
The Discovery of Novel α2a Adrenergic Receptor Agonists Only Coupling to Gαi/O Proteins by Virtual Screening

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Analysis of the Molecular Docking Results
2.2. Properties of the Selected Compounds
2.3. The Effects of Compounds on the Lorr and Locomotor Activity in Mice
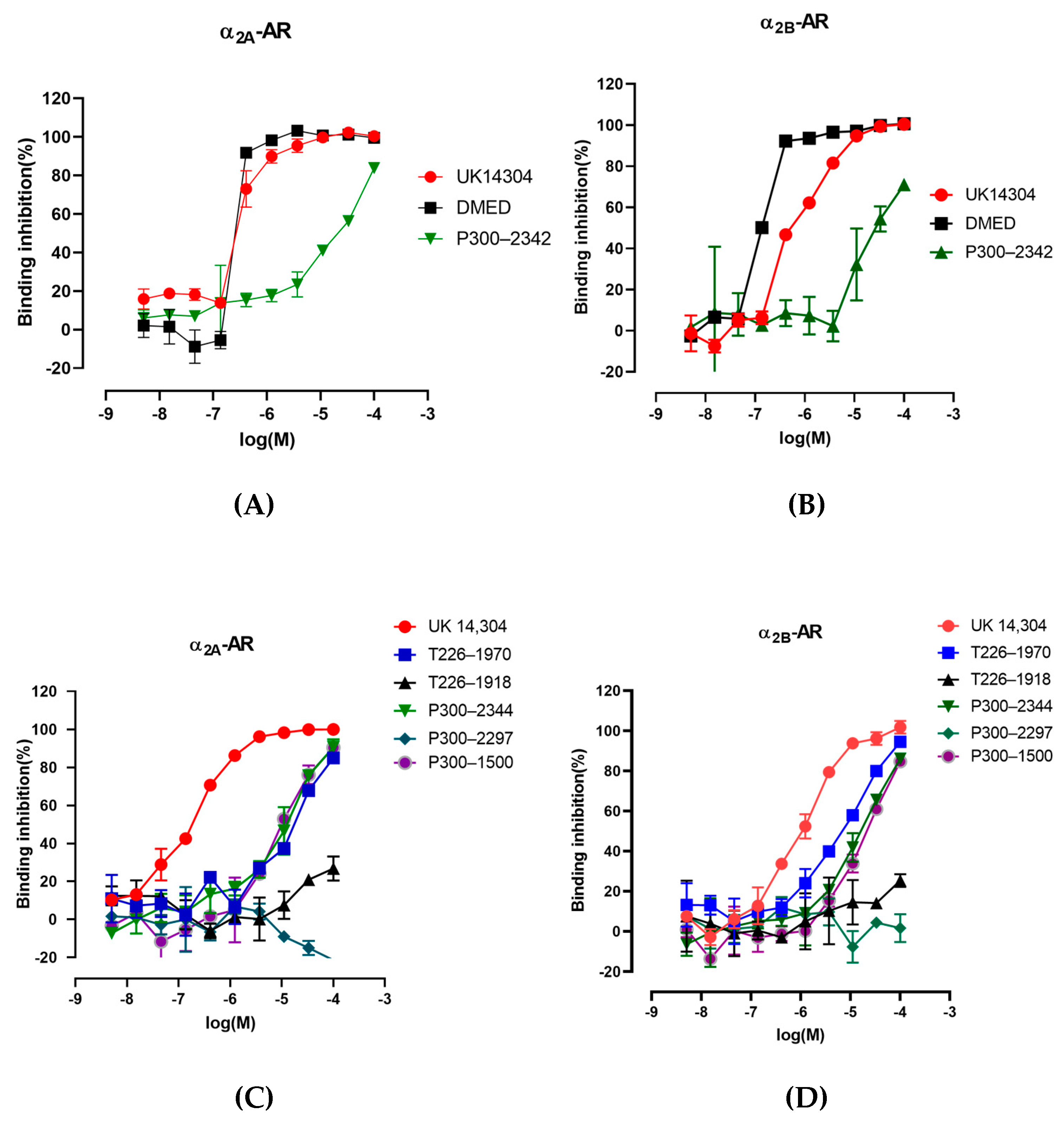
2.4. The Interaction of P300–2342 or Analogs with A2a-Ar or A2b-Ar
2.5. Cyclic Adenosine Monophosphate Assay
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Protein Preparation for Humanα2-AR Proteins and Binding Pocket Identification
5.2. Virtual Screening
5.3. Loss of Righting Reflex (LORR) and Locomotor Activity
5.3.1. Materials
5.3.2. LORR and Locomotor Activity
5.4. Ligand-Binding Assay
5.5. cAMP Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Delmas, P.; Abogadie, F.C.; Milligan, G.; Buckley, N.J.; Brown, D.A. Betagamma dimers derived from Go and Gi proteins contribute different components of adrenergic inhibition of Ca2+ channels in rat sympathetic neurons. J. Physiol. 1999, 518, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Scheinin, M.; Lomasney, J.W.; Hayden-Hixson, D.M.; Schambra, U.B.; Caron, M.G.; Lefkowitz, R.J.; Fremeau, R., Jr. Distribution of a2-adrenergic receptor subtype gene expression in rat brain. Brain Res. Mol. Brain Res. 1994, 21, 133–149. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, E.; Kobilka, B.K.; Scheinin, M. Gene targeting–homing in on alpha 2-adrenoceptor-subtype function. Trends Pharmacol. Sci. 1997, 18, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Gavras, I.; Manolis, A.J.; Gavras, H. The alpha2 -adrenergic receptors in hypertension and heart failure: Experimental and clinical studies. J. Hypertens. 2001, 19, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Timmermans, P.B.; van Zwieten, P.A. Mini-review. The postsynaptic alpha 2-adrenoreceptor. J. Auton. Pharmacol. 1981, 1, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Van Meel, J.C.; De Jonge, A.; Kalkman, H.O.; Wilffert, B.; Timmermans, P.B.; Van Zwieten, P.A. Vascular smooth muscle contraction intiated by postsynaptic alpha2-adrenoceptor activation is induced by an influx of extracellular calcium. Eur. J. Pharmacol. 1981, 69, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Link, R.E.; Desai, K.; Hein, L.; Stevens, M.E.; Chruscinski, A.; Bernstein, D.; Barsh, G.S.; Kobilka, B.K. Cardiovascular regulation in mice lacking alpha2-adrenergic receptor subtypes b and c. Science 1996, 273, 803–805. [Google Scholar] [CrossRef] [PubMed]
- Makaritsis, K.P.; Handy, D.E.; Johns, C.; Kobilka, B.; Gavras, I.; Gavras, H. Role of the alpha2B-adrenergic receptor in the development of salt-induced hypertension. Hypertension 1999, 33, 14–17. [Google Scholar] [CrossRef]
- Ekaterina Kintsurashvili, E.; Johns, C.; Ignjacev, I.; Gavras, I.; Gavras, H. Central alpha2B-adrenergic receptor antisense in plasmid vector prolongs reversal of salt-dependent hypertension. J. Hypertens. 2003, 21, 961–967. [Google Scholar] [CrossRef]
- Hein, L.; Altman, J.D.; Kobilka, B.K. Two functionally distinct alpha2-adrenergic receptors regulate sympathetic neurotransmission. Nature 1999, 402, 181–184. [Google Scholar] [CrossRef]
- Eason, M.G.; Liggett, S.B. Identification of a Gs coupling domain in the amino terminus of the third intracellular loop of the alpha 2A-adrenergic receptor. Evidence for distinct structural determinants that confer Gs versus Gi coupling. J. Biol. Chem. 1995, 270, 24753–24760. [Google Scholar] [CrossRef] [PubMed]
- Correa-Sales, C.; Reid, K.; Maze, M. Pertussis toxin-mediated ribosylation of G proteins blocks the hypnotic response to an alpha 2-agonist in the locus coeruleus of the rat. Pharmacol. Biochem. Behav. 1992, 43, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.D.; Buck, M.A.; Fraser, C.M. Site-directed mutagenesis of alpha 2A-adrenergic receptors: Identification of amino acids involved in ligand binding and receptor activation by agonists. Mol. Pharmacol. 1991, 40, 168–179. [Google Scholar] [PubMed]
- Qu, L.; Zhou, Q.T.; Xu, Y.M.; Guo, Y.; Xiaoyu Chen, X.Y.; Yao, D.Q.; Gye Won Han, G.H.; Zhi-Jie Liu, Z.J.; Stevens, R.C.; Zhong, G.S.; et al. Structural Basis of the Diversity of Adrenergic Receptors. Cell Rep. 2019, 29, 2929–2935. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Liu, Z.; Kaindl, J.; Maeda, S.; Zhao, J.; Sun, X.; Xu, J.; Gmeiner, P.; Wang, H.W.; Kobilka, B.K. Activation of the α 2B adrenoceptor by the sedative sympatholytic dexmedetomidine. Nat. Chem. Biol. 2020, 16, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Lakhlani, P.P.; MacMillan, L.B.; Guo, T.Z.; McCool, B.A.; Lovinger, D.M.; Maze, M.; Limbird, L.E. Substitution of a mutant alpha2a-adrenergic receptor via “hit and run” gene targeting reveals the role of this subtype in sedative, analgesic, and anesthetic-sparing responses in vivo. Proc. Natl. Acad. Sci. USA 1997, 94, 9950–9955. [Google Scholar] [CrossRef] [PubMed]
- Limbird, L.E. Receptors linked to inhibition of adenylate cyclase: Additional signaling mechanisms. FASEB J. 1988, 2, 2686–2695. [Google Scholar] [CrossRef] [PubMed]
- Insel, P.A. Seminars in medicine of the Beth Israel Hospital, Boston. Adrenergic receptors–evolving concepts and clinical implications. N. Engl. J. Med. 1996, 334, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Michino, M.; Beuming, T.; Donthamsetti, P.; Newman, A.H.; Javitch, J.A.; Shi, L. What can crystal structures of aminergic receptors tell us about designing subtype-selective ligands? Pharmacol. Rev. 2015, 67, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Marjamaki, A.; Pihlavisto, M.; Cockcroft, V.; Heinonen, P.; Savola, J.M.; Scheinin, M. Chloroethylclonidine binds irreversibly to exposed cysteines in the fifth membrane-spanning domain of the human alpha2A-adrenergic receptor. Mol. Pharmacol. 1998, 53, 370–376. [Google Scholar] [CrossRef]
- Eason, M.G.; Kurose, H.; Holt, B.D.; Raymond, J.R.; Liggett, S.B. Simultaneous coupling of alpha 2-adrenergic receptors to two G-proteins with opposing effects. Subtype-selective coupling of alpha 2C10, alpha 2C4, and alpha 2C2 adrenergic receptors to Gi and Gs. J. Biol. Chem. 1992, 267, 15795–15801. [Google Scholar] [CrossRef] [PubMed]
- Masureel, M.; Zou, Y.; Picard, L.P.; van derWesthuizen, E.; Mahoney, J.P.; Rodrigues, J.P.G.L.M.; Mildorf, T.J.; Dror, R.O.; Shaw, D.E.; Bouvier, M.; et al. Structural insights into binding specificity, efficacy and bias of a b2AR partial agonist. Nat. Chem. Biol. 2018, 14, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen, S.G.F.; Thian, F.S.; Kobilka, T.S.; Choi, H.J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K.; et al. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 2007, 318, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.M.; Cherezov, V.; Hanson, M.A.; Rasmussen, S.G.F.; Thian, F.S.; Kobilka, T.S.; Choi, H.J.; Yao, X.J.; Weis, W.I.; Stevens, R.C.; et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science 2007, 318, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Kuybeda, O.; de Waal, P.W.; Mukherjee, S.; Van Eps, N.; Dutka, P.; Zhou, X.E.; Bartesaghi, A.; Erramilli, S.; Morizumi, T.; et al. Cryo-EM structure of human rhodopsin bound to an inhibitory G protein. Nature 2018, 558, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Draper-Joyce, C.J.; Khoshouei, M.; Thal, D.M.; Liang, Y.L.; Nguyen, A.T.N.; Furness, S.G.B.; Venugopal, H.; Baltos, J.A.; Plitzko, J.M.; Danev, R.; et al. Structure of the adenosine-bound human adenosine A1 receptor-Gicomplex. Nature 2018, 558, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Kuybeda, O.; de Waal, P.W.; Mukherjee, S.; Van Eps, N.; Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; et al. Structure of the mu-opioid receptor-Gi protein complex. Nature 2018, 558, 547–552. [Google Scholar]
- Warne, T.; Moukhametzianov, R.; Baker, J.G.; Nehme’, R.; Edwards, P.C.; Le-slie, A.G.; Schertler, G.F.; Tate, C.G. The structural basis foragonist and partial agonist action on a β(1)-adrenergic receptor. Nature 2011, 469, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Moukhametzianov, R.; Warne, T.; Edwards, P.C.; Serrano-Vega, M.J.; Leslie, A.G.; Tate, C.G.; Schertler, G.F. Two distinct conformations of helix 6 observed in antagonist-bound structures of a beta1-adrenergic receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 8228–8232. [Google Scholar] [CrossRef]
- Ostopovici-Halip, L.; Curpăn, R.; Mracec, M.; Bologa, C.G. Structural determinants of the alpha2 adrenoceptor subtype selectivity. J. Mol. Graph. Model. 2011, 29, 1030–1038. [Google Scholar] [CrossRef]
- Liu, M.; Yang, Y.; Tan, B.; Li, Y.L.; Zhou, P.L.; Su, R.B. G αi and G βγ subunits have opposing effects on dexmedetomidine-induced sedation. Eur. J. Pharmacol. 2018, 831, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Liu, X.; Zhang, M.; Su, R. Effects of Ketanserin, M100907 and Olanzapine on hallucinogenic like action induced by 2, 5-dimethoxy-4-methylamphetamine. Behav. Pharmacol. 2023, 34, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Liu, X.; Wang, X.; Li, Y.; Ma, F.; Tan, B.; Zhou, P.; Fu, F.; Su, R. Gβγ subunits inhibitor decreased head twitch response induced by DOM through the PLCβ/IP3/Ca2+/ERK and cAMP signaling pathways. Eur. J. Pharmacol. 2023, 957, 176038. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, P.; Lu, F.; Zhu, H.; Shi, B.; Wang, X.; Sun, S.; Li, Y.; Su, R. The Discovery of Novel α2a Adrenergic Receptor Agonists Only Coupling to Gαi/O Proteins by Virtual Screening. Int. J. Mol. Sci. 2024, 25, 7233. https://doi.org/10.3390/ijms25137233
Zhou P, Lu F, Zhu H, Shi B, Wang X, Sun S, Li Y, Su R. The Discovery of Novel α2a Adrenergic Receptor Agonists Only Coupling to Gαi/O Proteins by Virtual Screening. International Journal of Molecular Sciences. 2024; 25(13):7233. https://doi.org/10.3390/ijms25137233
Chicago/Turabian StyleZhou, Peilan, Fengfeng Lu, Huili Zhu, Beibei Shi, Xiaoxuan Wang, Shiyang Sun, Yulei Li, and Ruibin Su. 2024. "The Discovery of Novel α2a Adrenergic Receptor Agonists Only Coupling to Gαi/O Proteins by Virtual Screening" International Journal of Molecular Sciences 25, no. 13: 7233. https://doi.org/10.3390/ijms25137233
APA StyleZhou, P., Lu, F., Zhu, H., Shi, B., Wang, X., Sun, S., Li, Y., & Su, R. (2024). The Discovery of Novel α2a Adrenergic Receptor Agonists Only Coupling to Gαi/O Proteins by Virtual Screening. International Journal of Molecular Sciences, 25(13), 7233. https://doi.org/10.3390/ijms25137233