

Production of Spinocerebellar Ataxia Type 3 Model Mice by Intravenous Injection of AAV-PHP.B Vectors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
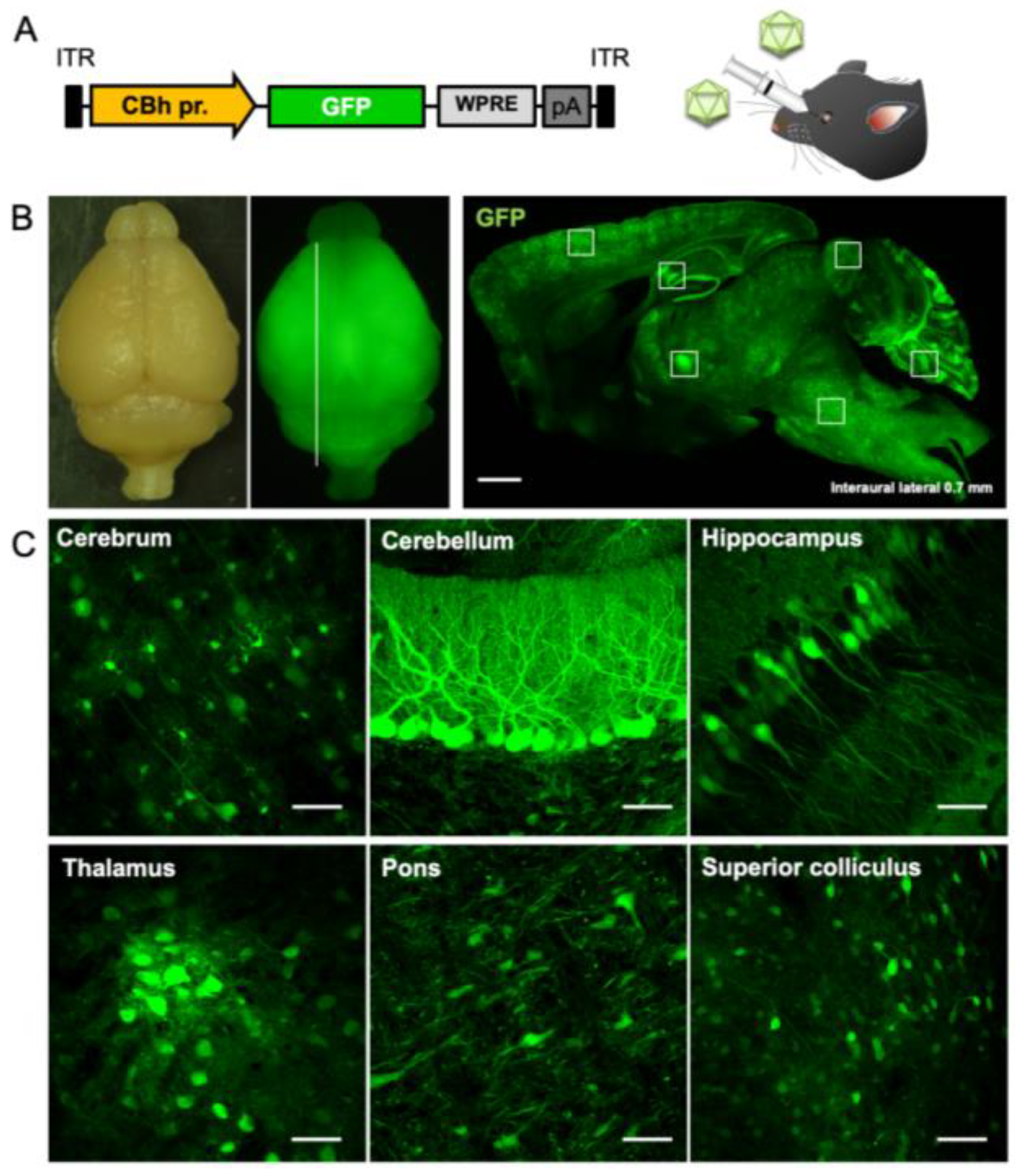
2.1. Efficient and Broad Transgene Expression in the Brain by Intravenous Injection of AAV-PHP.B
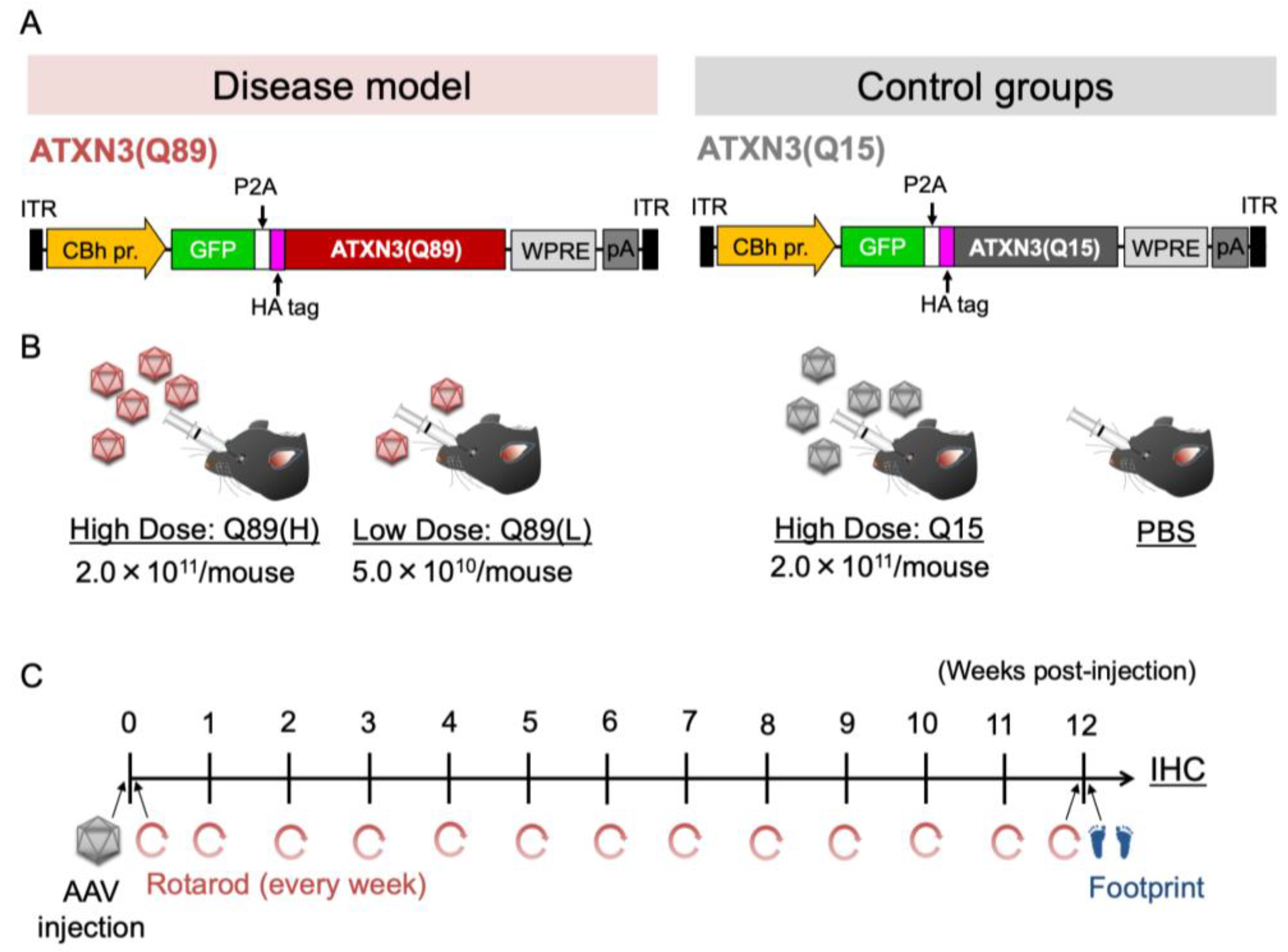
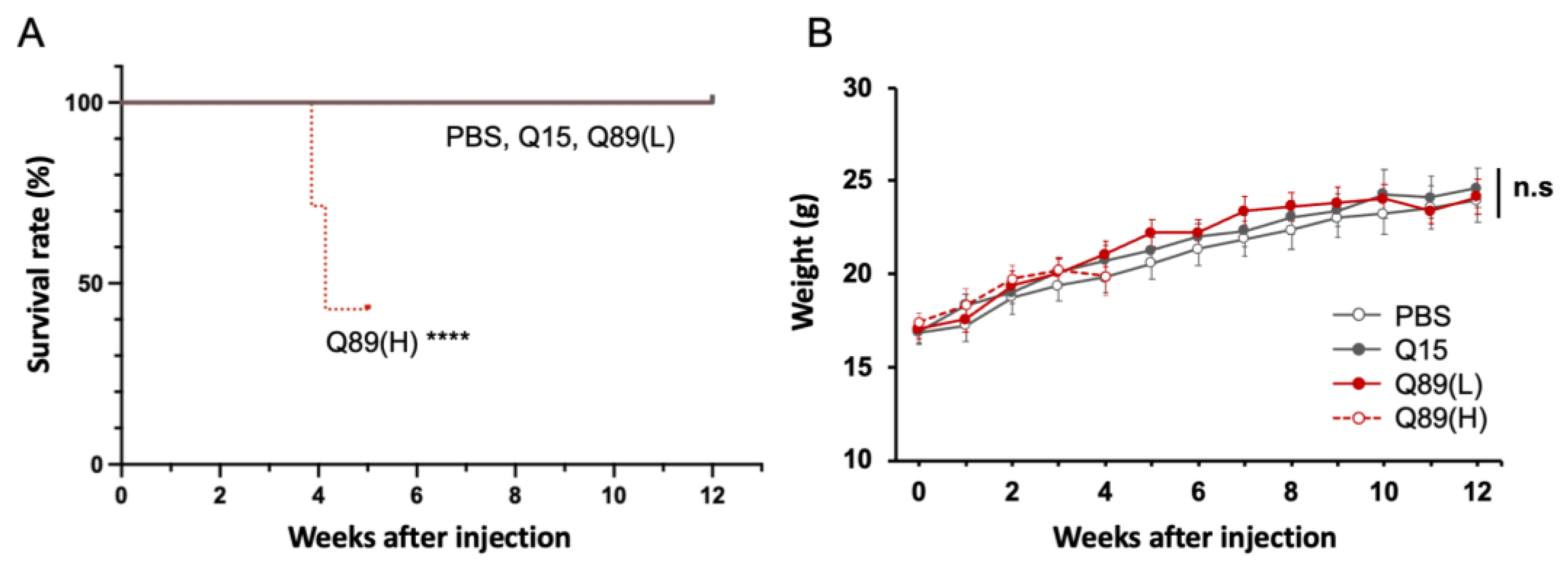
2.2. Early Death in Mice That Received High Doses of AAV Encoding ATXN3 with Abnormally Expanded Polyglutamine Tract
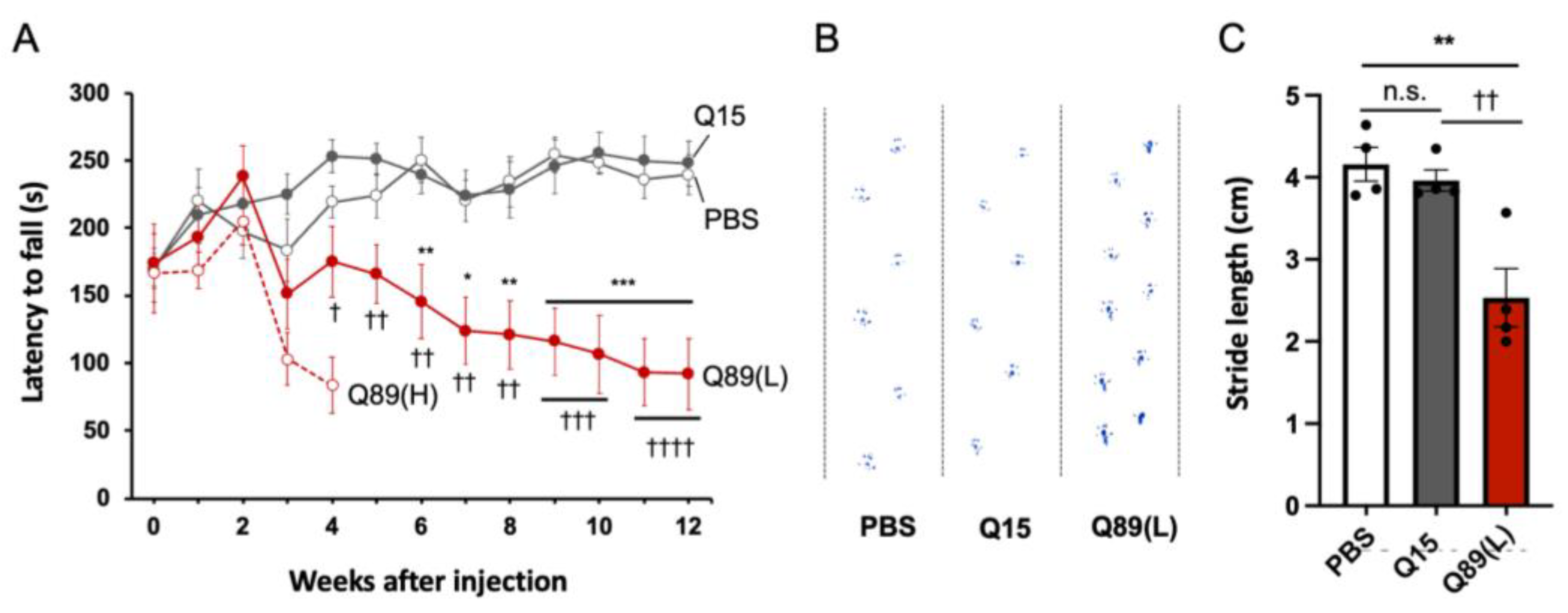
2.3. Motor Defects of Mice That Received AAV Encoding ATXN3(Q89)
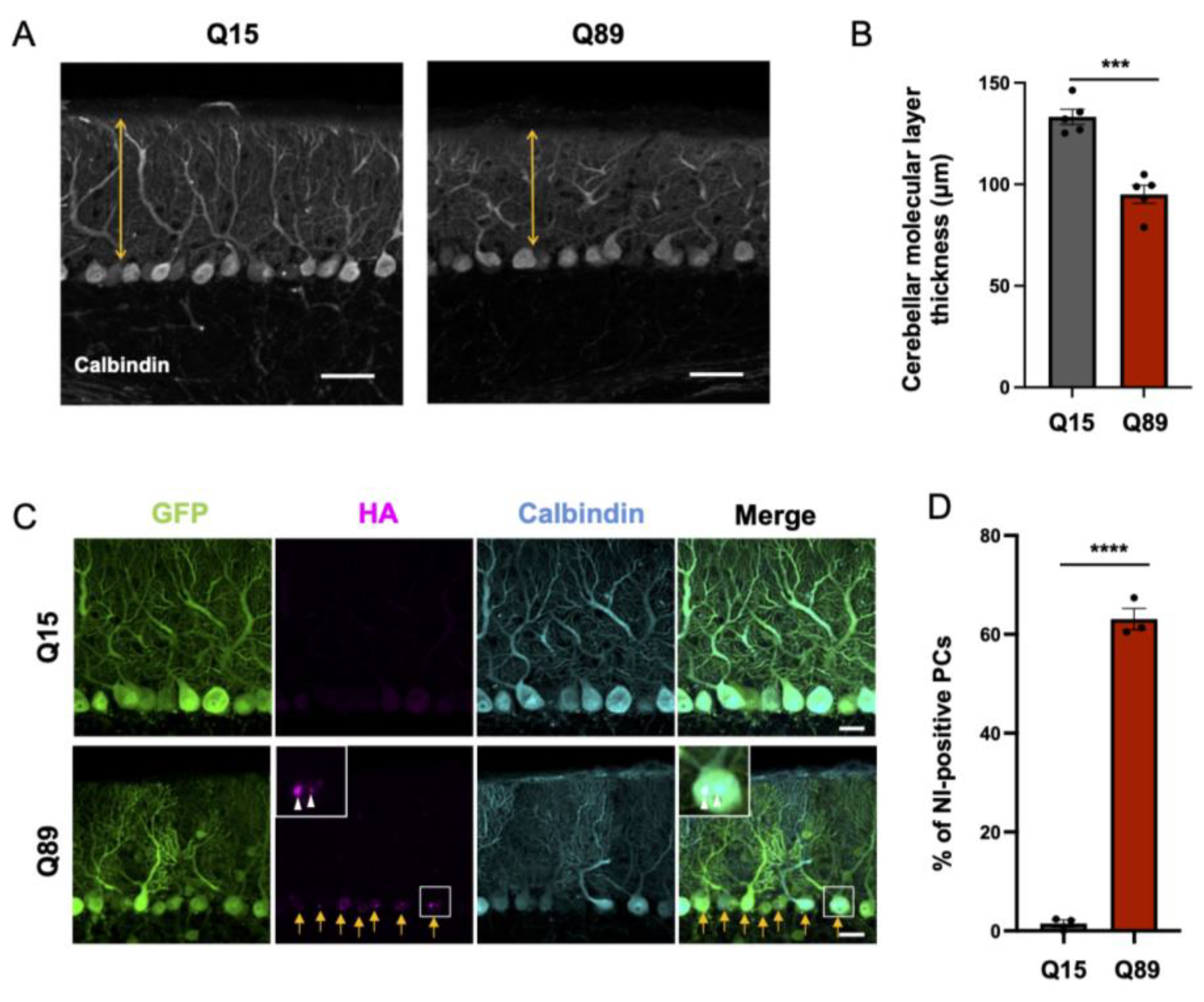
2.4. Significantly Reduced Thickness of the Molecular Layer of the Cerebellar Cortex in Mice Injected with AAV-PHP.B Encoding ATXN3(Q89)
2.5. Nuclear Inclusion Body Formation in Purkinje Cells of Mice Expressing ATXN3(Q89)
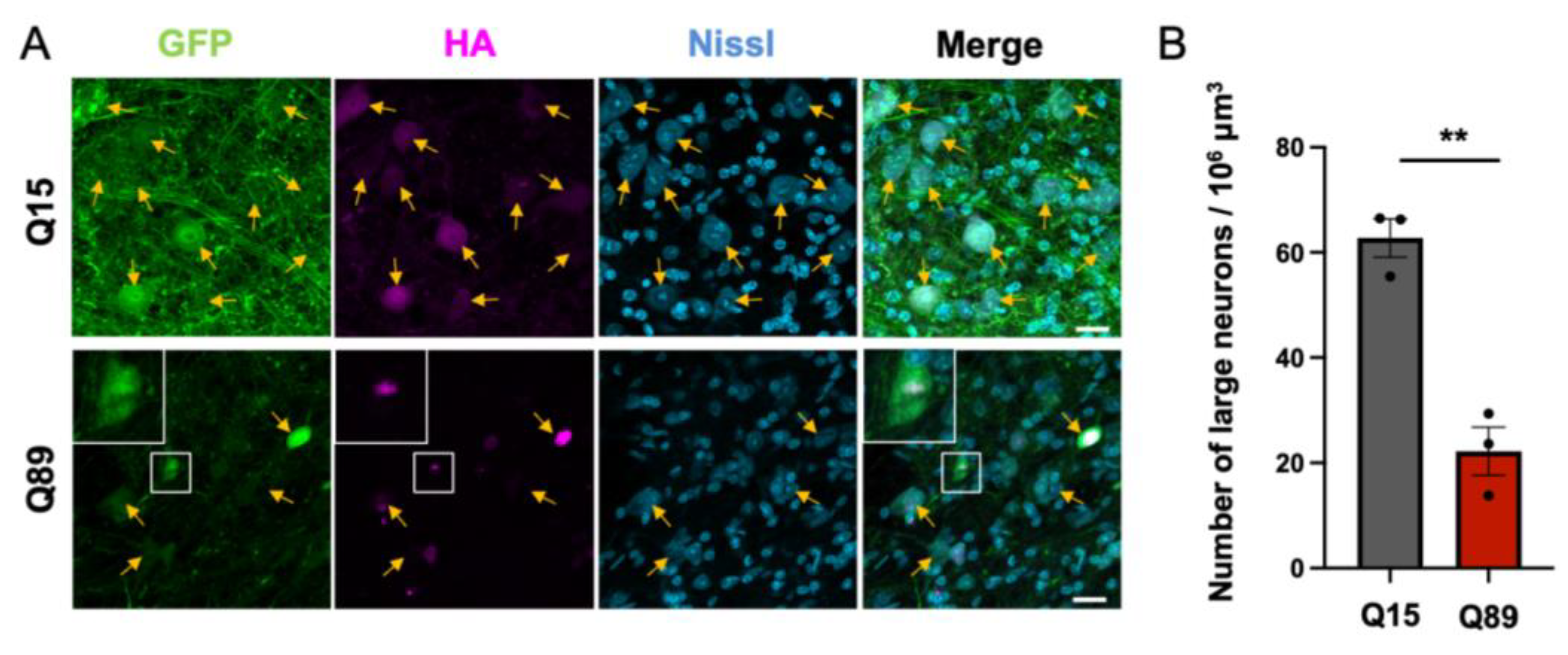
2.6. Significant Decrease in Large Projection Neurons in the Deep Cerebellar Nuclei of Mice Injected with Low Doses of AAV-PHP.B Encoding ATXN3(Q89)
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. AAV Vector Preparation
4.3. AAV Injection through Retro-Orbital Sinus
4.4. Behavioral Tests
4.5. Immunohistochemistry
4.6. Imaging Analysis
4.7. Data and Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McLoughlin, H.S.; Moore, L.R.; Paulson, H.L. Pathogenesis of SCA3 and implications for other polyglutamine diseases. Neurobiol. Dis. 2020, 134, 104635. [Google Scholar] [CrossRef]
- Li, X.; Liu, H.; Fischhaber, P.L.; Tang, T.S. Toward therapeutic targets for SCA3: Insight into the role of Machado-Joseph disease protein ataxin-3 in misfolded proteins clearance. Prog. Neurobiol. 2015, 132, 34–58. [Google Scholar] [CrossRef]
- Ren, H.; Hao, Z.; Wang, G. Autophagy and Polyglutamine Disease. Adv. Exp. Med. Biol. 2020, 1207, 149–161. [Google Scholar]
- Ingram, M.A.; Orr, H.T.; Clark, H.B. Genetically engineered mouse models of the trinucleotide-repeat spinocerebellar ataxias. Brain Res. Bull. 2012, 88, 33–42. [Google Scholar] [CrossRef]
- Nóbrega, C.; Nascimento-Ferreira, I.; Onofre, I.; Albuquerque, D.; Conceição, M.; Déglon, N.; de Almeida, L.P. Overexpression of mutant ataxin-3 in mouse cerebellum induces ataxia and cerebellar neuropathology. Cerebellum 2013, 12, 441–455. [Google Scholar] [CrossRef]
- Jansen-West, K.; Todd, T.W.; Daughrity, L.M.; Yue, M.; Tong, J.; Carlomagno, Y.; Del Rosso, G.; Kurti, A.; Jones, C.Y.; Dunmore, J.A.; et al. Plasma PolyQ-ATXN3 Levels Associate with Cerebellar Degeneration and Behavioral Abnormalities in a New AAV-Based SCA3 Mouse Model. Front. Cell Dev. Biol. 2022, 10, 863089. [Google Scholar] [CrossRef]
- Deverman, B.E.; Pravdo, P.L.; Simpson, B.P.; Kumar, S.R.; Chan, K.Y.; Banerjee, A.; Wu, W.L.; Yang, B.; Huber, N.; Pasca, S.P.; et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 2016, 34, 204–209. [Google Scholar] [CrossRef]
- Matsuzaki, Y.; Tanaka, M.; Hakoda, S.; Masuda, T.; Miyata, R.; Konno, A.; Hirai, H. Neurotropic Properties of AAV-PHP.B Are Shared among Diverse Inbred Strains of Mice. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 700–704. [Google Scholar] [CrossRef]
- Boy, J.; Schmidt, T.; Wolburg, H.; Mack, A.; Nuber, S.; Böttcher, M.; Schmitt, I.; Holzmann, C.; Zimmermann, F.; Servadio, A.; et al. Reversibility of symptoms in a conditional mouse model of spinocerebellar ataxia type 3. Hum. Mol. Genet. 2009, 18, 4282–4295. [Google Scholar] [CrossRef]
- Nóbrega, C.; Nascimento-Ferreira, I.; Onofre, I.; Albuquerque, D.; Hirai, H.; Déglon, N.; de Almeida, L.P. Silencing mutant ataxin-3 rescues motor deficits and neuropathology in Machado-Joseph disease transgenic mice. PLoS ONE 2013, 8, e52396. [Google Scholar] [CrossRef]
- Paulson, H.L.; Perez, M.K.; Trottier, Y.; Trojanowski, J.Q.; Subramony, S.H.; Das, S.S.; Vig, P.; Mandel, J.L.; Fischbeck, K.H.; Pittman, R.N. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 1997, 19, 333–344. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Scherzed, W.; Brunt, E.R.; Heinsen, H.; de Vos, R.A.; Seidel, K.; Bürk, K.; Schöls, L.; Auburger, G.; Del Turco, D.; Deller, T.; et al. Pathoanatomy of cerebellar degeneration in spinocerebellar ataxia type 2 (SCA2) and type 3 (SCA3). Cerebellum 2012, 11, 749–760. [Google Scholar] [CrossRef]
- Stefanescu, M.R.; Dohnalek, M.; Maderwald, S.; Thürling, M.; Minnerop, M.; Beck, A.; Schlamann, M.; Diedrichsen, J.; Ladd, M.E.; Timmann, D. Structural and functional MRI abnormalities of cerebellar cortex and nuclei in SCA3, SCA6 and Friedreich’s ataxia. Brain 2015, 138 Pt. 5, 1182–1197. [Google Scholar] [CrossRef]
- Bagley, J.; LaRocca, G.; Jimenez, D.A.; Urban, N.N. Adult neurogenesis and specific replacement of interneuron subtypes in the mouse main olfactory bulb. BMC Neurosci. 2007, 8, 92. [Google Scholar] [CrossRef]
- Uusisaari, M.; Obata, K.; Knöpfel, T. Morphological and electrophysiological properties of GABAergic and non-GABAergic cells in the deep cerebellar nuclei. J. Neurophysiol. 2007, 97, 901–911. [Google Scholar] [CrossRef]
- Chan, K.Y.; Jang, M.J.; Yoo, B.B.; Greenbaum, A.; Ravi, N.; Wu, W.L.; Sánchez-Guardado, L.; Lois, C.; Mazmanian, S.K.; Deverman, B.E.; et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017, 20, 1172–1179. [Google Scholar] [CrossRef]
- Radhiyanti, P.T.; Konno, A.; Matsuzaki, Y.; Hirai, H. Comparative study of neuron-specific promoters in mouse brain transduced by intravenously administered AAV-PHP.eB. Neurosci. Lett. 2021, 756, 135956. [Google Scholar] [CrossRef]
- Hanlon, K.S.; Meltzer, J.C.; Buzhdygan, T.; Cheng, M.J.; Sena-Esteves, M.; Bennett, R.E.; Sullivan, T.P.; Razmpour, R.; Gong, Y.; Ng, C.; et al. Selection of an Efficient AAV Vector for Robust CNS Transgene Expression. Mol. Ther. Methods Clin. Dev. 2019, 15, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, H.; Konno, A.; Matsuzaki, Y.; Sato, Y.; Kawachi, M.; Aoki, R.; Tsutsumi, S.; Togai, S.; Kobayashi, R.; Horii, T.; et al. Improving cell-specific recombination using AAV vectors in the murine CNS by capsid and expression cassette optimization. Mol. Ther. Methods Clin. Dev. 2024, 32, 101185. [Google Scholar] [CrossRef]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J.M. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018, 29, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Palazzi, X.; Pardo, I.D.; Sirivelu, M.P.; Newman, L.; Kumpf, S.W.; Qian, J.; Franks, T.; Lopes, S.; Liu, J.; Monarski, L.; et al. Biodistribution and Tolerability of AAV-PHP.B-CBh-SMN1 in Wistar Han Rats and Cynomolgus Macaques Reveal Different Toxicologic Profiles. Hum. Gene Ther. 2022, 33, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, H.; Konno, A.; Matsuzaki, Y.; Hirai, H. A blood-brain barrier-penetrating AAV2 mutant created by a brain microvasculature endothelial cell-targeted AAV2 variant. Mol. Ther. Methods Clin. Dev. 2023, 29, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Hordeaux, J.; Yuan, Y.; Clark, P.M.; Wang, Q.; Martino, R.A.; Sims, J.J.; Bell, P.; Raymond, A.; Stanford, W.L.; Wilson, J.M. The GPI-Linked Protein LY6A Drives AAV-PHP.B Transport across the Blood-Brain Barrier. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Chan, K.Y.; Tobey, I.G.; Chan, Y.A.; Poterba, T.; Boutros, C.L.; Balazs, A.B.; Daneman, R.; Bloom, J.M.; Seed, C.; et al. Delivering genes across the blood-brain barrier: LY6A, a novel cellular receptor for AAV-PHP.B capsids. PLoS ONE 2019, 14, e0225206. [Google Scholar] [CrossRef] [PubMed]
- Chuapoco, M.R.; Flytzanis, N.C.; Goeden, N.; Christopher Octeau, J.; Roxas, K.M.; Chan, K.Y.; Scherrer, J.; Winchester, J.; Blackburn, R.J.; Campos, L.J.; et al. Adeno-associated viral vectors for functional intravenous gene transfer throughout the non-human primate brain. Nat. Nanotechnol. 2023, 18, 1241–1251. [Google Scholar] [CrossRef]
- Moyer, T.C.; Hoffman, B.A.; Chen, W.; Shah, I.; Ren, X.-Q.; Knox, T.; Liu, J.; Wang, W.; Li, J.; Khalid, H.; et al. Highly conserved brain vascular receptor ALPL mediates transport of engineered viral vectors across the blood-brain barrier. bioRxiv 2024. [Google Scholar]
- Huang, Q.; Chan, K.Y.; Wu, J.; Botticello-Romero, N.R.; Zheng, Q.; Lou, S.; Keyes, C.; Svanbergsson, A.; Johnston, J.; Mills, A.; et al. An AAV capsid reprogrammed to bind human transferrin receptor mediates brain-wide gene delivery. Science 2024, 384, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.J.; Foti, S.B.; Schwartz, J.W.; Bachaboina, L.; Taylor-Blake, B.; Coleman, J.; Ehlers, M.D.; Zylka, M.J.; McCown, T.J.; Samulski, R.J. Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Hum. Gene Ther. 2011, 22, 1143–1153. [Google Scholar] [CrossRef]
- Laccone, F.; Maiwald, R.; Bingemann, S. A fast polymerase chain reaction-mediated strategy for introducing repeat expansions into CAG-repeat containing genes. Hum. Mutat. 1999, 13, 497–502. [Google Scholar] [CrossRef]
- Konno, A.; Hirai, H. Efficient whole brain transduction by systemic infusion of minimally purified AAV-PHP.eB. J. Neurosci. Methods 2020, 346, 108914. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konno, A.; Shinohara, Y.; Hirai, H. Production of Spinocerebellar Ataxia Type 3 Model Mice by Intravenous Injection of AAV-PHP.B Vectors. Int. J. Mol. Sci. 2024, 25, 7205. https://doi.org/10.3390/ijms25137205
Konno A, Shinohara Y, Hirai H. Production of Spinocerebellar Ataxia Type 3 Model Mice by Intravenous Injection of AAV-PHP.B Vectors. International Journal of Molecular Sciences. 2024; 25(13):7205. https://doi.org/10.3390/ijms25137205
Chicago/Turabian StyleKonno, Ayumu, Yoichiro Shinohara, and Hirokazu Hirai. 2024. "Production of Spinocerebellar Ataxia Type 3 Model Mice by Intravenous Injection of AAV-PHP.B Vectors" International Journal of Molecular Sciences 25, no. 13: 7205. https://doi.org/10.3390/ijms25137205
APA StyleKonno, A., Shinohara, Y., & Hirai, H. (2024). Production of Spinocerebellar Ataxia Type 3 Model Mice by Intravenous Injection of AAV-PHP.B Vectors. International Journal of Molecular Sciences, 25(13), 7205. https://doi.org/10.3390/ijms25137205