
Transcriptome Analysis Reveals the Role of Sucrose in the Production of Latilactobacillus sakei L3 Exopolysaccharide



Abstract
:1. Introduction
2. Results and Discussion
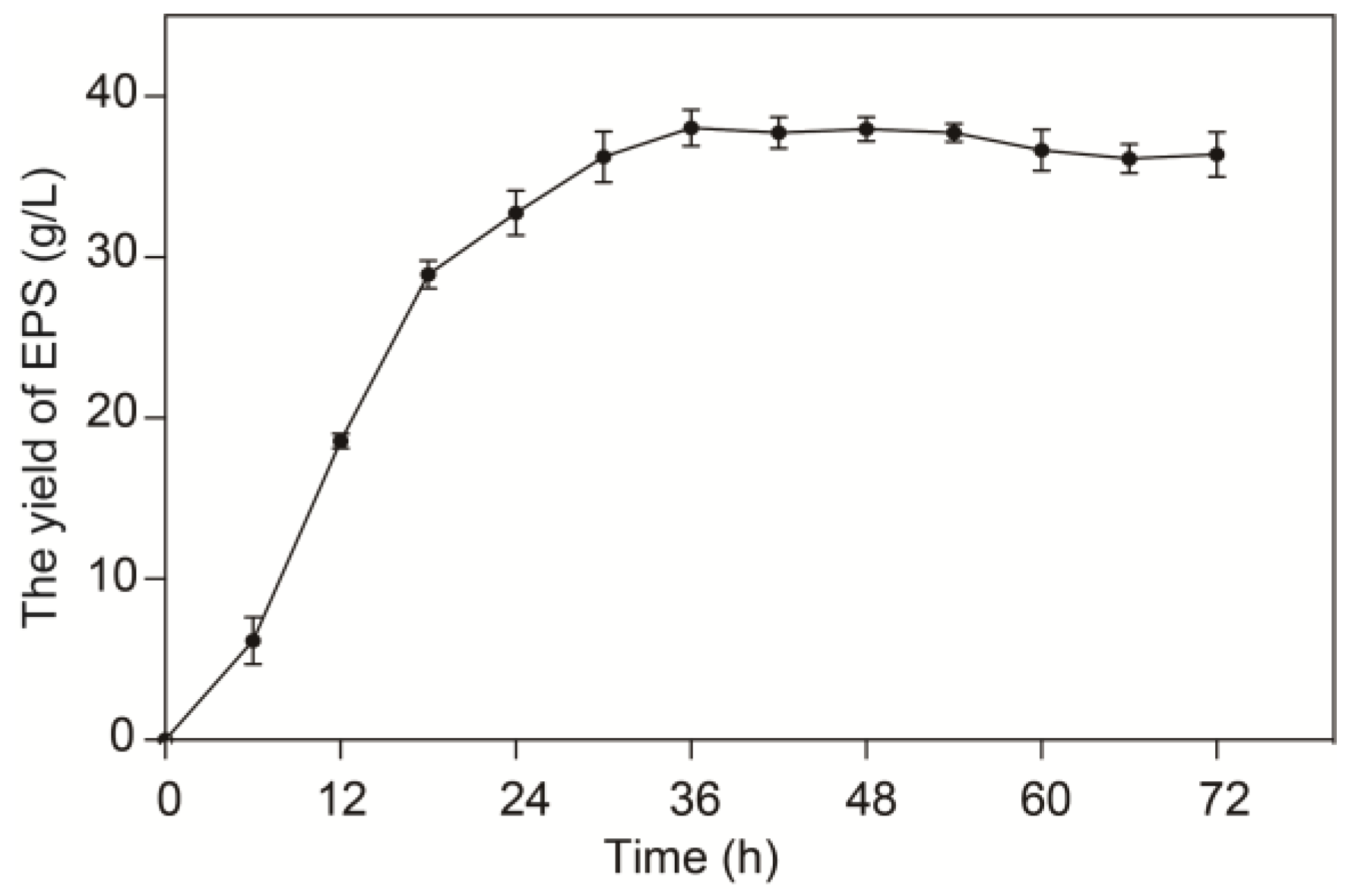
2.1. Growth Characteristics of L. sakei L3
2.2. Sequencing Quality Assessment and Comparison with Reference Genomes
2.3. Screening of DEGs between MRS and MRS-S Groups
2.4. GO Annotations Analysis
2.5. KEGG Pathway Analysis
2.6. Critical DEG Analysis under Sucrose-Added Fermentation
2.6.1. The Positive Regulation of Genes Involved in Sucrose Metabolism in the MRS-S Group
2.6.2. The Negative Regulation of Genes Involved in the Uridine Monophosphate (UMP) Pathway in the MRS-S Group
2.6.3. The Negative Regulation of Genes Involved in the Fatty Acid Synthetic Pathway in the MRS-S Group
2.7. The Analysis of ncRNA and Its Target Genes during Sucrose-Added Fermentation
2.8. Validation of Typical DEGs Using RT-qPCR
3. Materials and Methods
3.1. Bacterial Strains and Culture Conditions
3.2. Biological Characteristics Analysis of L. sakei L3
3.2.1. Morphological Analysis
3.2.2. Biochemical Characteristics Tests
3.3. Transcriptomic Sequencing Analysis
3.3.1. Sample Preparation
3.3.2. Transcriptome Sequencing Data Processing
3.3.3. Functional Analysis of DEGs
3.3.4. Molecular Docking Analysis
3.3.5. ncRNA Target Genes and Interaction Network Analysis
3.3.6. Bioinformatic Analysis of TTP Encoded by LCA_RS09375
3.3.7. Real-Time Fluorescence Quantitative PCR (RT-qPCR) Verification of DEGs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peng, K.; Koubaa, M.; Bals, O.; Vorobiev, E. Recent insights in the impact of emerging technologies on lactic acid bacteria: A review. Food Res. Int. 2020, 137, 109544. [Google Scholar] [CrossRef]
- Leroy, F.; De Vuyst, L. Lactic acid bacteria as functional starter cultures for the food fermentation industry. Trends Food Sci. Technol. 2004, 15, 67–78. [Google Scholar] [CrossRef]
- Tarique, M.; Ali, A.H.; Kizhakkayil, J.; Gan, R.-Y.; Liu, S.-Q.; Kamal-Eldin, A.; Ayyash, M. Investigating the biological activities and prebiotic potential of exopolysaccharides Produced by Lactobacillus delbrueckii and Lacticaseibacillus rhamnosus: Implications for gut microbiota modulation and rheological properties in fermented milk. Food Hydrocoll. Health 2023, 4, 100162. [Google Scholar] [CrossRef]
- Laiño, J.; Villena, J.; Kanmani, P.; Kitazawa, H. Immunoregulatory effects triggered by lactic acid bacteria exopolysaccharides: New insights into molecular interactions with host cells. Microorganisms 2016, 4, 27. [Google Scholar] [CrossRef]
- Sharma, H.; Ozogul, F.; Bartkiene, E.; Rocha, J.M. Impact of lactic acid bacteria and their metabolites on the techno-functional properties and health benefits of fermented dairy products. Crit. Rev. Food Sci. 2023, 63, 4819–4841. [Google Scholar] [CrossRef] [PubMed]
- Zagorec, M.; Champomier-Vergès, M.-C. Lactobacillus sakei: A starter for sausage fermentation, a protective culture for meat products. Microorganisms 2017, 5, 56. [Google Scholar] [CrossRef]
- Bajpai, V.K.; Rather, I.A.; Park, Y.-H. Partially purified exopolysaccharide from Lactobacillus Sakei Probio 65 with antioxidant, α-glucosidase and tyrosinase inhibitory potential. J. Food Biochem. 2016, 40, 264–274. [Google Scholar] [CrossRef]
- Boels, I.C.; van Kranenburg, R.; Hugenholtz, J.; Kleerebezem, M.; de Vos, W.M. Sugar catabolism and its impact on the biosynthesis and engineering of exopolysaccharide production in lactic acid bacteria. Int. Dairy J. 2001, 11, 723–732. [Google Scholar] [CrossRef]
- Abd-Alla, M.H.; Bashandy, S.R.; Nafady, N.A.; Hassan, A.A. Enhancement of exopolysaccharide production by Stenotrophomonas maltophilia and Brevibacillus parabrevis isolated from root nodules of Cicer arietinum L. and Vigna unguiculata L. (Walp.) plants. Rend. Lincei. Sci. Fis. Nat. 2018, 29, 117–129. [Google Scholar] [CrossRef]
- Cheng, X.; Huang, L.; Li, K.-t. Antioxidant activity changes of exopolysaccharides with different carbon sources from Lactobacillus plantarum LPC-1 and its metabolomic analysis. World J. Microb. Biot. 2019, 35, 68. [Google Scholar] [CrossRef]
- Oshlack, A.; Robinson, M.D.; Young, M.D. From RNA-seq reads to differential expression results. Genome Biol. 2010, 11, 220. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Peng, Q.; Liu, Y.; Wu, N.; He, Y.; Cui, X.; Dan, T. Genomic and transcriptomic analysis of genes involved in exopolysaccharide biosynthesis by Streptococcus thermophilus IMAU20561 grown on different sources of nitrogen. Front. Microbiol. 2024, 14, 1328824. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, A.; Tong, Y.; Wu, Q.; Zhang, J.; Shah, N.P. Transcriptomic insights into the growth phase- and sugar-associated changes in the exopolysaccharide production of a high eps-producing Streptococcus thermophilus ASCC 1275. Front. Microbiol. 2018, 9, 1919. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Song, Q.; Zhao, F.; Zhang, L.; Han, Y.; Zhou, Z. Isolation and characterization of dextran produced by Lactobacillus sakei L3 from Hubei sausage. Carbohyd. Polym. 2019, 223, 115111. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Song, Q.; Zhao, F.; Han, Y.; Zhou, Z. Production optimization, partial characterization and properties of an exopolysaccharide from Lactobacillus sakei L3. Int. J. Biol. Macromol. 2019, 141, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Jawan, R.; Abbasiliasi, S.; Mustafa, S.; Kapri, M.R.; Halim, M.; Ariff, A.B. In vitro evaluation of potential probiotic strain Lactococcus lactis Gh1 and its bacteriocin-like inhibitory substances for potential use in the food industry. Probiotics Antimicrob. Proteins 2021, 13, 422–440. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Park, M.H.; Kim, S.H. Selection and characterization of probiotic bacteria exhibiting antiadipogenic potential in 3T3-L1 preadipocytes. Probiotics Antimicrob. Proteins 2022, 14, 72–86. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tian, Y.; Cao, Y.; Li, J.; Guo, H.; Su, Y.; Tian, Y.; Wang, C.; Wang, T.; Zhang, L. Probiotic properties of Lactobacillus paracasei subsp. paracasei L1 and its growth performance-promotion in chicken by improving the intestinal microflora. Front. Physiol. 2019, 10, 937. [Google Scholar] [CrossRef]
- Mao, Q.; Sun, X.; Sun, J.; Zhang, F.; Lv, A.; Hu, X.; Guo, Y. A candidate probiotic strain of Enterococcus faecium from the intestine of the crucian carp Carassius auratus. AMB Express 2020, 10, 40. [Google Scholar] [CrossRef]
- McLeod, A. Lactobacillus sakei Metabolism and Diversity. Ph.D. Thesis, Norwegian University of Life Sciences, As, Norway, 2010. [Google Scholar]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2022, 51, D587–D592. [Google Scholar] [CrossRef]
- Costa Oliveira, B.E.; Ricomini Filho, A.P.; Burne, R.A.; Zeng, L. The route of sucrose utilization by Streptococcus mutans affects intracellular polysaccharide metabolism. Front. Microbiol. 2021, 12, 636684. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Ren, R.; Sun, Y.; Xu, J.; Yang, H.; Li, X.; Xiao, Y.; Li, J.; Lyu, W. The combination of metagenome and metabolome to compare the differential effects and mechanisms of fructose and sucrose on the metabolic disorders and gut microbiota in vitro and in vivo. Food Funct. 2023, 14, 7284–7298. [Google Scholar] [CrossRef] [PubMed]
- Mataragas, M. Investigation of genomic characteristics and carbohydrates’ metabolic activity of Lactococcus lactis subsp. lactis during ripening of a Swiss-type cheese. Food Microbiol. 2020, 87, 103392. [Google Scholar] [CrossRef] [PubMed]
- Sreelekshmy, B.R.; Basheer, R.; Shibli, S.M.A. Exploration of bifurcated electron transfer mechanism in Bacillus cereus for enhanced power generation in double-chambered microbial fuel cells. J. Environ. Chem. Eng. 2022, 10, 107601. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, Q.; Chi, Y.; Liu, Z.; Liu, H.; Li, C.; Feng, X. Constructing an intensified UDP recycling system for the glycosylation of natural products by phosphorylation of byproduct fructose. J. Agric. Food Chem. 2023, 71, 9441–9450. [Google Scholar] [CrossRef]
- Hunsucker, S.A.; Mitchell, B.S.; Spychala, J. The 5′-nucleotidases as regulators of nucleotide and drug metabolism. Pharmacol. Therapeut. 2005, 107, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Abt, E.R.; Rosser, E.W.; Durst, M.A.; Lok, V.; Poddar, S.; Le, T.M.; Cho, A.; Kim, W.; Wei, L.; Song, J.; et al. Metabolic modifier screen reveals secondary targets of protein kinase inhibitors within nucleotide metabolism. Cell Chem. Biol. 2020, 27, 197–205.e6. [Google Scholar] [CrossRef]
- Fialho, D.M.; Roche, T.P.; Hud, N.V. Prebiotic syntheses of noncanonical nucleosides and nucleotides. Chem. Rev. 2020, 120, 4806–4830. [Google Scholar] [CrossRef]
- Goncheva, M.I.; Chin, D.; Heinrichs, D.E. Nucleotide biosynthesis: The base of bacterial pathogenesis. Trends Microbiol. 2022, 30, 793–804. [Google Scholar] [CrossRef]
- Guo, J.; Song, X.; Zou, L.-F.; Zou, H.-S.; Chen, G.-Y. The small and large subunits of carbamoyl-phosphate synthase exhibit diverse contributions to pathogenicity in Xanthomonas citri subsp. Citri. J. Integr. Agric. 2015, 14, 1338–1347. [Google Scholar] [CrossRef]
- Alqahtani, S.S.; Koltai, T.; Ibrahim, M.E.; Bashir, A.H.H.; Alhoufie, S.T.S.; Ahmed, S.B.M.; Molfetta, D.D.; Carvalho, T.M.A.; Cardone, R.A.; Reshkin, S.J.; et al. Role of pH in regulating cancer pyrimidine synthesis. J. Xenobiotics 2022, 12, 158–180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhu, S.; Zhang, C.; Soliman, M.M.; Li, H.; Liu, X. Transcriptome analysis revealing the mechanism of soybean protein isolates and soybean peptides on Lacticaseibacillus rhamnosus Lra05. Food Biosci. 2022, 47, 101681. [Google Scholar] [CrossRef]
- Fujita, Y.; Matsuoka, H.; Hirooka, K. Regulation of fatty acid metabolism in bacteria. Mol. Microbiol. 2007, 66, 829–839. [Google Scholar] [CrossRef]
- Rock, C.O. Chapter 3—Fatty acid and phospholipid metabolism in prokaryotes. In Biochemistry of Lipids, Lipoproteins and Membranes, 5th ed.; Vance, D.E., Vance, J.E., Eds.; Elsevier: San Diego, CA, USA, 2008; pp. 59–96. [Google Scholar]
- Brück, F.M.; Brummel, M.; Schuch, R.; Spener, F. In-vitro evidence for feed-back regulation of β-ketoacyl-acyl carrier protein synthase III in medium-chain fatty acid biosynthesis. Planta 1996, 198, 271–278. [Google Scholar] [CrossRef]
- Abbadi, A.; Brummel, M.; SchüTt, B.S.; Slabaugh, M.B.; Schuch, R.; Spener, F. Reaction mechanism of recombinant 3-oxoacyl-(acyl-carrier-protein) synthase III from Cuphea wrightii embryo, a fatty acid synthase type II condensing enzyme. Biochem. J. 1999, 345, 153–160. [Google Scholar] [CrossRef]
- Chen, X.; Huang, L. Computational model for ncRNA research. Brief. Bioinform. 2022, 23, bbac472. [Google Scholar] [CrossRef]
- Leitão, A.L.; Costa, M.C.; Gabriel, A.F.; Enguita, F.J. Interspecies communication in holobionts by non-coding RNA exchange. Int. J. Mol. Sci. 2020, 21, 2333. [Google Scholar] [CrossRef] [PubMed]
- Cody, V.; Luft, J.R.; Pangborn, W.; Toy, J.; Bognar, A.L. Purification and crystallization of Lactobacillus casei folylpolyglutamate synthetase expressed in Escherichia coli. J. Mol. Biol. 1992, 224, 1179–1180. [Google Scholar] [CrossRef]
- Srivastava, A.C.; Thompson, Y.G.; Singhal, J.; Stellern, J.; Srivastava, A.; Du, J.; O’Connor, T.R.; Riggs, A.D. Elimination of human folypolyglutamate synthetase alters programming and plasticity of somatic cells. FASEB J. 2019, 33, 13747–13761. [Google Scholar] [CrossRef]
- Okawa, F.; Hama, Y.; Zhang, S.; Morishita, H.; Yamamoto, H.; Levine, T.P.; Mizushima, N. Evolution and insights into the structure and function of the DedA superfamily containing TMEM41B and VMP1. J. Cell Sci. 2021, 134, jcs255877. [Google Scholar] [CrossRef]
- Schwarzenbacher, R.; McMullan, D.; Krishna, S.S.; Xu, Q.; Miller, M.D.; Canaves, J.M.; Elsliger, M.A.; Floyd, R.; Grzechnik, S.K.; Jaroszewski, L.; et al. Crystal structure of a glycerate kinase (TM1585) from Thermotoga maritima at 2.70 Å resolution reveals a new fold. Proteins 2006, 65, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Reher, M.; Bott, M.; Schönheit, P. Characterization of glycerate kinase (2-phosphoglycerate forming), a key enzyme of the nonphosphorylative Entner–Doudoroff pathway, from the thermoacidophilic euryarchaeon Picrophilus torridus. FEMS Microbiol. Lett. 2006, 259, 113–119. [Google Scholar] [CrossRef]
- Chistoserdova, L.; Lidstrom, M.E. Identification and mutation of a gene required for glycerate kinase activity from a facultative methylotroph, Methylobacterium extorquens AM1. J. Bacteriol. 1997, 179, 4946–4948. [Google Scholar] [CrossRef]
- Hansen Robert, W.; Hayashi James, A. Glycolate metabolism in Escherichia coli. J. Bacteriol. 1962, 83, 679–687. [Google Scholar] [CrossRef]
- DuBois, M.; Gilles, K.A.; Hamilton, J.K.; Rebers, P.A.; Smith, F. Colorimetric method for determination of sugars and related substances. Anal. Chem. 1956, 28, 350–356. [Google Scholar] [CrossRef]
- Moyes, R.B.; Reynolds, J.; Breakwell, D.P. Differential staining of bacteria: Gram stain. Curr. Protoc. Microbiol. 2009, 15, A.3C.1–A.3C.8. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model 1999, 17, 57–61. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Tafer, H.; Hofacker, I.L. RNAplex: A fast tool for RNA–RNA interaction search. Bioinformatics 2008, 24, 2657–2663. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Chai, J.; Yuan, S.; Mai, C.; Cai, L.; Murphy, R.W.; Zhou, W.; Luo, J. VennPainter: A Tool for the comparison and identification of candidate genes based on venn diagrams. PLoS ONE 2016, 11, e0154315. [Google Scholar] [CrossRef] [PubMed]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2018, 47, D607–D613. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Raw Reads (Mb) | Total Clean Reads (Mb) | Total Clean Bases (Gb) | Clean Reads Q20 (%) | Clean Reads Q30 (%) | Clean Reads Ratio (%) |
|---|---|---|---|---|---|---|
| MRS | 24.88 ± 0 | 24.28 ± 0.07 | 2.43 ± 0.01 | 98.98 ± 0.08 | 96.92 ± 0.20 | 97.58 ± 0.30 |
| MRS-S | 24.88 ± 0 | 24.21 ± 0.16 | 2.42 ± 0.01 | 99.01 ± 0.01 | 96.99 ± 0.02 | 97.29 ± 0.63 |
| Sample | Total Clean Reads (Mb) | Total Mapping (%) | Unique Mapping (%) |
|---|---|---|---|
| MRS | 24.28 ± 0.07 | 78.36 ± 0.81 | 62.88 ± 6.99 |
| MRS-S | 24.21 ± 0.16 | 82.13 ± 0.08 | 48.15 ± 2.00 |
| Gene ID | log2(Fold-Change) | Description |
|---|---|---|
| LCA_RS05240 | 2.09 | PTS fructose transporter subunit IIC |
| LCA_RS08975 | 6.66 | Fructokinase |
| LCA_RS08985 | 7.95 | PTS sucrose transporter subunit IIABC |
| LCA_RS08990 | 4.84 | Sucrose-6-phosphate hydrolase |
| LCA_RS04780 | −1.55 | Carbamoyl-phosphate synthase large chain |
| LCA_RS04775 | −1.53 | Carbamoyl-phosphate synthase small subunit |
| LCA_RS04765 | −1.58 | Aspartate carbamoyltransferase |
| LCA_RS04770 | −2.03 | Dihydroorotase |
| LCA_RS04790 | −2.27 | Dihydroorotate dehydrogenase |
| LCA_RS04785 | −1.73 | Dihydroorotate dehydrogenase electron transfer subunit |
| LCA_RS04800 | −1.96 | Orotate phosphoribosyltransferase |
| LCA_RS04795 | −2.85 | Orotidine-5′-phosphate decarboxylase |
| LCA_RS04115 | −1.12 | Hypothetical protein |
| LCA_RS04105 | −1.79 | 3-oxoacyl-ACP synthase III |
| LCA_RS04125 | −1.57 | Beta-ketoacyl-[acyl-carrier-protein] synthase II |
| LCA_RS00775 | −1.6 | Oxidoreductase |
| LCA_RS04120 | −1.65 | Beta-ketoacyl-ACP reductase |
| LCA_RS02325 | −1.58 | NAD(P)-dependent dehydrogenase |
| LCA_RS04135 | −1.54 | Beta-hydroxyacyl-ACP dehydratase |
| LCA_RS03600 | −1.69 | Glycerate kinase |
| LCA_RS05480 | −1.32 | Folylpolyglutamate synthase |
| LCA_RS09375 | −1.04 | TVP38/TMEM64 family protein |
| LCA_RS05495 | 1.38 | Dihydroneopterin aldolase |
| LCA_RS05475 | −1.48 | Dihydropteroate synthase |
| LCA_RS05000 | −1.31 | Dihydrofolate reductase |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, B.; Wu, B.; Xu, M.; Zuo, K.; Han, Y.; Zhou, Z. Transcriptome Analysis Reveals the Role of Sucrose in the Production of Latilactobacillus sakei L3 Exopolysaccharide. Int. J. Mol. Sci. 2024, 25, 7185. https://doi.org/10.3390/ijms25137185
Wang B, Wu B, Xu M, Zuo K, Han Y, Zhou Z. Transcriptome Analysis Reveals the Role of Sucrose in the Production of Latilactobacillus sakei L3 Exopolysaccharide. International Journal of Molecular Sciences. 2024; 25(13):7185. https://doi.org/10.3390/ijms25137185
Chicago/Turabian StyleWang, Binbin, Baomei Wu, Min Xu, Kaiyue Zuo, Ye Han, and Zhijiang Zhou. 2024. "Transcriptome Analysis Reveals the Role of Sucrose in the Production of Latilactobacillus sakei L3 Exopolysaccharide" International Journal of Molecular Sciences 25, no. 13: 7185. https://doi.org/10.3390/ijms25137185