
A Novel Class I HDAC Inhibitor, AW01178, Inhibits Epithelial–Mesenchymal Transition and Metastasis of Breast Cancer
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
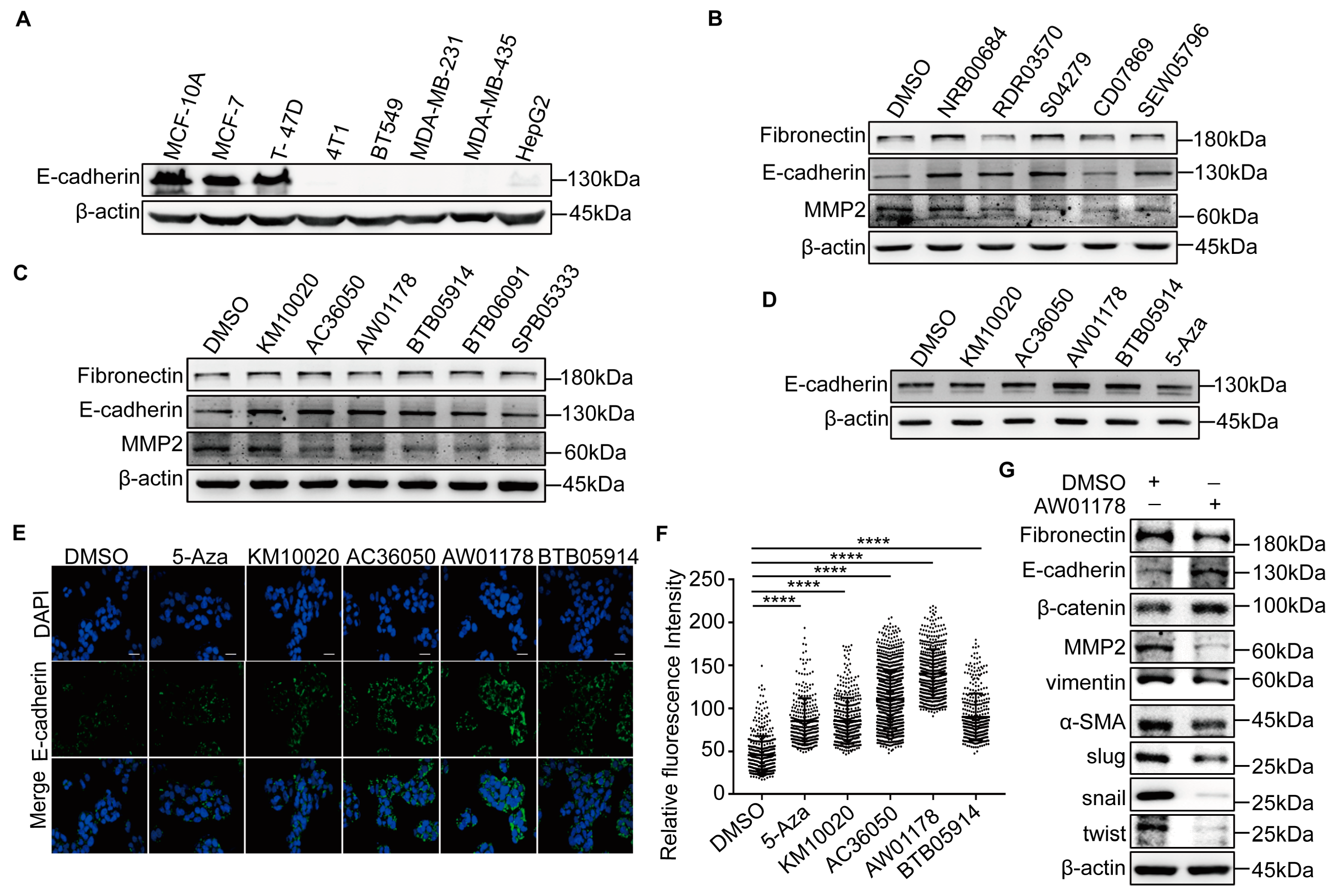
2.1. A Novel Small-Molecule Compound, AW01178, Upregulates E-Cadherin
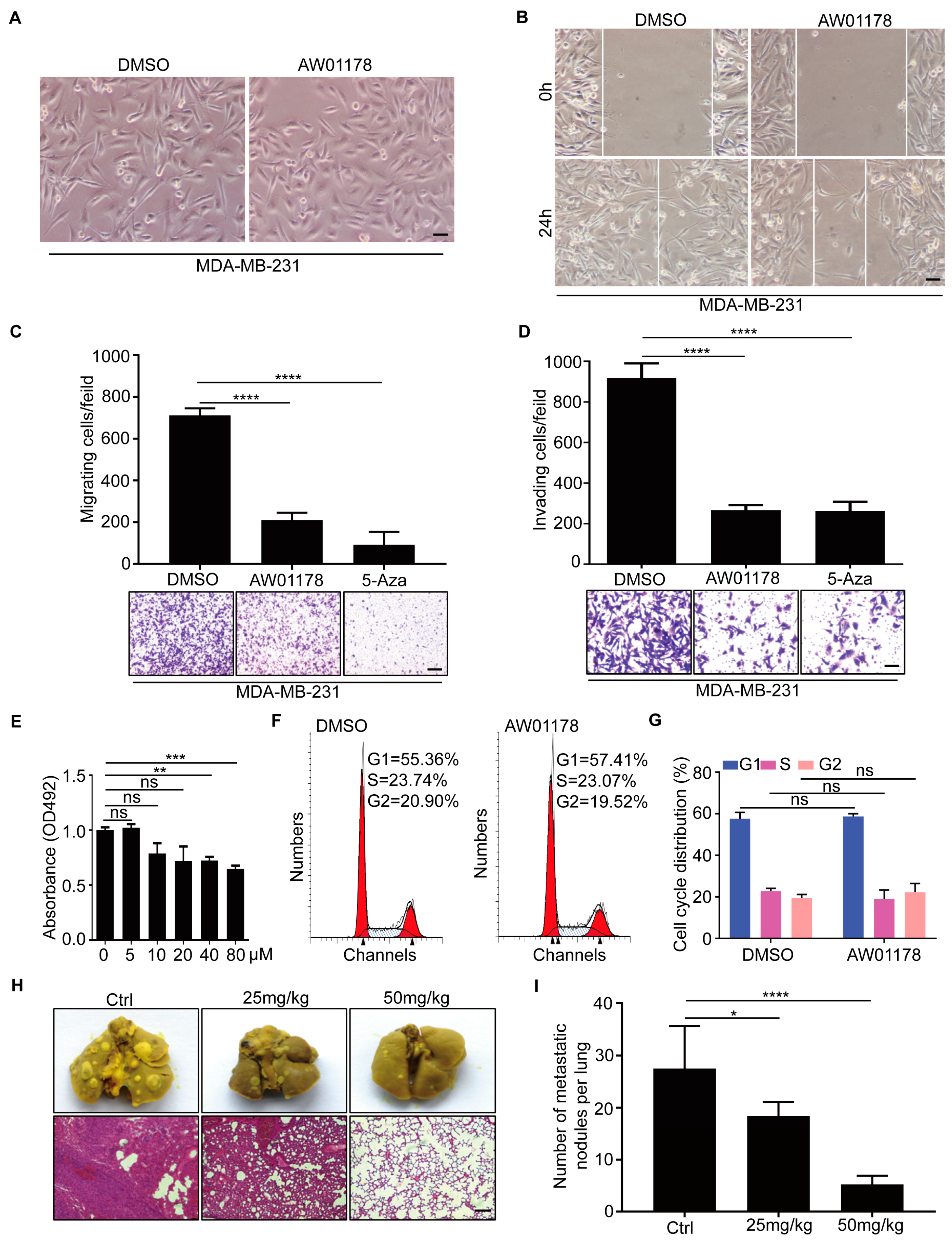
2.2. Small-Molecule Compound AW01178 Has the Ability to Inhibit Breast Cancer Migration and Invasion
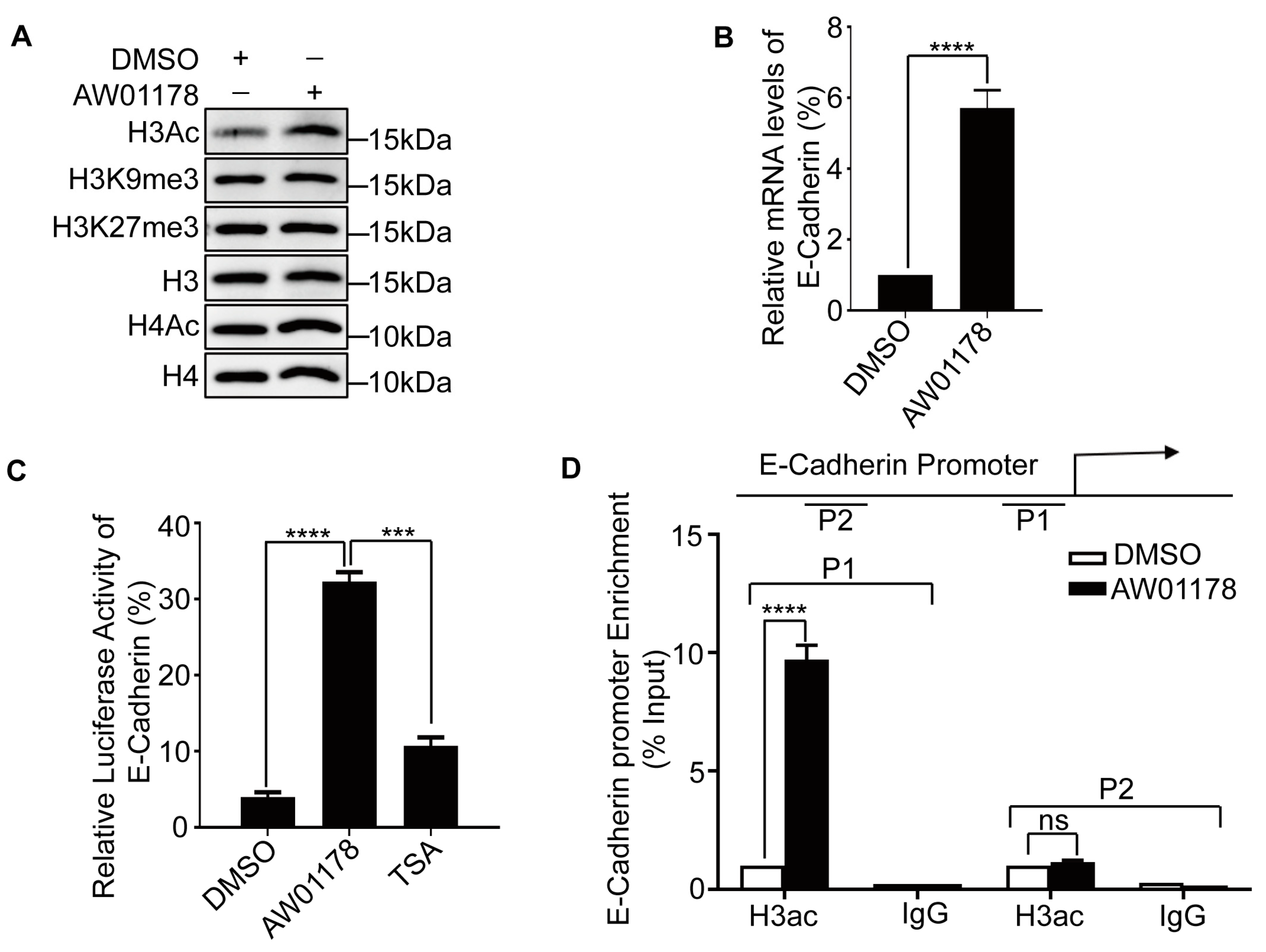
2.3. AW01178 Upregulates Acetylation of Histone H3 at E-Cadherin Promoter
2.4. AW01178 Is a Novel Class I HDAC Inhibitor
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Reverse Transcription, PCR, and Real-Time PCR Analysis
4.3. Western Blotting
4.4. Immunofluorescence
4.5. Wound-Healing, Transwell Migration, and Invasion Assays
4.6. In-Cell Western Assays
4.7. Luciferase Reporter Assay
4.8. Chromatin Immunoprecipitation
4.9. Cell Cycle
4.10. MTT Assay
4.11. Enzyme Activity Assay
4.12. HDAC Binding Analysis Assay
4.13. In Vivo Mouse Lung Metastasis Assay
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Massagué, J.; Ganesh, K. Metastasis-Initiating Cells and Ecosystems. Cancer Discov. 2021, 11, 971–994. [Google Scholar] [CrossRef] [PubMed]
- Farkas, A.H.; Nattinger, A.B. Breast Cancer Screening and Prevention. Ann. Intern. Med. 2023, 176, ITC161–ITC176. [Google Scholar] [CrossRef] [PubMed]
- Barzaman, K.; Karami, J.; Zarei, Z.; Hosseinzadeh, A.; Kazemi, M.H.; Moradi-Kalbolandi, S.; Safari, E.; Farahmand, L. Breast cancer: Biology, biomarkers, and treatments. Int. Immunopharmacol. 2020, 84, 106535. [Google Scholar] [CrossRef] [PubMed]
- Celià-Terrassa, T.; Kang, Y. How important is EMT for cancer metastasis? PLoS Biol. 2024, 22, e3002487. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.; Jiang, H.; Ma, Y.; Wang, L.; Wang, L.; Du, J.; Hou, P.; Gao, Y.; Zhao, L.; Wang, G.; et al. PRMT7 Induces Epithelial-to-Mesenchymal Transition and Promotes Metastasis in Breast Cancer. Cancer Res. 2014, 74, 5656–5667. [Google Scholar] [CrossRef]
- Zhuyan, J.; Chen, M.; Zhu, T.; Bao, X.; Zhen, T.; Xing, K.; Wang, Q.; Zhu, S. Critical steps to tumor metastasis: Alterations of tumor microenvironment and extracellular matrix in the formation of pre-metastatic and metastatic niche. Cell Biosci. 2020, 10, 89. [Google Scholar] [CrossRef]
- Li, Y.; Li, M.; Su, K.; Zong, S.; Zhang, H.; Xiong, L. Pre-metastatic niche: From revealing the molecular and cellular mechanisms to the clinical applications in breast cancer metastasis. Theranostics. 2023, 13, 2301–2318. [Google Scholar] [CrossRef]
- Wang, N.; He, Y.L.; Pang, L.J.; Zou, H.; Liu, C.X.; Zhao, J.; Hu, J.M.; Zhang, W.J.; Qi, Y.; Li, F. Down-Regulated E-Cadherin Expression Is Associated with Poor Five-Year Overall Survival in Bone and Soft Tissue Sarcoma: Results of a Meta-Analysis. PLoS ONE 2015, 10, e0121448. [Google Scholar] [CrossRef]
- Louie, E.; Chen, X.F.; Coomes, A.; Ji, K.; Tsirka, S.; I Chen, E. Neurotrophin-3 modulates breast cancer cells and the microenvironment to promote the growth of breast cancer brain metastasis. Oncogene 2012, 32, 4064–4077. [Google Scholar] [CrossRef]
- Lu, J.; Kornmann, M.; Traub, B. Role of Epithelial to Mesenchymal Transition in Colorectal Cancer. Int. J. Mol. Sci. 2023, 24, 14815. [Google Scholar] [CrossRef]
- Bolós, V.; Peinado, H.; Perez-Moreno, M.A.; Fraga, M.F.; Esteller, M.; Cano, A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: A comparison with Snail and E47 repressors. J. Cell Sci. 2003, 116, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Canel, M.; Serrels, A.; Frame, M.C.; Brunton, V.G. E-cadherin–integrin crosstalk in cancer invasion and metastasis. J. Cell Sci. 2013, 126, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Vesuna, F.; van Diest, P.; Chen, J.H.; Raman, V. Twist is a transcriptional repressor of E-cadherin gene expression in breast cancer. Biochem. Biophys. Res. Commun. 2007, 367, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Aghdassi, A.; Sendler, M.; Guenther, A.; Mayerle, J.; Behn, C.-O.; Heidecke, C.-D.; Friess, H.; Büchler, M.; Evert, M.; Lerch, M.M.; et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut 2011, 61, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef] [PubMed]
- Min, S.-W.; Cho, S.-H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of Tau Inhibits Its Degradation and Contributes to Tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Santoro, F.; Botrugno, O.A.; Zuffo, R.D.; Pallavicini, I.; Matthews, G.M.; Cluse, L.; Barozzi, I.; Senese, S.; Fornasari, L.; Moretti, S.; et al. A dual role for Hdac1: Oncosuppressor in tumorigenesis, oncogene in tumor maintenance. Blood 2013, 121, 3459–3468. [Google Scholar] [CrossRef]
- Shakespear, M.R.; Halili, M.A.; Irvine, K.M.; Fairlie, D.P.; Sweet, M.J. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011, 32, 335–343. [Google Scholar] [CrossRef]
- Coradini, D.; Zorzet, S.; Rossin, R.; Scarlata, I.; Pellizzaro, C.; Turrin, C.; Bello, M.; Cantoni, S.; Speranza, A.; Sava, G.; et al. Inhibition of Hepatocellular Carcinomas in vitro and Hepatic Metastases in vivo in Mice by the Histone Deacetylase Inhibitor HA-But. Clin. Cancer Res. 2004, 10, 4822–4830. [Google Scholar] [CrossRef]
- Xue, K.; Gu, J.J.; Zhang, Q.; Mavis, C.; Hernandez-Ilizaliturri, F.J.; Czuczman, M.S.; Guo, Y. Vorinostat, a histone deacetylase (HDAC) inhibitor, promotes cell cycle arrest and re-sensitizes rituximab- and chemo-resistant lymphoma cells to chemotherapy agents. J. Cancer Res. Clin. Oncol. 2015, 142, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Yadav, A.; Lang, J.C.; Teknos, T.N.; Kumar, P. Suberoylanilide hydroxamic acid (SAHA) reverses chemoresistance in head and neck cancer cells by targeting cancer stem cells via the downregulation of nanog. Genes Cancer 2015, 6, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, K.; Kishikawa, F.; Tanaka, M.; Sakamoto, T.; Tanimura, S.; Kohno, M. Histone deacetylase inhibitors enhance the chemosensitivity of tumor cells with cross-resistance to a wide range of DNA-damaging drugs. Cancer Sci. 2008, 99, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.; Easley, C.; Kirkpatrick, P. Vorinostat. Nat. Rev. Drug Discov. 2007, 6, 21–22. [Google Scholar] [CrossRef] [PubMed]
- Gojo, I.; Jiemjit, A.; Trepel, J.B.; Sparreboom, A.; Figg, W.D.; Rollins, S.; Tidwell, M.L.; Greer, J.; Chung, E.J.; Lee, M.-J.; et al. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood 2006, 109, 2781–2790. [Google Scholar] [CrossRef] [PubMed]
- Yeruva, S.L.H.; Zhao, F.; Miller, K.D.; Tevaarwerk, A.J.; Wagner, L.I.; Gray, R.J.; Sparano, J.A.; Connolly, R.M. E2112: Randomized phase iii trial of endocrine therapy plus entinostat/placebo in patients with hormone receptor-positive advanced breast cancer. npj Breast Cancer 2018, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- E Gryder, B.; Sodji, Q.H.; Oyelere, A.K. Targeted Cancer Therapy: Giving Histone Deacetylase Inhibitors All They Need To Succeed. Future Med. Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef] [PubMed]
- Hesham, H.M.; Lasheen, D.S.; Abouzid, K.A. Chimeric HDAC inhibitors: Comprehensive review on the HDAC-based strategies developed to combat cancer. Med. Res. Rev. 2018, 38, 2058–2109. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.; Chang, A.; Ho, C.; Tsai, T.; Chen, H.; Chen, P.; Hwang, T.I. Thrombospondin-4 promotes bladder cancer cell migration and invasion via MMP2 production. J. Cell. Mol. Med. 2021, 25, 6046–6055. [Google Scholar] [CrossRef]
- Peinado, H.; Ballestar, E.; Esteller, M.; Cano, A. Snail Mediates E-Cadherin Repression by the Recruitment of the Sin3A/Histone Deacetylase 1 (HDAC1)/HDAC2 Complex. Mol. Cell. Biol. 2004, 24, 306–319. [Google Scholar] [CrossRef]
- Lin, T.; Ponn, A.; Hu, X.; Law, B.K.; Lu, J. Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene 2010, 29, 4896–4904. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Feng, J.; Li, L.; Lin, L.; Ji, J.; Lin, C.; Liu, L.; Zhang, N.; Duan, D.; Li, Z.; et al. Arginine methylation-dependent LSD1 stability promotes invasion and metastasis of breast cancer. Embo Rep. 2019, 21, e48597. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Tan, R.; Cao, J.; Yang, Y.; Kong, C.; Du, J.; Zhu, S.; Zhang, Y.; Lu, J.; Huang, B.; et al. Discovery of polyoxometalate-based HDAC inhibitors with profound anticancer activity in vitro and in vivo. Eur. J. Med. Chem. 2011, 46, 2477–2484. [Google Scholar] [CrossRef] [PubMed]
- Kolli, R.T.; Glenn, T.C.; Brown, B.T.; Kaur, S.P.; Barnett, L.M.; Lash, L.H.; Cummings, B.S. Bromate-induced Changes in p21 DNA Methylation and Histone Acetylation in Renal Cells. Toxicol. Sci. 2019, 168, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Xie, R.; Tian, T.; Zheng, L.; Tang, L.; Cai, S.; Ma, Z.; Yang, T.; Han, B.; Yang, Q. Suberoylanilide hydroxamic acid upregulates histone acetylation and activates endoplasmic reticulum stress to induce apoptosis in HepG2 liver cancer cells. Oncol. Lett. 2019, 18, 3537–3544. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K. DNMT1: A key drug target in triple-negative breast cancer. Semin. Cancer Biol. 2020, 72, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Topper, M.J.; Vaz, M.; Chiappinelli, K.B.; Shields, C.E.D.; Niknafs, N.; Yen, R.-W.C.; Wenzel, A.; Hicks, J.; Ballew, M.; Stone, M.; et al. Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell 2017, 171, 1284–1300.e21. [Google Scholar] [CrossRef] [PubMed]
- Drummond, D.C.; Noble, C.O.; Kirpotin, D.B.; Guo, Z.; Scott, G.K.; Benz, C.C. Clinical Development of Histone Deacetylase Inhibitors as Anticancer Agents. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 495–528. [Google Scholar] [CrossRef]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef]
- Huang, W.-Y.; Yang, P.-M.; Chang, Y.-F.; Marquez, V.E.; Chen, C.-C. Methotrexate induces apoptosis through p53/p21-dependent pathway and increases E-cadherin expression through downregulation of HDAC/EZH2. Biochem. Pharmacol. 2011, 81, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.-T.; Cai, M.-Y.; Wang, X.-G.; Kong, L.-L.; Mai, S.-J.; Liu, Y.-H.; Zhang, H.-B.; Liao, Y.-J.; Zheng, F.; Zhu, W.; et al. EZH2 supports nasopharyngeal carcinoma cell aggressiveness by forming a co-repressor complex with HDAC1/HDAC2 and Snail to inhibit E-cadherin. Oncogene 2011, 31, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-N.; Liu, Y.; Lee, H.-J.; Hsu, Y.-H.; Chen, J.-H. Activated Androgen Receptor Downregulates E-Cadherin Gene Expression and Promotes Tumor Metastasis. Mol. Cell. Biol. 2008, 28, 7096–7108. [Google Scholar] [CrossRef] [PubMed]
- Gaughan, L. Regulation of androgen receptor and histone deacetylase 1 by Mdm2-mediated ubiquitylation. Nucleic Acids Res. 2005, 33, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Tóth, K.F.; Knoch, T.A.; Wachsmuth, M.; Frank-Stöhr, M.; Stöhr, M.; Bacher, C.P.; Müller, G.; Rippe, K. Trichostatin A-induced histone acetylation causes decondensation of interphase chromatin. J. Cell Sci. 2004, 117, 4277–4287. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Shang, M.; Han, Y.; Liu, J.; Liu, J.; Kong, S.H.; Hou, J.; Huang, B.; Lu, J.; Zhang, Y. EZH2–CCF–cGAS Axis Promotes Breast Cancer Metastasis. Int. J. Mol. Sci. 2022, 23, 1788. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liang, Q.; Lei, Y.; Yao, M.; Li, L.; Gao, X.; Feng, J.; Zhang, Y.; Gao, H.; Liu, D.-X.; et al. SOX4 Induces Epithelial–Mesenchymal Transition and Contributes to Breast Cancer Progression. Cancer Res. 2012, 72, 4597–4608. [Google Scholar] [CrossRef]
- Feng, J.; Li, L.; Zhang, N.; Liu, J.; Zhang, L.; Gao, H.; Wang, G.; Li, Y.; Zhang, Y.; Li, X.; et al. Androgen and AR contribute to breast cancer development and metastasis: An insight of mechanisms. Oncogene 2016, 36, 2775–2790. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Chen, Y.; Li, Y.; Shen, Y.; Dong, S.; Tan, J. A Novel Class I HDAC Inhibitor, AW01178, Inhibits Epithelial–Mesenchymal Transition and Metastasis of Breast Cancer. Int. J. Mol. Sci. 2024, 25, 7234. https://doi.org/10.3390/ijms25137234
Liu X, Chen Y, Li Y, Shen Y, Dong S, Tan J. A Novel Class I HDAC Inhibitor, AW01178, Inhibits Epithelial–Mesenchymal Transition and Metastasis of Breast Cancer. International Journal of Molecular Sciences. 2024; 25(13):7234. https://doi.org/10.3390/ijms25137234
Chicago/Turabian StyleLiu, Xiangxiang, Yawen Chen, Yang Li, Ying Shen, Shasha Dong, and Jiang Tan. 2024. "A Novel Class I HDAC Inhibitor, AW01178, Inhibits Epithelial–Mesenchymal Transition and Metastasis of Breast Cancer" International Journal of Molecular Sciences 25, no. 13: 7234. https://doi.org/10.3390/ijms25137234