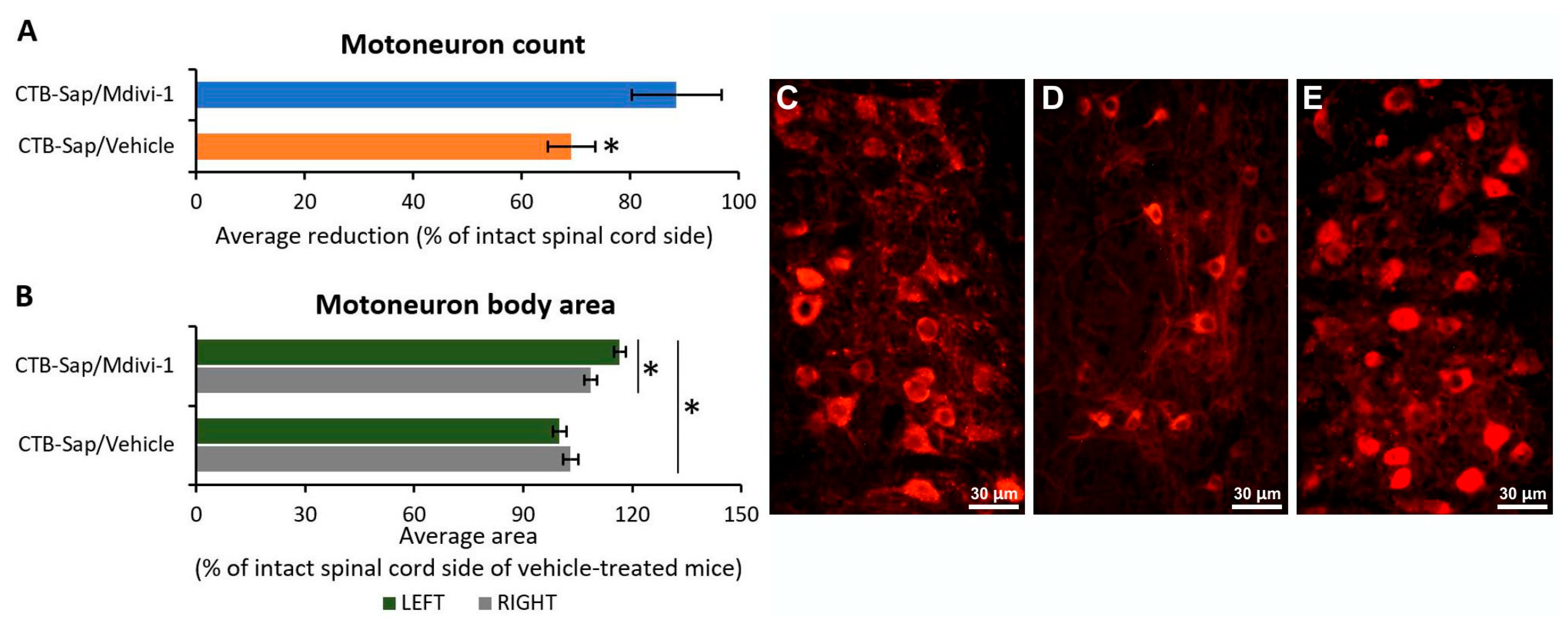
2.3. Mdivi-1 Treatment Has Neuroprotective Effects on Motoneurons and Promotes Their Plasticity
2.4. Effects of CTB-Sap Lesion and Drug Treatment on Synaptic Function
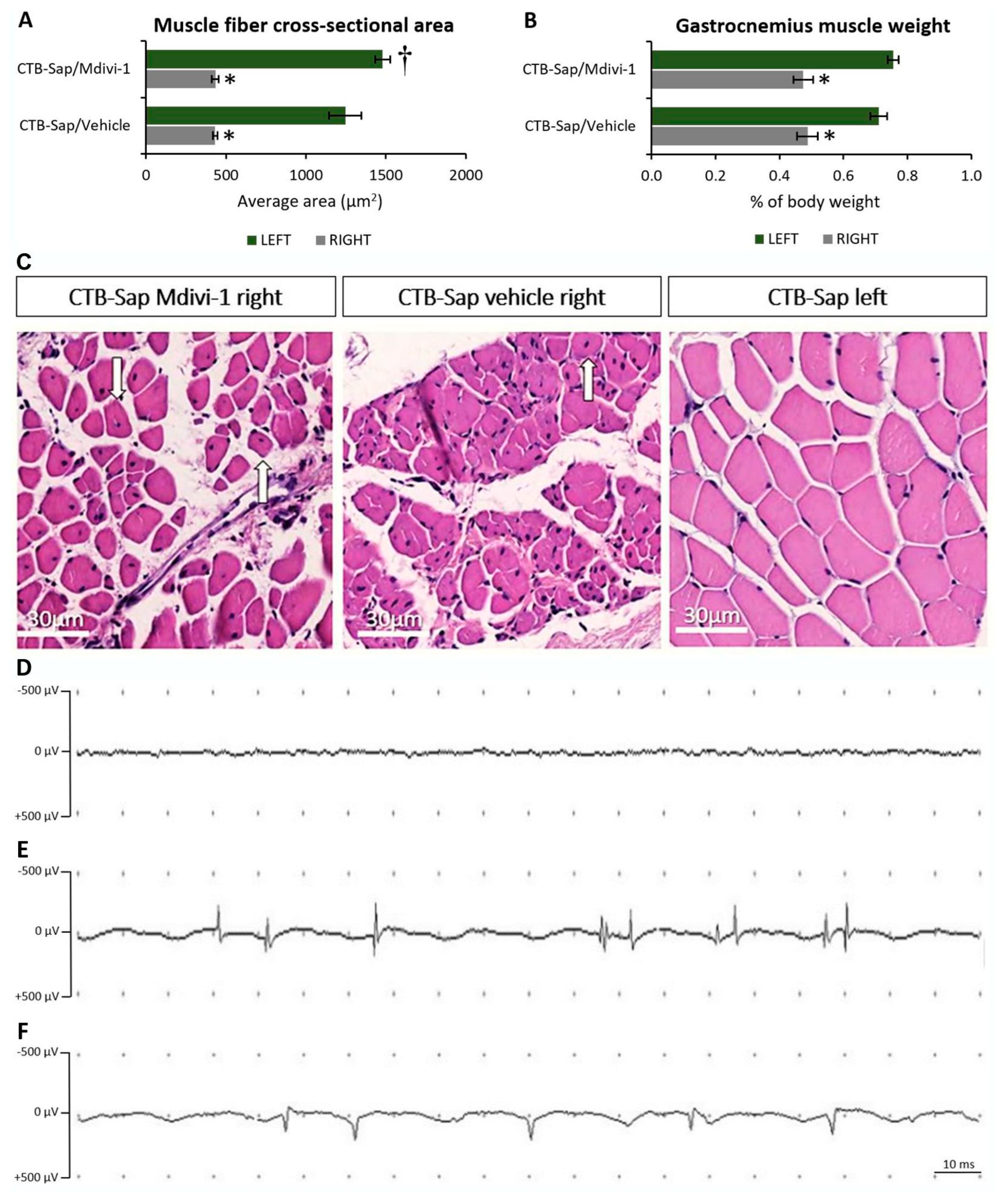
2.5. Muscle Denervation Leads to Atrophy
3. Discussion
4. Materials and Methods
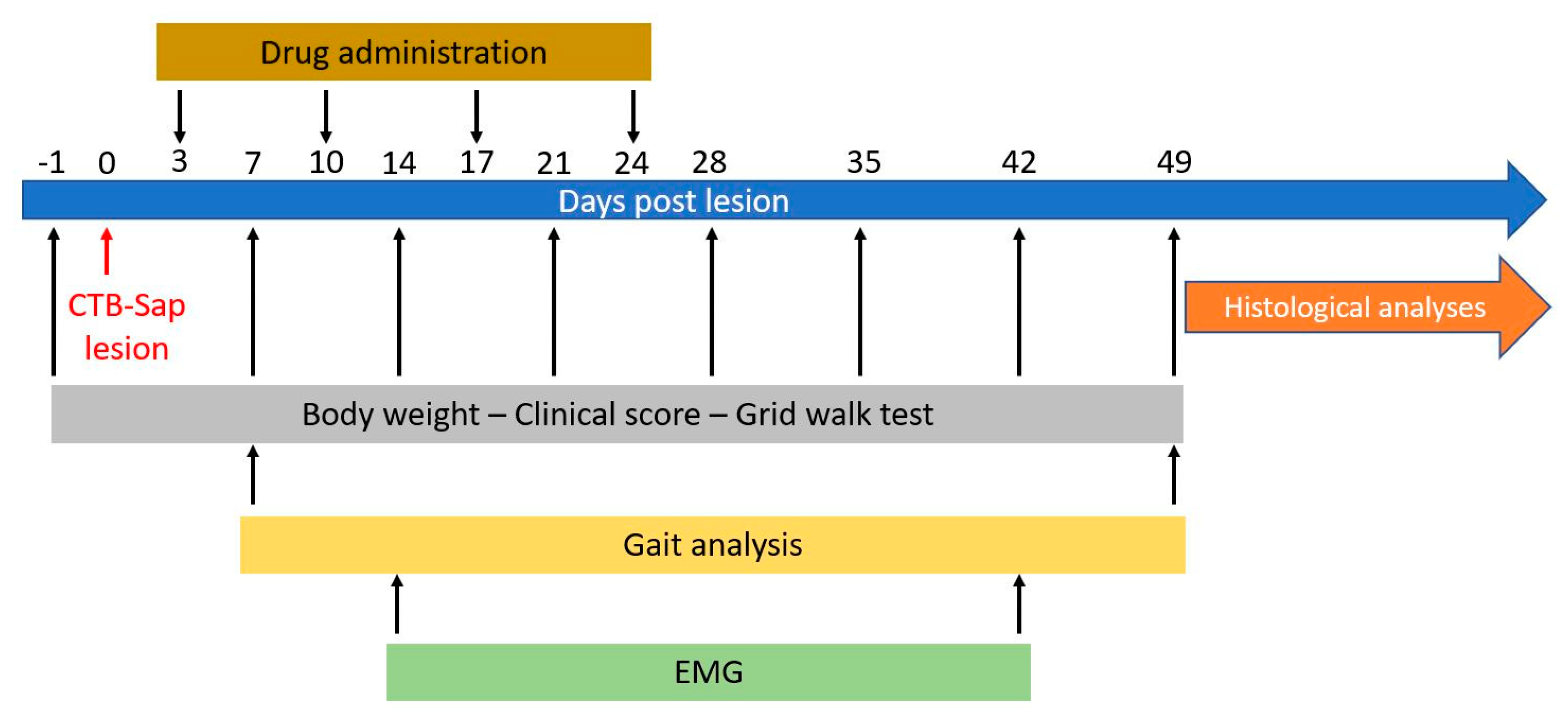
4.1. Animal Model
4.2. Behavioral Tests
- -
- score 0 = healthy;
- -
- score 1 = during the tail suspension test, collapse or partial collapse of leg extension towards the lateral midline;
- -
- score 2 = toes curl under at least twice during walking of 30 cm, or any part of the foot is dragging along the table;
- -
- score 3 = rigid paralysis or minimal joint movement, or foot not being used for generating forward motion.
4.3. Electromyography
4.4. Immunofluorescence Staining
4.5. Measurements of Motoneuron Number and Body Size
4.6. Hematoxylin and Eosin Staining
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chiò, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G.; Eurals Consortium. Prognostic factors in ALS: A critical review. Amyotroph. Lateral Scler. 2009, 10, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Mazzini, L.; D’Alfonso, S.; Corrado, L.; Canosa, A.; Moglia, C.; Manera, U.; Bersano, E.; Brunetti, M.; Barberis, M.; et al. The multistep hypothesis of ALS revisited: The role of genetic mutations. Neurology 2018, 91, e635–e642. [Google Scholar] [CrossRef] [PubMed]
- Ciuro, M.; Sangiorgio, M.; Leanza, G.; Gulino, R. A Meta-Analysis Study of SOD1-Mutant Mouse Models of ALS to Analyse the Determinants of Disease Onset and Progression. Int. J. Mol. Sci. 2022, 24, 216. [Google Scholar] [CrossRef] [PubMed]
- Gordon, P.H. Amyotrophic lateral sclerosis: An update for 2013 clinical features, pathophysiology, management and therapeutic trials. Aging Dis. 2013, 4, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Shatunov, A.; Al-Chalabi, A. The genetic architecture of ALS. Neurobiol. Dis. 2021, 147, 105156. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Calvo, A.; Chio, A.; Colville, S.; Ellis, C.M.; Hardiman, O.; Heverin, M.; Howard, R.S.; Huisman, M.H.B.; Keren, N.; et al. Analysis of amyotrophic lateral sclerosis as a multistep process: A population-based modelling study. Lancet Neurol. 2014, 13, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; Al-Chalabi, A.; Chiò, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Körner, S.; Kollewe, K.; Fahlbusch, M.; Zapf, A.; Dengler, R.; Krampfl, K.; Petri, S. Onset and spreading patterns of upper and lower motor neuron symptoms in amyotrophic lateral sclerosis. Muscle Nerve 2011, 43, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef]
- Verma, S.; Khurana, S.; Vats, A.; Sahu, B.; Ganguly, N.K.; Chakraborti, P.; Gourie-Devi, M.; Taneja, V. Neuromuscular junction dysfunction in amyotrophic lateral sclerosis. Mol. Neurobiol. 2022, 59, 1502–1527. [Google Scholar] [CrossRef] [PubMed]
- Tröger, M.; Dengler, R. The role of electromyography (EMG) in the diagnosis of ALS. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1 (Suppl. 2), S33–S40. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn-Smith, I.J.; Martin, C.L.; Arnolda, L.F.; Minson, J.B. Tracer-toxins: Cholera toxin B-saporin as a model. J. Neurosci. Method 2000, 103, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Lind, L.A.; Murphy, E.R.; Lever, T.E.; Nichols, N.L. Hypoglossal motor neuron death via intralingual CTB-saporin (CTB-SAP) injections mimic aspects of amyotrophic lateral sclerosis (ALS) related to dysphagia. Neuroscience 2018, 390, 303–316. [Google Scholar] [CrossRef]
- Murphy, E.R.; Thompson, R.; Osman, K.L.; Haxton, C.; Brothers, M.; Lee, L.; Warncke, K.; Smith, C.L.; Keilholz, A.N.; Hamad, A.; et al. A strength endurance exercise paradigm mitigates deficits in hypoglossal-tongue axis function, strength, and structure in a rodent model of hypoglossal motor neuron degeneration. Front. Neurosci. 2022, 16, 869592. [Google Scholar] [CrossRef]
- Borkowski, L.; Keilholz, A.N.; Smith, C.L.; Canda, K.A.; Nichols, N.L. Nonsteroidal anti-inflammatory drug (ketoprofen)delivery differentially impacts phrenic long-term facilitation in rats with motor neuron death induced by intrapleural CTB-SAP injections. Exp. Neurol. 2022, 347, 113892. [Google Scholar] [CrossRef]
- Nichols, N.L.; Craig, T.A.; Tanner, M.A. Phrenic long-term facilitation following intrapleural CTB-SAP-induced respiratory motor neuron death. Respir. Physiol. Neurobiol. 2018, 256, 43–49. [Google Scholar] [CrossRef]
- Gulino, R.; Gulisano, M. Involvement of brain-derived neurotrophic factor and sonic hedgehog in the spinal cord plasticity after neurotoxic partial removal of lumbar motoneurons. Neurosci. Res. 2012, 72, 238–247. [Google Scholar] [CrossRef]
- Gulino, R.; Gulisano, M. Noggin and Sonic hedgehog are involved in compensatory changes within the motoneuron-depleted mouse spinal cord. J. Neurol. Sci. 2013, 332, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Gulino, R.; Perciavalle, V.; Gulisano, M. Expression of cell fate determinants and plastic changes after neurotoxic lesion of adult mice spinal cord by cholera toxin-B saporin. Eur. J. Neurosci. 2010, 31, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Gulino, R.; Forte, S.; Parenti, R.; Gulisano, M. TDP-43 as a Modulator of Synaptic Plasticity in a Mouse Model of Spinal Motoneuron Degeneration. CNS Neurol. Disord. Drug Targets 2015, 14, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Gulino, R.; Vicario, N.; Giunta, M.A.S.; Spoto, G.; Calabrese, G.; Vecchio, M.; Gulisano, M.; Leanza, G.; Parenti, R. Neuromuscular Plasticity in a Mouse Neurotoxic Model of Spinal Motoneuronal Loss. Int. J. Mol. Sci. 2019, 20, E1500. [Google Scholar] [CrossRef] [PubMed]
- Vicario, N.; Spitale, F.M.; Tibullo, D.; Giallongo, C.; Amorini, A.M.; Scandura, G.; Spoto, G.; Saab, M.W.; D’Aprile, S.; Alberghina, C.; et al. Clobetasol promotes neuromuscular plasticity in mice after motoneuronal loss via sonic hedgehog signaling, immunomodulation and metabolic rebalancing. Cell Death Dis. 2021, 12, 625. [Google Scholar] [CrossRef]
- Gulino, R. Neuroplasticity and repair in rodent neurotoxic models of spinal motoneuron disease. Neural Plast. 2016, 2016, 2769735. [Google Scholar] [CrossRef] [PubMed]
- Giusto, E.; Codrich, M.; de Leo, G.; Francardo, V.; Coradazzi, M.; Parenti, R.; Gulisano, M.; Vicario, N.; Gulino, R.; Leanza, G. Compensatory changes in degenerating spinal motoneurons sustain functional sparing in the SOD1-G93A mouse model of amyotrophic lateral sclerosis. J. Comp. Neurol. 2020, 528, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Gulino, R. Synaptic dysfunction and plasticity in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2023, 24, 4613. [Google Scholar] [CrossRef]
- Taylor, H.B.C.; Jeans, A.F. Friend or foe? The varied faces of homeostatic synaptic plasticity in neurodegenerative disease. Front. Cell Neurosci. 2021, 15, 782768. [Google Scholar] [CrossRef]
- Klemmensen, M.M.; Borrowman, S.H.; Pearce, C.; Pyles, B.; Chandra, B. Mitochondrial dysfunction in neurodegenerative disorders. Neurotherapeutics 2024, 21, e00292. [Google Scholar] [CrossRef]
- Ratano, P.; Cocozza, G.; Pinchera, C.; Busdraghi, L.M.; Cantando, I.; Martinello, K.; Scioli, M.; Rosito, M.; Bezzi, P.; Fucile, S.; et al. Reduction of inflammation and mitochondrial degeneration in mutant SOD1 mice through inhibition of voltage-gated potassium channel Kv1.3. Front. Mol. Neurosci. 2024, 16, 1333745. [Google Scholar] [CrossRef]
- Salvatori, I.; Nesci, V.; Spalloni, A.; Marabitti, V.; Muzzi, M.; Zenuni, H.; Scaricamazza, S.; Rosina, M.; Fenili, G.; Goglia, M.; et al. Trimetazidine improves mitochondrial dysfunction in SOD1G93A cellular models of amyotrophic lateral sclerosis through autophagy activation. Int. J. Mol. Sci. 2024, 25, 3251. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Yi, J.; Ma, C.; Xiao, Y.; Yi, F.; Yu, T.; Zhou, J. Defective mitochondrial dynamics is an early event in skeletal muscle of an amyotrophic lateral sclerosis mouse model. PLoS ONE 2013, 8, e82112. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Huang, Z.; Xie, F.; Chen, L. Dynamin-related protein 1: A critical protein in the pathogenesis of neural system dysfunctions and neurodegenerative diseases. J. Cell Physiol. 2019, 234, 10032–10046. [Google Scholar] [CrossRef]
- Genin, E.C.; Abou-Ali, M.; Paquis-Flucklinger, V. Mitochondria, a Key Target in Amyotrophic Lateral Sclerosis Pathogenesis. Genes 2023, 14, 1981. [Google Scholar] [CrossRef] [PubMed]
- Bido, S.; Soria, F.N.; Fan, R.Z.; Bezard, E.; Tieu, K. Mitochondrial division inhibitor-1 is neuroprotective in the A53T-α-synuclein rat model of Parkinson’s disease. Sci. Rep. 2017, 7, 7495. [Google Scholar] [CrossRef]
- Liu, X.; Song, L.; Yu, J.; Huang, F.; Li, Y.; Ma, C. Mdivi-1: A promising drug and its underlying mechanisms in the treatment of neurodegenerative diseases. Histol. Histopathol. 2022, 37, 505–512. [Google Scholar] [CrossRef]
- Albano, R.; Liu, X.; Lobner, D. Regulation of system x(c)- in the SOD1-G93A mouse model of ALS. Exp. Neurol. 2013, 250, 69–73. [Google Scholar] [CrossRef]
- Mendes, C.S.; Bartos, I.; Márka, Z.; Akay, T.; Márka, S.; Mann, R.S. Quantification of gait parameters in freely walking rodents. BMC Biol. 2015, 22, 13–50. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Wang, B.; Hu, Y.; Wang, S.; Zhang, X. Abnormal Mitochondrial Quality Control in Neurodegenerative Diseases. Front. Cell Neurosci. 2020, 14, 138. [Google Scholar] [CrossRef] [PubMed]
- Dang, X.; Zhang, L.; Franco, A.; Dorn, G.W., II. Activating mitofusins interrupts mitochondrial degeneration and delays disease progression in SOD1 mutant amyotrophic lateral sclerosis. Hum. Mol. Genet. 2023, 32, 1208–1222. [Google Scholar] [CrossRef]
- Kodavati, M.; Wang, H.; Hegde, M.L. Altered Mitochondrial Dynamics in Motor Neuron Disease: An Emerging Perspective. Cells 2020, 9, 1065. [Google Scholar] [CrossRef] [PubMed]
- Petrozziello, T.; Bordt, E.A.; Mills, A.N.; Kim, S.E.; Sapp, E.; Devlin, B.A.; Obeng-Marnu, A.A.; Farhan, S.M.K.; Amaral, A.C.; Dujardin, S.; et al. Targeting tau mitigates mitochondrial fragmentation and oxidative stress in amyotrophic lateral sclerosis. Mol. Neurobiol. 2022, 59, 683–702. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Huang, Y.; Li, L. Drp1-Dependent Mitochondrial Fission Plays Critical Roles in Physiological and Pathological Progresses in Mammals. Int. J. Mol. Sci. 2017, 18, 144. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.U.; Saw, N.L.; Vogel, H.; Cunningham, A.D.; Shamloo, M.; Mochly-Rosen, D. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol. Med. 2018, 10, e8166. [Google Scholar] [CrossRef] [PubMed]
- Nemtsova, Y.; Steinert, B.L.; Wharton, K.A. Compartment specific mitochondrial dysfunction in Drosophila knock-in model of ALS reversed by altered gene expression of OXPHOS subunits and pro-fission factor Drp1. Mol. Cell Neurosci. 2023, 125, 103834. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, X.; Huo, Z.; Chen, Y.; Liu, J.; Zhao, Z.; Meng, F.; Su, Q.; Bao, W.; Zhang, L.; et al. The Impact of Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis. Cells 2022, 11, 2049. [Google Scholar] [CrossRef]
- Dukkipati, S.S.; Garrett, T.L.; Elbasiouny, S.M. The vulnerability of spinal motoneurons and soma size plasticity in a mouse model of amyotrophic lateral sclerosis. J. Physiol. 2018, 596, 1723–1745. [Google Scholar] [CrossRef]
- Mishra, A.; Singh, S.; Tiwari, V.; Bano, S.; Shukla, S. Dopamine D1 receptor agonism induces dynamin related protein-1 inhibition to improve mitochondrial biogenesis and dopaminergic neurogenesis in rat model of Parkinson’s disease. Behav. Brain Res. 2020, 378, 112304. [Google Scholar] [CrossRef]
- Zhang, X.L.; Huang, W.M.; Tang, P.C.; Sun, Y.; Zhang, X.; Qiu, L.; Yu, B.C.; Zhang, X.Y.; Hong, Y.X.; He, Y.; et al. Anti-inflammatory and neuroprotective effects of natural cordycepin in rotenone-induced PD models through inhibiting Drp1-mediated mitochondrial fission. Neurotoxicology 2021, 84, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Edgerton, V.R.; Tillakaratne, N.J.K.; Bigbee, A.J.; de Leon, R.D.; Roy, R.R. Plasticity of the spinal neural circuitry after injury. Annu. Rev. Neurosci. 2004, 27, 145–167. [Google Scholar] [CrossRef] [PubMed]
- Turrigiano, G. Homeostatic synaptic plasticity: Local and global mechanisms for stabilizing neuronal function. Cold Spring Harb. Perspect. Biol. 2012, 4, a005736. [Google Scholar] [CrossRef] [PubMed]
- Von Berhardi, R.; Eugenín-von Bernhardi, L.; Eugenín, J. What is neural plasticity? Adv. Exp. Med. Biol. 2017, 1015, 1–15. [Google Scholar] [CrossRef]
- Windhorst, U.; Dibaj, P. Plastic spinal motor circuits in health and disease. J. Integr. Neurosci. 2023, 22, 167. [Google Scholar] [CrossRef]
- Wolpaw, J.R.; Tennissen, A.M. Activity-dependent spinal cord plasticity in health and disease. Annu. Rev. Neurosci. 2001, 24, 807–843. [Google Scholar] [CrossRef]
- Folker, E.S.; Baylies, M.K. Nuclear positioning in muscle development and disease. Front. Physiol. 2013, 4, 363. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciuro, M.; Sangiorgio, M.; Cacciato, V.; Cantone, G.; Fichera, C.; Salvatorelli, L.; Magro, G.; Leanza, G.; Vecchio, M.; Valle, M.S.; et al. Mitigating the Functional Deficit after Neurotoxic Motoneuronal Loss by an Inhibitor of Mitochondrial Fission. Int. J. Mol. Sci. 2024, 25, 7059. https://doi.org/10.3390/ijms25137059
Ciuro M, Sangiorgio M, Cacciato V, Cantone G, Fichera C, Salvatorelli L, Magro G, Leanza G, Vecchio M, Valle MS, et al. Mitigating the Functional Deficit after Neurotoxic Motoneuronal Loss by an Inhibitor of Mitochondrial Fission. International Journal of Molecular Sciences. 2024; 25(13):7059. https://doi.org/10.3390/ijms25137059
Chicago/Turabian StyleCiuro, Maria, Maria Sangiorgio, Valeria Cacciato, Giuliano Cantone, Carlo Fichera, Lucia Salvatorelli, Gaetano Magro, Giampiero Leanza, Michele Vecchio, Maria Stella Valle, and et al. 2024. "Mitigating the Functional Deficit after Neurotoxic Motoneuronal Loss by an Inhibitor of Mitochondrial Fission" International Journal of Molecular Sciences 25, no. 13: 7059. https://doi.org/10.3390/ijms25137059