
Quercetin Modulates Ferroptosis via the SIRT1/Nrf−2/HO−1 Pathway and Attenuates Cartilage Destruction in an Osteoarthritis Rat Model
Abstract
:1. Introduction
2. Results
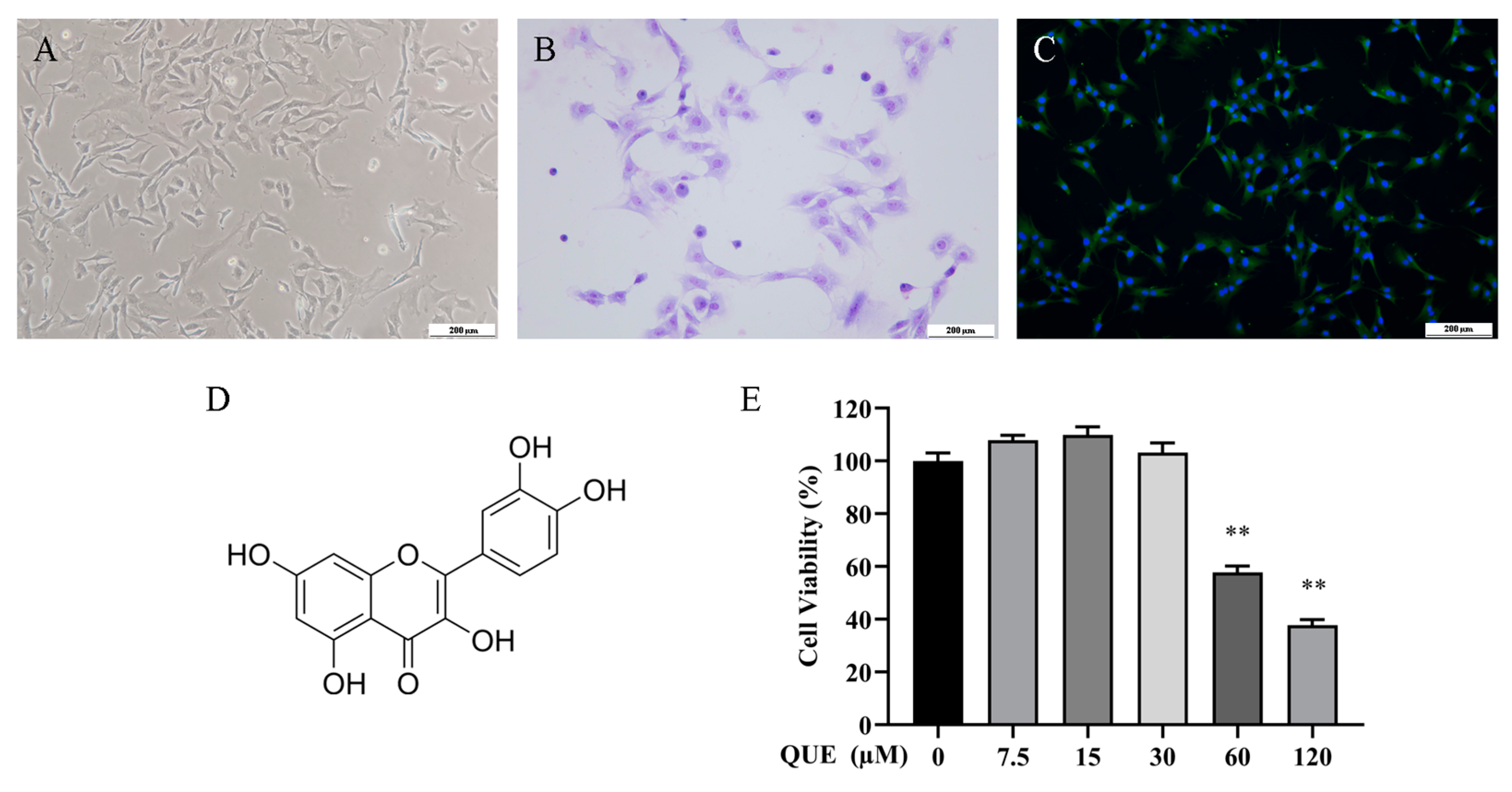
2.1. Effect of QUE on Chondrocyte Activity
2.2. QUE Protects Chondrocytes from IL−1β-Induced Inflammation and Cartilage Degeneration
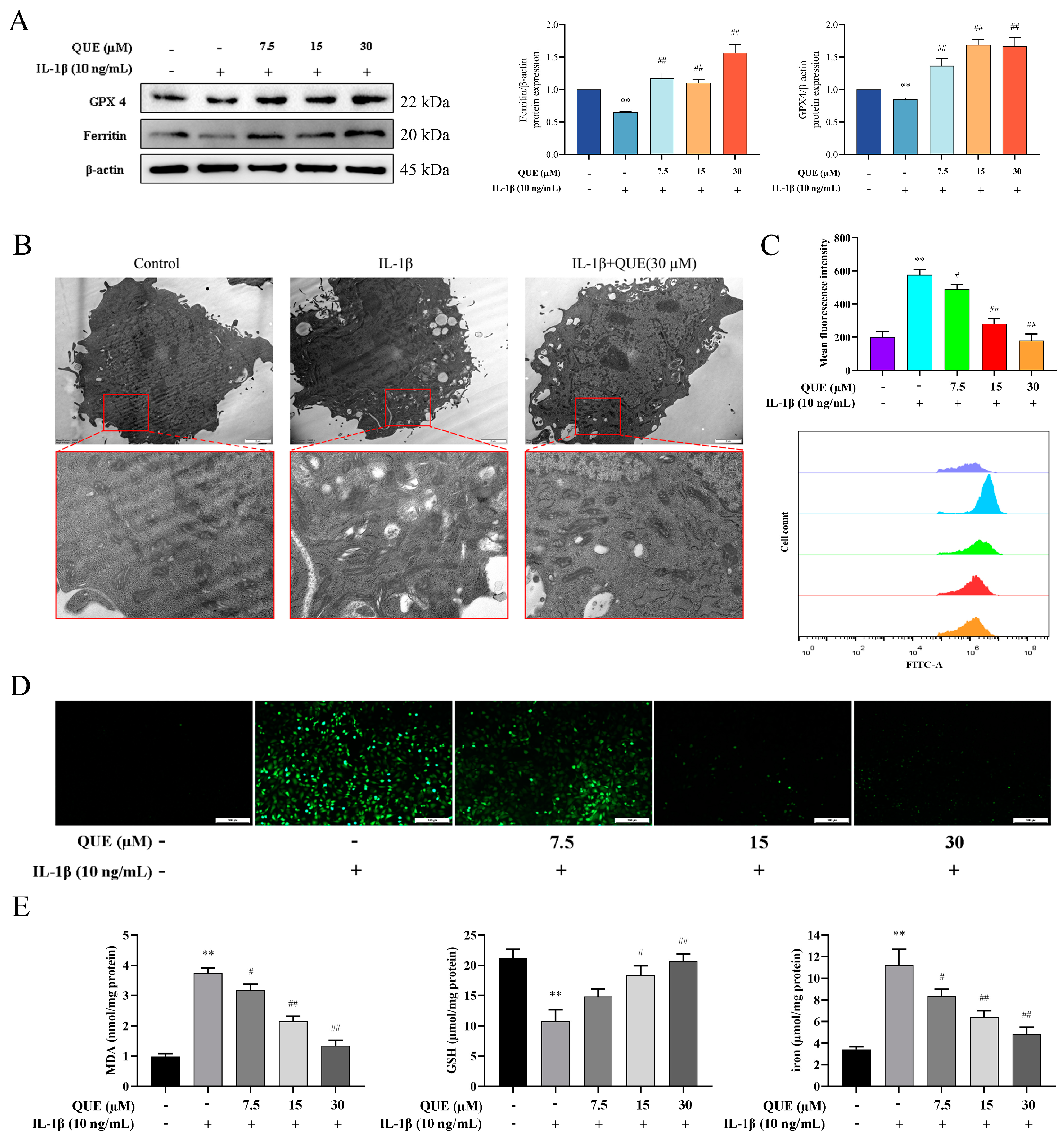
2.3. QUE Reduces IL−1β-Induced Ferroptosis and Oxidative Damage in Chondrocytes
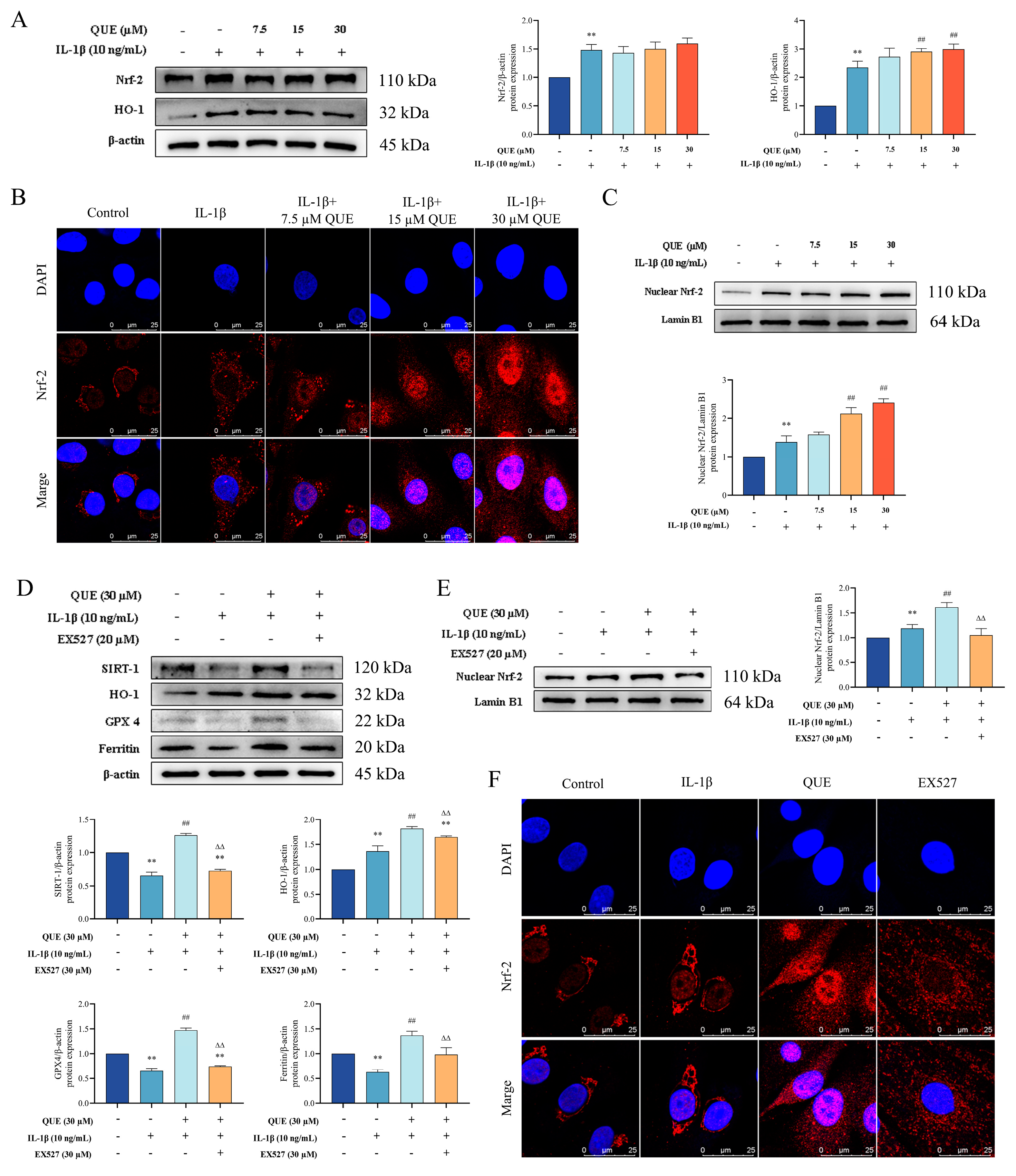
2.4. QUE Improves IL−1β-Induced Chondrocyte Oxidative Damage and Ferroptosis by Regulating the Nrf−2/HO−1 Pathway through SIRT1
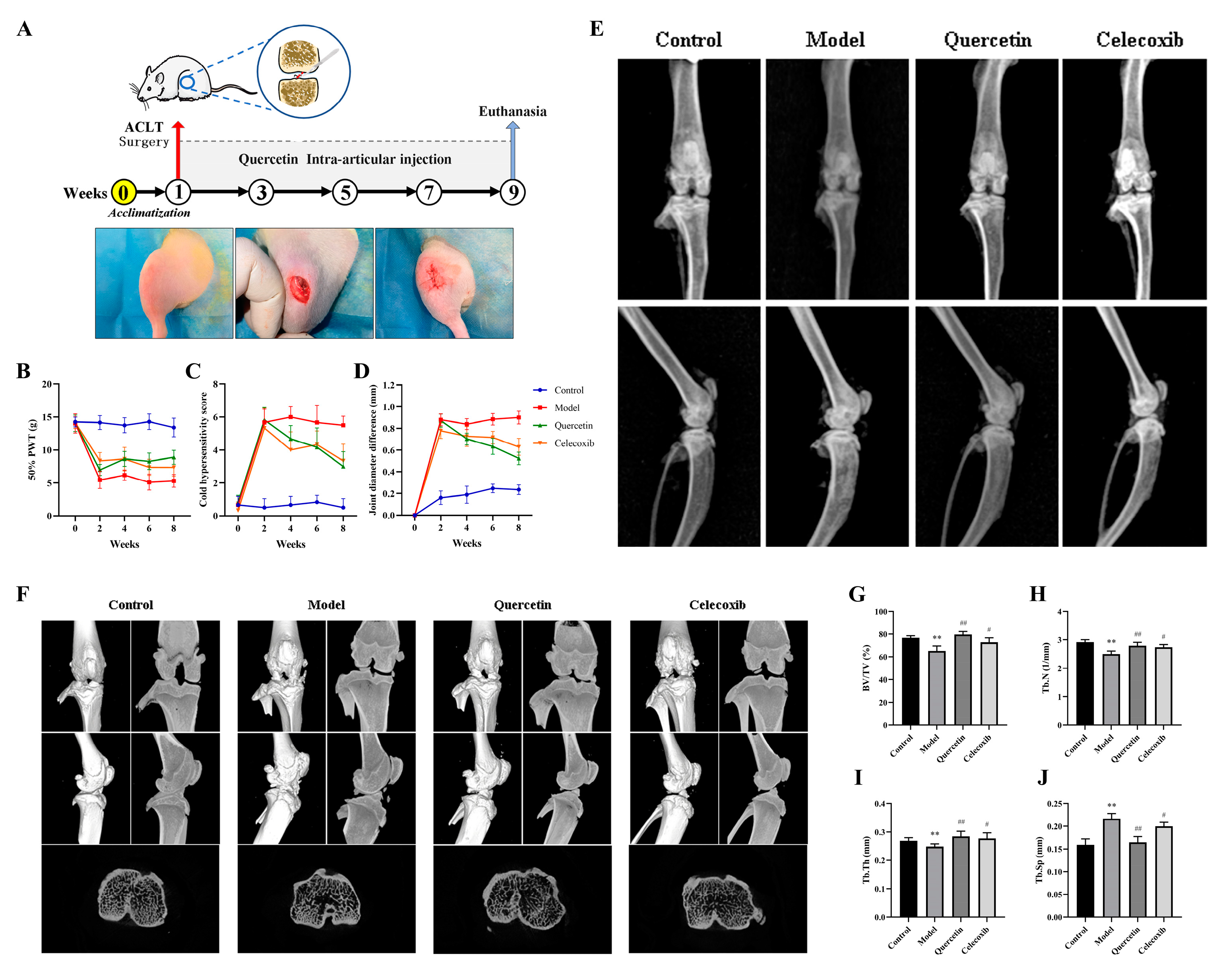
2.5. Que Improves Joint Pain in OA Rats
2.6. Que Improves Joint Imaging Features in OA Rats
2.7. QUE Improves Cartilage Damage in OA Rats
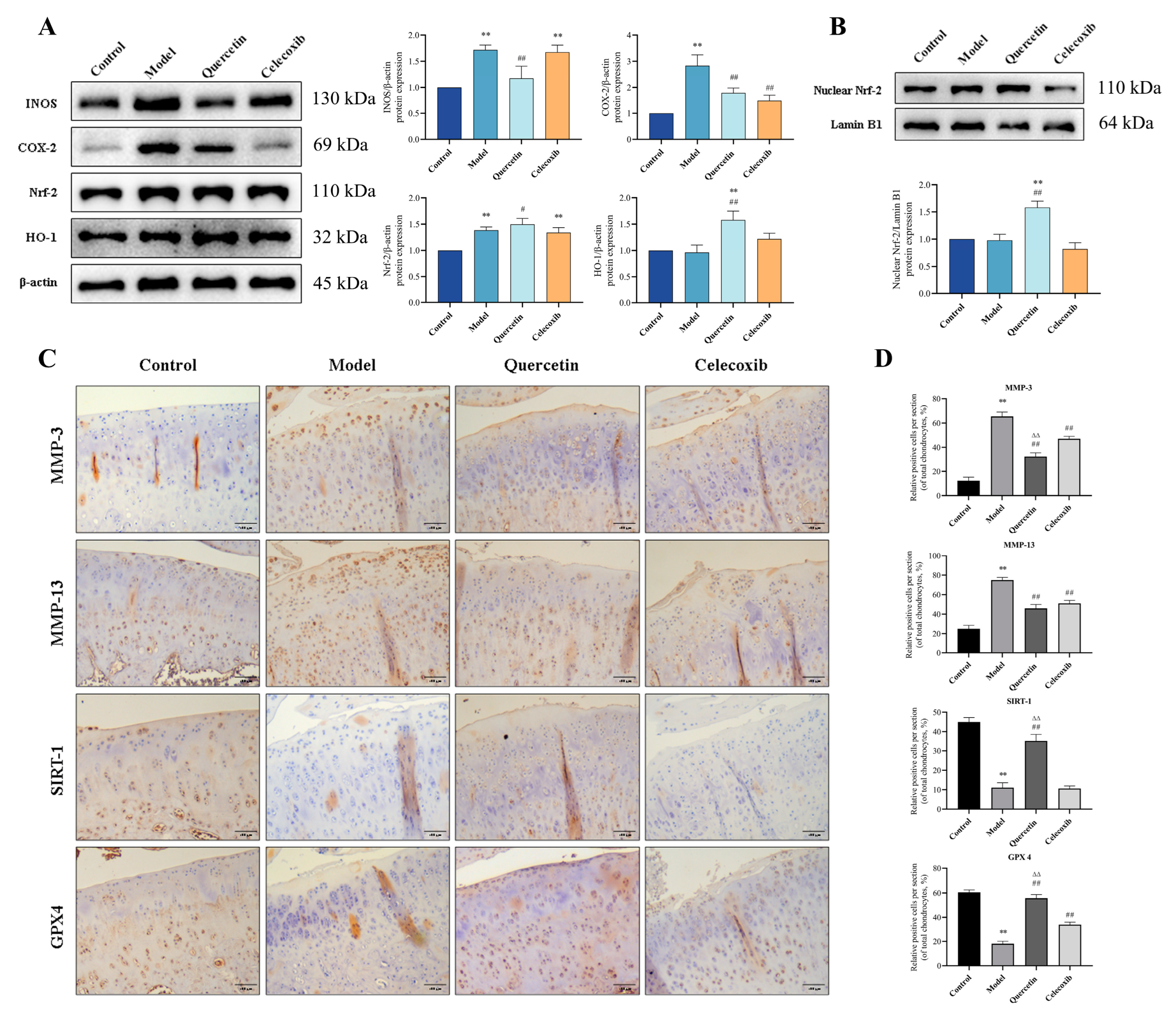
2.8. QUE Inhibits Cartilage ECM Degradation and Chondrocyte Ferroptosis in OA Rats via Sirt1/Nrf−2/HO−1 Pathway
3. Discussion
4. Materials and Methods
4.1. Isolation of Chondrocytes
4.2. CCK-8 Cell Viability Assay
4.3. Detection of Intracellular ROS
4.4. MDA, GSH, and Iron Assay
4.5. Observation of Mitochondrial Morphology
4.6. RNA Isolation and Reverse Transcription-qPCR
4.7. Immunofluorescence
4.8. OA Induction and Treatment
4.9. Animal Behavior Tests
4.10. Imaging Examination
4.11. Macroscopic Observation of Articular Cartilage
4.12. Histological Analysis
4.13. Immunohistochemical Analysis of Cartilage Tissue
4.14. Enzyme-Linked Immunosorbent Assay
4.15. Western Blot Analysis
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martel-Pelletier, J.; Barr, A.J.; Cicuttini, F.M.; Conaghan, P.G.; Cooper, C.; Goldring, M.B.; Goldring, S.R.; Jones, G.; Teichtahl, A.J.; Pelletier, J.P. Osteoarthritis. Nat. Rev. Dis. Primers 2016, 2, 16072. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.J.; March, L.; Chew, M. Osteoarthritis in 2020 and beyond: A Lancet Commission. Lancet 2020, 396, 1711–1712. [Google Scholar] [CrossRef] [PubMed]
- Abramoff, B.; Caldera, F.E. Osteoarthritis: Pathology, Diagnosis, and Treatment Options. Med. Clin. N. Am. 2020, 104, 293–311. [Google Scholar] [CrossRef] [PubMed]
- Magni, A.; Agostoni, P.; Bonezzi, C.; Massazza, G.; Menè, P.; Savarino, V.; Fornasari, D. Management of Osteoarthritis: Expert Opinion on NSAIDs. Pain Ther. 2021, 10, 783–808. [Google Scholar] [CrossRef] [PubMed]
- Lanas, A.; Garcia-Tell, G.; Armada, B.; Oteo-Alvaro, A. Prescription patterns and appropriateness of NSAID therapy according to gastrointestinal risk and cardiovascular history in patients with diagnoses of osteoarthritis. BMC Med. 2011, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Zhai, G. Clinical relevance of biochemical and metabolic changes in osteoarthritis. Adv. Clin. Chem. 2021, 101, 95–120. [Google Scholar] [CrossRef] [PubMed]
- Belluzzi, E.; Todros, S.; Pozzuoli, A.; Ruggieri, P.; Carniel, E.L.; Berardo, A. Human Cartilage Biomechanics: Experimental and Theoretical Approaches towards the Identification of Mechanical Properties in Healthy and Osteoarthritic Conditions. Processes 2023, 11, 1014. [Google Scholar] [CrossRef]
- Sandell, L.J.; Aigner, T. Articular cartilage and changes in arthritis. An introduction: Cell biology of osteoarthritis. Arthritis Res. 2001, 3, 107–113. [Google Scholar] [CrossRef]
- Zhu, R.; Wang, Y.; Ouyang, Z.; Hao, W.; Zhou, F.; Lin, Y.; Cheng, Y.; Zhou, R.; Hu, W. Targeting regulated chondrocyte death in osteoarthritis therapy. Biochem. Pharmacol. 2023, 215, 115707. [Google Scholar] [CrossRef]
- Chen, B.; Wang, L.; Xie, D.; Wang, Y. Exploration and breakthrough in the mode of chondrocyte death—A potential new mechanism for osteoarthritis. Biomed. Pharmacother. Biomed. Pharmacother. 2024, 170, 115990. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Burton, L.H.; Radakovich, L.B.; Marolf, A.J.; Santangelo, K.S. Systemic iron overload exacerbates osteoarthritis in the strain 13 guinea pig. Osteoarthr. Cartil. 2020, 28, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Lin, J.; Du, T.; Jiang, Z.; Li, T.; Wang, G.; Liu, X.; Cui, X.; Sun, K. Iron Overload Is Associated With Accelerated Progression of Osteoarthritis: The Role of DMT1 Mediated Iron Homeostasis. Front. Cell Dev. Biol. 2020, 8, 594509. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Wang, C.; Zhang, Y.; Sun, J. Brevilin A attenuates cartilage destruction in osteoarthritis mouse model by inhibiting inflammation and ferroptosis via SIRT1/Nrf2/GPX4 signaling pathway. Int. Immunopharmacol. 2023, 124, 110924. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Sun, K.; Yu, S.; Luo, J.; Guo, J.; Lin, J.; Wang, G.; Guo, Z.; Ye, Y.; Guo, F. Chondrocyte ferroptosis contribute to the progression of osteoarthritis. J. Orthop. Transl. 2021, 27, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Chen, Y.; Xue, F.; Liu, K.; Zhu, B.; Gao, J.; Yin, J.; Zhang, C.; Li, G. Contribution of ferroptosis and GPX4’s dual functions to osteoarthritis progression. EBioMedicine 2022, 76, 103847. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, W.; Zhang, P.; Wang, Z.; Ma, X.; Liu, C.; Vasilev, K.; Zhang, L.; Zhou, X.; Liu, L.; et al. Mechanical overloading induces GPX4-regulated chondrocyte ferroptosis in osteoarthritis via Piezo1 channel facilitated calcium influx. J. Adv. Res. 2022, 41, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Jia, Z.; Wang, J.; Huang, S.; Yang, S.; Xiao, S.; Xia, D.; Zhou, Y. Curcumin reverses erastin-induced chondrocyte ferroptosis by upregulating Nrf2. Heliyon 2023, 9, e20163. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Qi, G.; Lu, W.; Wang, H.; Li, D.; Chen, W.; Ding, L.; Yang, X.; Yuan, H.; Zeng, Q. Omaveloxolone inhibits IL-1β-induced chondrocyte apoptosis through the Nrf2/ARE and NF-κB signalling pathways in vitro and attenuates osteoarthritis in vivo. Front. Pharmacol. 2022, 13, 952950. [Google Scholar] [CrossRef]
- Qiang, Z.; Dong, H.; Xia, Y.; Chai, D.; Hu, R.; Jiang, H. Nrf2 and STAT3 Alleviates Ferroptosis-Mediated IIR-ALI by Regulating SLC7A11. Oxidative Med. Cell. Longev. 2020, 2020, 5146982. [Google Scholar] [CrossRef]
- Xu, C.; Ni, S.; Xu, N.; Yin, G.; Yu, Y.; Zhou, B.; Zhao, G.; Wang, L.; Zhu, R.; Jiang, S.; et al. Theaflavin-3,3߰-Digallate Inhibits Erastin-Induced Chondrocytes Ferroptosis via the Nrf2/GPX4 Signaling Pathway in Osteoarthritis. Oxidative Med. Cell. Longev. 2022, 2022, 3531995. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Lin, J.; Sun, K.; Guo, J.; Yao, X.; Wang, G.; Hou, L.; Xu, J.; Guo, J.; Guo, F. Deferoxamine Alleviates Osteoarthritis by Inhibiting Chondrocyte Ferroptosis and Activating the Nrf2 Pathway. Front. Pharmacol. 2022, 13, 791376. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, M.R.; Nabavi, S.M.; Braidy, N.; Setzer, W.N.; Ahmed, T.; Nabavi, S.F. Quercetin and the mitochondria: A mechanistic view. Biotechnol. Adv. 2016, 34, 532–549. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Agrawal, N. Quercetin: A Potential Candidate for the Treatment of Arthritis. Curr. Mol. Med. 2022, 22, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.P.; Xie, W.P.; Bi, Y.F.; Wang, B.A.; Song, H.B.; Wang, S.L.; Bi, R.X. Quercetin suppresses apoptosis of chondrocytes induced by IL-1β via inactivation of p38 MAPK signaling pathway. Exp. Ther. Med. 2021, 21, 468. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yin, J.; Zhao, D.; Wang, C.; Zhang, Y.; Wang, Y.; Li, T. Therapeutic effect and mechanism of action of quercetin in a rat model of osteoarthritis. J. Int. Med. Res. 2020, 48, 300060519873461. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, Y.; Tang, Y.; Lu, H.; Qi, Y.; Li, G.; He, H.; Lu, F.; Yang, Y.; Sun, H. Quercetin Alleviates Osteoarthritis Progression in Rats by Suppressing Inflammation and Apoptosis via Inhibition of IRAK1/NLRP3 Signaling. J. Inflamm. Res. 2021, 14, 3393–3403. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Luo, Y.; Chen, X. Quercetin attenuates mitochondrial dysfunction and biogenesis via upregulated AMPK/SIRT1 signaling pathway in OA rats. Biomed. Pharmacother. Biomed. Pharmacother. 2018, 103, 1585–1591. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.; Chen, Z.; Pengcheng, L.; Zhang, S.; Wang, X. Quercetin attenuates oxidative stress-induced apoptosis via SIRT1/AMPK-mediated inhibition of ER stress in rat chondrocytes and prevents the progression of osteoarthritis in a rat model. J. Cell. Physiol. 2019, 234, 18192–18205. [Google Scholar] [CrossRef]
- Hu, Y.; Gui, Z.; Zhou, Y.; Xia, L.; Lin, K.; Xu, Y. Quercetin alleviates rat osteoarthritis by inhibiting inflammation and apoptosis of chondrocytes, modulating synovial macrophages polarization to M2 macrophages. Free Radic. Biol. Med. 2019, 145, 146–160. [Google Scholar] [CrossRef]
- Li, D.; Jiang, C.; Mei, G.; Zhao, Y.; Chen, L.; Liu, J.; Tang, Y.; Gao, C.; Yao, P. Quercetin Alleviates Ferroptosis of Pancreatic β Cells in Type 2 Diabetes. Nutrients 2020, 12, 2954. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Quan, F.; Cao, Q.; Lin, Y.; Yue, C.; Bi, R.; Cui, X.; Yang, H.; Yang, Y.; Birnbaumer, L.; et al. Quercetin alleviates acute kidney injury by inhibiting ferroptosis. J. Adv. Res. 2021, 28, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Zhao, W.; Lowe, S.; Bentley, R.; Hu, G.; Mei, H.; Jiang, X.; Sun, C.; Wu, Y.; Yueying, L. Quercetin alleviates kainic acid-induced seizure by inhibiting the Nrf2-mediated ferroptosis pathway. Free Radic. Biol. Med. 2022, 191, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.W.; Zhao, G.J.; Li, X.L.; Hong, G.L.; Li, M.F.; Qiu, Q.M.; Wu, B.; Lu, Z.Q. SIRT1 exerts protective effects against paraquat-induced injury in mouse type II alveolar epithelial cells by deacetylating NRF2 in vitro. Int. J. Mol. Med. 2016, 37, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Gagarina, V.; Gabay, O.; Dvir-Ginzberg, M.; Lee, E.J.; Brady, J.K.; Quon, M.J.; Hall, D.J. SirT1 enhances survival of human osteoarthritic chondrocytes by repressing protein tyrosine phosphatase 1B and activating the insulin-like growth factor receptor pathway. Arthritis Rheum. 2010, 62, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ying, L.; Wei, B.; Ji, Y.; Xu, Y. Effects of quercetin on apoptosis and extracellular matrix degradation of chondrocytes induced by oxidative stress-mediated pyroptosis. J. Biochem. Mol. Toxicol. 2022, 36, e22951. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yan, Y.; Pathak, J.L.; Hong, W.; Zeng, J.; Qian, D.; Hao, B.; Li, H.; Gu, J.; Jaspers, R.T.; et al. Quercetin prevents osteoarthritis progression possibly via regulation of local and systemic inflammatory cascades. J. Cell. Mol. Med. 2023, 27, 515–528. [Google Scholar] [CrossRef]
- Peng, Z.; Sun, H.; Bunpetch, V.; Koh, Y.; Wen, Y.; Wu, D.; Ouyang, H. The regulation of cartilage extracellular matrix homeostasis in joint cartilage degeneration and regeneration. Biomaterials 2021, 268, 120555. [Google Scholar] [CrossRef]
- Karila, T.; Tervahartiala, T.; Cohen, B.; Sorsa, T. The collagenases: Are they tractable targets for preventing cartilage destruction in osteoarthritis? Expert. Opin. Ther. Targets 2022, 26, 93–105. [Google Scholar] [CrossRef]
- Jenei-Lanzl, Z.; Meurer, A.; Zaucke, F. Interleukin-1β signaling in osteoarthritis-chondrocytes in focus. Cell. Signal. 2019, 53, 212–223. [Google Scholar] [CrossRef]
- Ma, T.; Ruan, H.; Lv, L.; Wei, C.; Yu, Y.; Jia, L.; Song, X.; Zhang, J.; Li, Y. Oleanolic acid, a small-molecule natural product, inhibits ECM degeneration in osteoarthritis by regulating the Hippo/YAP and Wnt/β-catenin pathways. Food Funct. 2023, 14, 9999–10013. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Zhang, Z.; Song, X.; Bai, H.; Li, Y.; Li, X.; Zhao, J.; Ma, Y.; Gao, L. Combined detection of COMP and CS846 biomarkers in experimental rat osteoarthritis: A potential approach for assessment and diagnosis of osteoarthritis. J. Orthop. Surg. Res. 2018, 13, 230. [Google Scholar] [CrossRef]
- Guo, H.; Yin, W.; Zou, Z.; Zhang, C.; Sun, M.; Min, L.; Yang, L.; Kong, L. Quercitrin alleviates cartilage extracellular matrix degradation and delays ACLT rat osteoarthritis development: An in vivo and in vitro study. J. Adv. Res. 2021, 28, 255–267. [Google Scholar] [CrossRef]
- Wood, M.J.; Miller, R.E.; Malfait, A.M. The Genesis of Pain in Osteoarthritis: Inflammation as a Mediator of Osteoarthritis Pain. Clin. Geriatr. Med. 2022, 38, 221–238. [Google Scholar] [CrossRef] [PubMed]
- Nees, T.A.; Rosshirt, N.; Reiner, T.; Schiltenwolf, M.; Moradi, B. [Inflammation and osteoarthritis-related pain]. Schmerz 2019, 33, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Ishijima, M.; Nakamura, T.; Shimizu, K.; Hayashi, K.; Kikuchi, H.; Soen, S.; Omori, G.; Yamashita, T.; Uchio, Y.; Chiba, J.; et al. Different changes in the biomarker C-terminal telopeptides of type II collagen (CTX-II) following intra-articular injection of high molecular weight hyaluronic acid and oral non-steroidal anti-inflammatory drugs in patients with knee osteoarthritis: A multi-center randomized controlled study. Osteoarthr. Cartil. 2022, 30, 852–861. [Google Scholar] [CrossRef]
- Duclos, M.E.; Roualdes, O.; Cararo, R.; Rousseau, J.C.; Roger, T.; Hartmann, D.J. Significance of the serum CTX-II level in an osteoarthritis animal model: A 5-month longitudinal study. Osteoarthr. Cartil. 2010, 18, 1467–1476. [Google Scholar] [CrossRef]
- Maly, K.; Andres Sastre, E.; Farrell, E.; Meurer, A.; Zaucke, F. COMP and TSP-4: Functional Roles in Articular Cartilage and Relevance in Osteoarthritis. Int. J. Mol. Sci. 2021, 22, 2245. [Google Scholar] [CrossRef] [PubMed]
- Williams, F.M.; Spector, T.D. Biomarkers in osteoarthritis. Arthritis Res. Ther. 2008, 10, 101. [Google Scholar] [CrossRef]
- Chen, B.Y.; Pathak, J.L.; Lin, H.Y.; Guo, W.Q.; Chen, W.J.; Luo, G.; Wang, L.J.; Sun, X.F.; Ding, Y.; Li, J.; et al. Inflammation Triggers Chondrocyte Ferroptosis in TMJOA via HIF-1α/TFRC. J. Dent. Res. 2024, 7, 712–722. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, H.; Chen, J.; Zuo, Q.; Liu, F. Baicalin inhibits IL-1β-induced ferroptosis in human osteoarthritis chondrocytes by activating Nrf-2 signaling pathway. J. Orthop. Surg. Res. 2024, 19, 23. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zheng, Y.; Sun, W.; Zhang, Z.; Liu, J.; Yang, W.; Yuan, W.; Yi, Y.; Wang, J.; Liu, J. D-mannose alleviates osteoarthritis progression by inhibiting chondrocyte ferroptosis in a HIF-2α-dependent manner. Cell Prolif. 2021, 54, e13134. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Zhao, X.; Amevor, F.K.; Du, X.; Wang, Y.; Li, D.; Shu, G.; Tian, Y.; Zhao, X. Therapeutic application of quercetin in aging-related diseases: SIRT1 as a potential mechanism. Front. Immunol. 2022, 13, 943321. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Shen, Y.J.; Chen, M.; Zhao, J.Y.; Chen, S.H.; Zhang, W.; Song, J.K.; Li, L.; Du, G.H. Quercetin attenuates ischemia reperfusion injury by protecting the blood-brain barrier through Sirt1 in MCAO rats. J. Asian Nat. Prod. Res. 2022, 24, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Qiongyue, Z.; Xin, Y.; Meng, P.; Sulin, M.; Yanlin, W.; Xinyi, L.; Xuemin, S. Post-treatment With Irisin Attenuates Acute Kidney Injury in Sepsis Mice Through Anti-Ferroptosis via the SIRT1/Nrf2 Pathway. Front. Pharmacol. 2022, 13, 857067. [Google Scholar] [CrossRef] [PubMed]
- Panahifar, A.; Jaremko, J.L.; Tessier, A.G.; Lambert, R.G.; Maksymowych, W.P.; Fallone, B.G.; Doschak, M.R. Development and reliability of a multi-modality scoring system for evaluation of disease progression in pre-clinical models of osteoarthritis: Celecoxib may possess disease-modifying properties. Osteoarthr. Cartil. 2014, 22, 1639–1650. [Google Scholar] [CrossRef] [PubMed]
- Nirmal, P.; Koppikar, S.; Bhondave, P.; Narkhede, A.; Nagarkar, B.; Kulkarni, V.; Wagh, N.; Kulkarni, O.; Harsulkar, A.; Jagtap, S. Influence of six medicinal herbs on collagenase-induced osteoarthritis in rats. Am. J. Chin. Med. 2013, 41, 1407–1425. [Google Scholar] [CrossRef]
- Chaplan, S.R.; Bach, F.W.; Pogrel, J.W.; Chung, J.M.; Yaksh, T.L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 1994, 53, 55–63. [Google Scholar] [CrossRef]
- Fang, C.; Guo, J.W.; Wang, Y.J.; Li, X.Q.; Zhang, H.; Cui, J.; Hu, Y.; Jing, Y.Y.; Chen, X.; Su, J.C. Diterbutyl phthalate attenuates osteoarthritis in ACLT mice via suppressing ERK/c-fos/NFATc1 pathway, and subsequently inhibiting subchondral osteoclast fusion. Acta Pharmacol. Sin. 2022, 43, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Katri, A.; Dąbrowska, A.; Löfvall, H.; Karsdal, M.A.; Andreassen, K.V.; Thudium, C.S.; Henriksen, K. A dual amylin and calcitonin receptor agonist inhibits pain behavior and reduces cartilage pathology in an osteoarthritis rat model. Osteoarthr. Cartil. 2019, 27, 1339–1346. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.P.; Jovanovic, D.; Fernandes, J.C.; Manning, P.; Connor, J.R.; Currie, M.G.; Di Battista, J.A.; Martel-Pelletier, J. Reduced progression of experimental osteoarthritis in vivo by selective inhibition of inducible nitric oxide synthase. Arthritis Rheum. 1998, 41, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Glasson, S.S.; Chambers, M.G.; Van Den Berg, W.B.; Little, C.B. The OARSI histopathology initiative-recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthr. Cartil. 2010, 18 (Suppl. S3), S17–S23. [Google Scholar] [CrossRef] [PubMed]
- Pritzker, K.P.; Gay, S.; Jimenez, S.A.; Ostergaard, K.; Pelletier, J.P.; Revell, P.A.; Salter, D.; van den Berg, W.B. Osteoarthritis cartilage histopathology: Grading and staging. Osteoarthr. Cartil. 2006, 14, 13–29. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′−3′) | Reverse Primer (5′−3′) |
|---|---|---|
| MMP3 (NM_133523.3) | TTTGGCCGTCTCTTCCATCC | GCATCGATCTTCTGGACGGT |
| MMP13 (NM_133530.1) | TTCTGGTCTTCTGGCACACG | TGGAGCTGCTTGTCCAGGT |
| INOS (NM_012611.4) | AAGAGACGCACAGGCAGAGG | AGCAGGCACACGCAATGATG |
| COX−2 (NM_017232.4) | AGAAGCGAGGACCTGGGTTCAC | ACACCTCTCCACCGATGACCTG |
| Collagen II (NM_001414896.1) | ACGAAGCGGCTGGCAACCTCA | CCCTCGGCCCTCATCTCTACATCA |
| β-Actin (NM_031144.3) | TCCCTGGAGAAGAGCTATGA | ATAGAGCCACCAATCCACAC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruan, H.; Zhu, T.; Wang, T.; Guo, Y.; Liu, Y.; Zheng, J. Quercetin Modulates Ferroptosis via the SIRT1/Nrf−2/HO−1 Pathway and Attenuates Cartilage Destruction in an Osteoarthritis Rat Model. Int. J. Mol. Sci. 2024, 25, 7461. https://doi.org/10.3390/ijms25137461
Ruan H, Zhu T, Wang T, Guo Y, Liu Y, Zheng J. Quercetin Modulates Ferroptosis via the SIRT1/Nrf−2/HO−1 Pathway and Attenuates Cartilage Destruction in an Osteoarthritis Rat Model. International Journal of Molecular Sciences. 2024; 25(13):7461. https://doi.org/10.3390/ijms25137461
Chicago/Turabian StyleRuan, Hongri, Tingting Zhu, Tiantian Wang, Yingchao Guo, Yun Liu, and Jiasan Zheng. 2024. "Quercetin Modulates Ferroptosis via the SIRT1/Nrf−2/HO−1 Pathway and Attenuates Cartilage Destruction in an Osteoarthritis Rat Model" International Journal of Molecular Sciences 25, no. 13: 7461. https://doi.org/10.3390/ijms25137461