
Role of Oxidative Stress and Inflammation in Doxorubicin-Induced Cardiotoxicity: A Brief Account



Abstract
1. Introduction
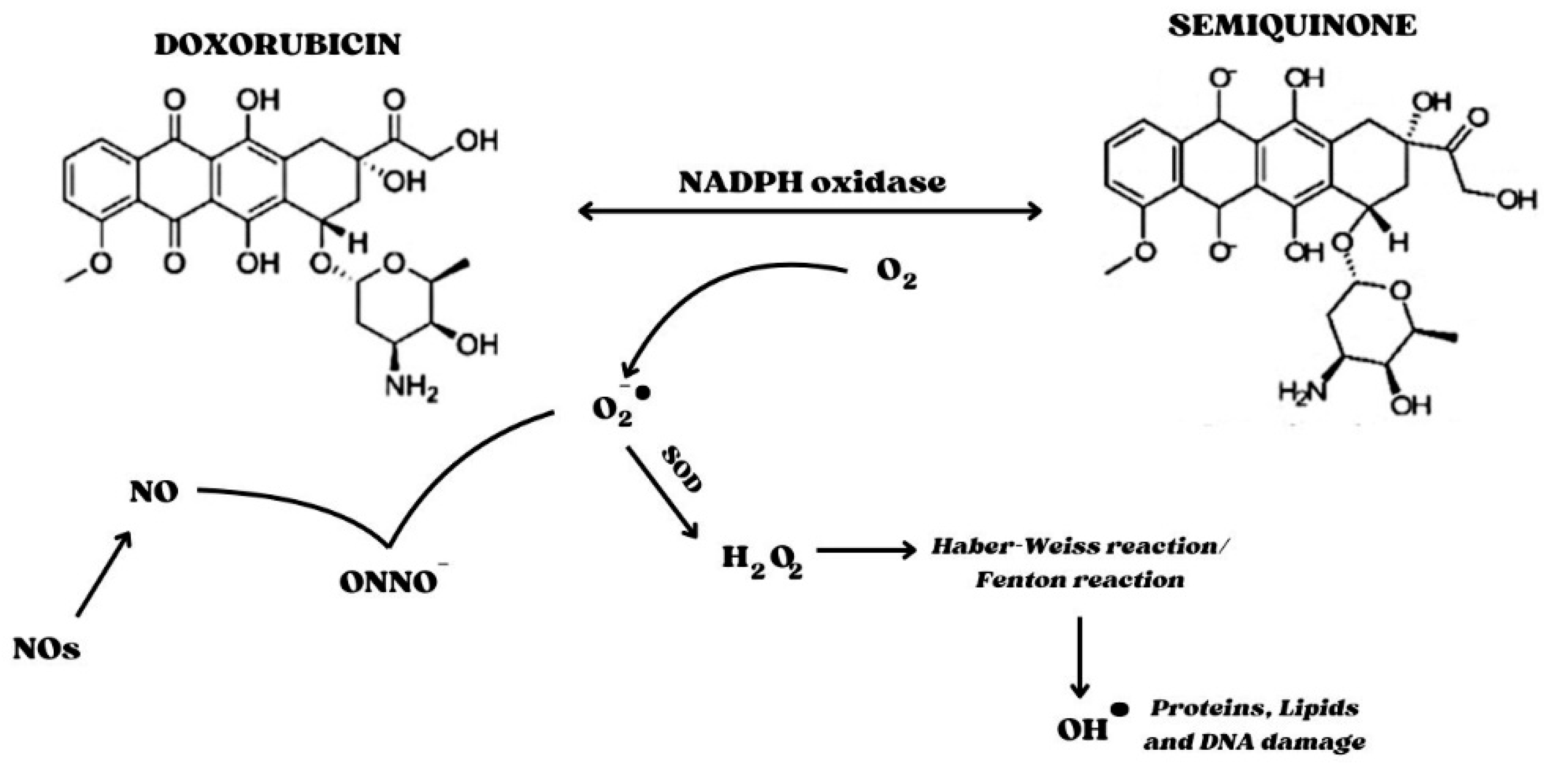
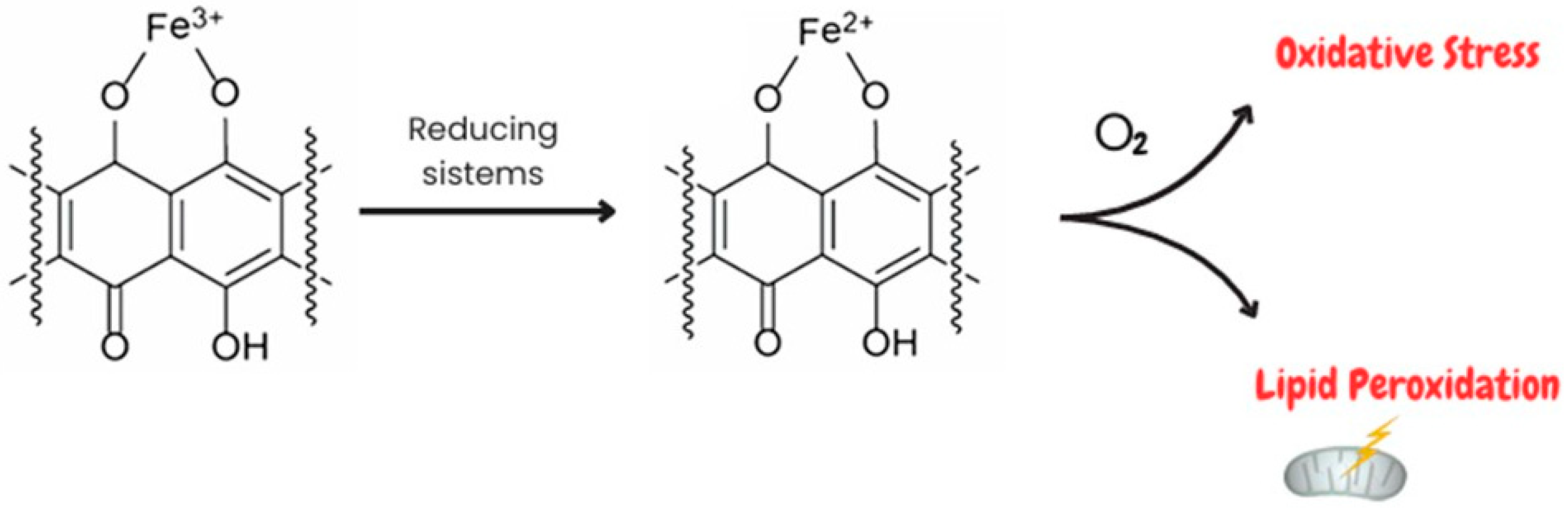
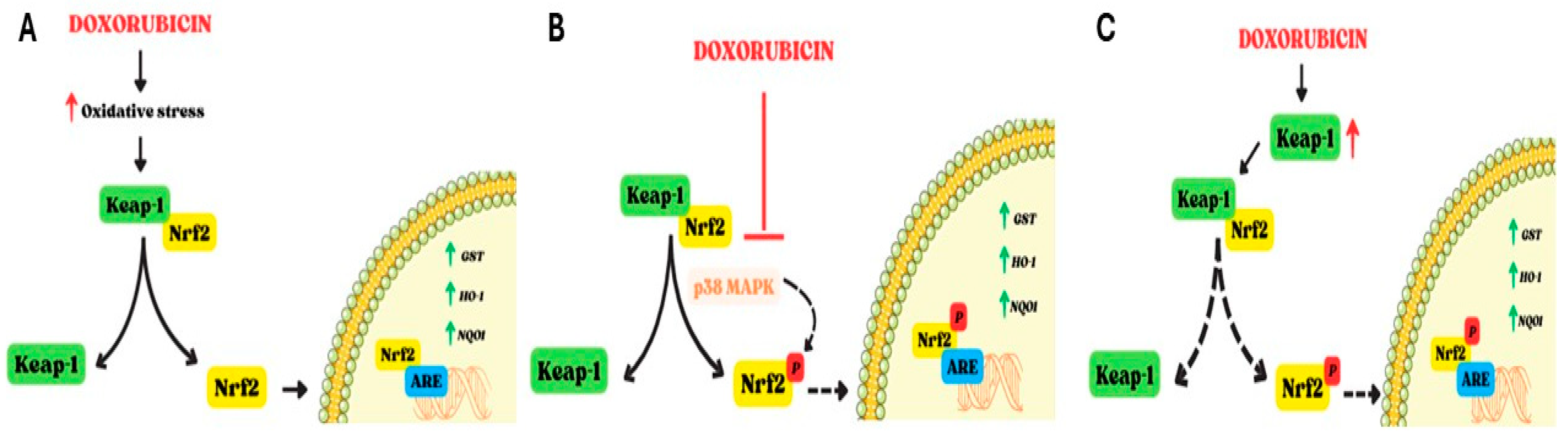
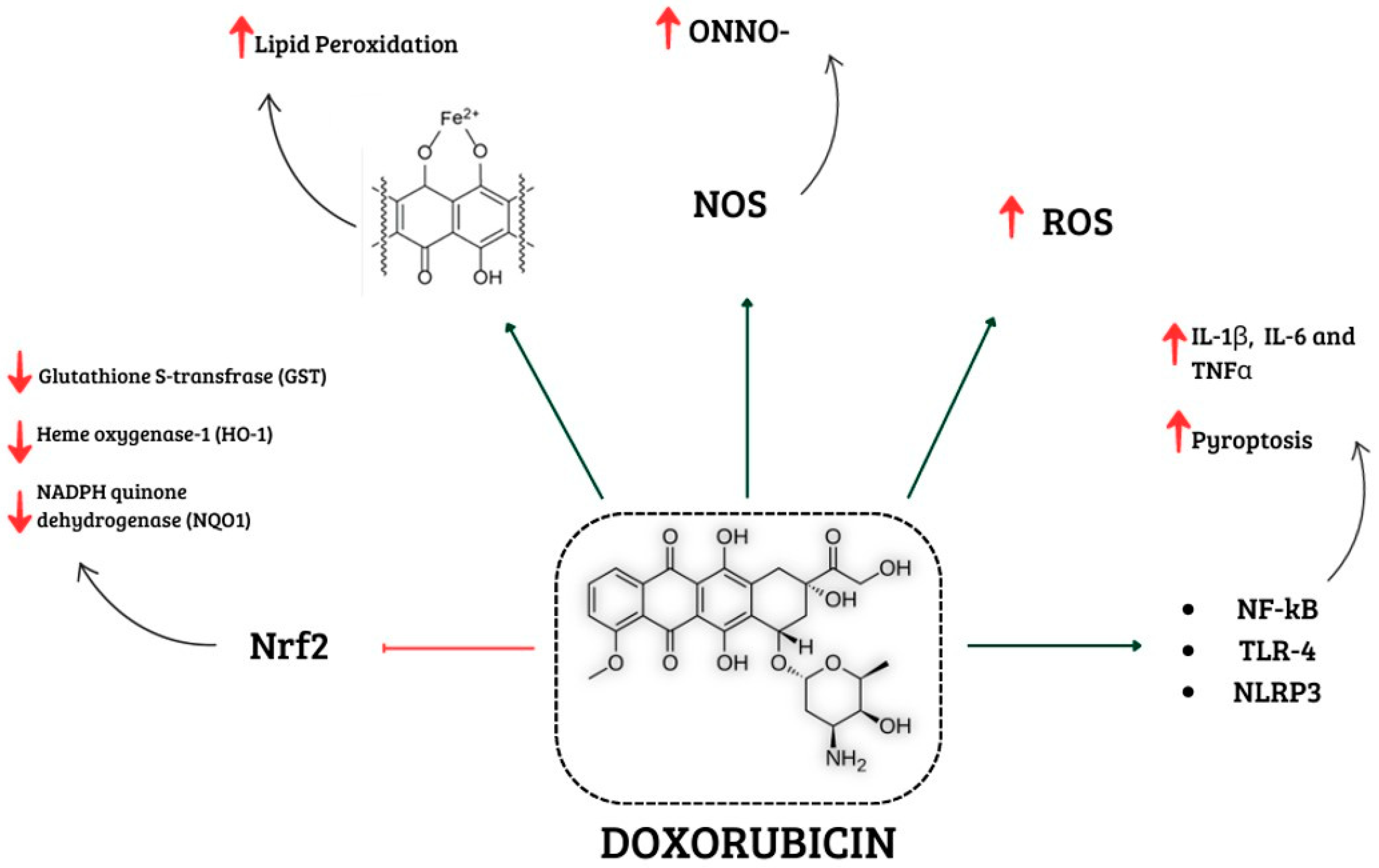
2. Involvement of Oxidative Stress in Doxorubicin-Induced Cardiotoxicity
3. Involvement of Inflammation in Doxorubicin-Induced Cardiotoxicity
4. Therapeutic Strategies to Counteract Doxorubicin-Induced Cardiotoxicity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial | Drug | Study Population | Results | Ref. |
|---|---|---|---|---|
| CarDHA trial | Carvedilol + DHA starting 2 and 7 days before chemotherapy | 32 breast cancer patients (aged 18–75 years) | Improved LVEF | [120] |
| Carvedilol Administration Can Prevent Doxorubicin-Induced Cardiotoxicity: A Double-Blind Randomized Trial | Carvedilol daily during chemotherapy | 70 breast cancer patients | Improved LVEF | [118] |
| Cardioprotective Effects of Carvedilol in Inhibiting Doxorubicin-induced Cardiotoxicity | Carvedilol daily starting 24 h before chemotherapy | 91 breast cancer patients (aged 21–69 years) | Reduced troponin I levels | [121] |
| Prophylactic use of carvedilol to prevent ventricular dysfunction in patients with cancer treated with doxorubicin | Carvedilol during chemotherapy | 154 cancer patients | Improved LVEF | [119] |
| A prospective study to evaluate the efficacy and safety of vitamin E and levocarnitine prophylaxis against doxorubicin-induced cardiotoxicity in adult breast cancer patients | Vitamin E three times daily Levocarnitine four times daily before chemotherapy | 74 breast cancer patients | Reduced CKMB No significant effects on LVEF | [122] |
| STOP CA | Atorvastatin daily starting before Doxo | 300 patients with lymphoma | Improved LVEF | [126] |
| PREVENT | Atorvastatin daily starting 48 h before Doxo | 279 cancer patients | No changes in LVEF Modest effects on oxidative and nitrosative stress biomarkers | [127] |
| Model | Treatment | Effects | Statistical Analysis | Ref. | |
|---|---|---|---|---|---|
| Male Swiss albino mice (10 weeks of age) | Allicin (10 and 20 mg/kg) once daily for 2 weeks Doxorubicin (10 mg/kg) on the 7th, 9th, and 11th days. | ⬇AST, LDH, CK, CK-MB ⬇IL-1β, TNF-α, 8-OHdG, ⬇COX-2, caspase-3 | ⬆GSH,CAT, SOD, GPx | One-way ANOVA followed by post-hoc Duncan’s test. | [130] |
| Male Sprangue Dawley rats (220–225 g) | Acacia hydaspica (200 and 400 mg/kg) once daily for 6 weeks Doxorubicin (3 mg/kg) single dose/week | ⬇AST, LDH, CK, CK-MB | ⬆POD,CAT, SOD, QR | One-way ANOVA followed by Tukey’s test. | [135] |
| Male C57BL/6J mice (22.5–23.5 g) | Resveratrol (20 mg/kg/day) two weeks before Doxo injection Doxorubicin (cumulative dose of 24 mg/kg) | ⬇PTGS2, ACSL4, NCOA4, ⬇p-ERK/ERK, p-p30/p38, ⬇pJNK/JNK,LDH | ⬆GSH, GPX4 | Shapiro–Wilk test and Brown–Forsythe test. Ordinary ANOVA or Welch ANOVA test. | [129] |
| H9C2 | Resveratrol (20 µM) 6 h before Doxorubicin Doxorubicin (1 µM) 24 h | ⬇p-ERK/ERK, p-38/p-38, p-JNK/JNK, ⬇LDH,PTGS2, ACSL4, NCOA4 | ⬆GSH, GPX4 | ||
| Male Sprangue Dawley rats (8 weeks) | Valsartan (20 mg/kg) daily for 6 weeks Doxorubicin (2.5 mg/kg) once per week for 6 weeks | ⬇mRNA expression levels of NOX1,NOX2, NOX4, Bax, Caspase-3, (MMP)2,MMP9, Collagen I, Beclin-1, BNP,ANP, β-MHC, GDF15, TPM1, BGN, POSTN. ⬇ALD, TNF-α, IL-6, BNP,ROS, MDA | ⬆BCL2, Collagen III ⬆SOD | One-way or two-way ANOVA followed by Bonferroni’s post-hoc comparisons | [137] |
| H9C2 | Valsartan (5 or 10 µM) 1 h of pre-treatment Doxorubicin (1 µM) | ⬇ROS | |||
| Male C57BL/6J mice (10–12 weeks) | Rosuvastatin (100 µg/kg) 1 week before Doxorubicin and for further 2 weeks. Doxorubicin (10 mg/kg) single dose | ⬇CAT ⬇NCX1, Ryr2 | ⬆p-PLN | Unpaired Student t-test. ANOVA and the Tukey–Kramer multiple comparison post-hoc test | [125] |
| Sprangue Dawley rats | Dapagliflozin (0.1 mg/kg) per day Doxorubicin (3 mg/kg) four weekly | ⬇p-Smad3, BNP, α-SMA, p-38, ⬇NF-kB-p65, IL-8 | One-way ANOVA followed by Bonferroni’s post-hoc comparisons | [138] | |
| H9C2 | Dapagliflozin (0–20 µM) Doxorubicin (10 µM) for 24 h | ⬇ROS, pSmad3/Smad3, ANP, BNP, α-SMA, Collagen I, Fibronectin, NF-kB-p65, IL-8 | ⬆p-AKT, HO-1, NQO1, SOD | ||
| C57BL/6J male mice | Resveratrol (10 mg/kg)+ Fibroblast growth factor-1 (0.5 mg/kg) for 7 consecutive days Doxorubicin (20 mg/kg) single injection | ⬇CK,LDH, cTnI, cleaved caspase-3 ⬇IL-1β,IL-1α, TNF-α,Mcp1, p-IKBα, p65, ROS | ⬆BCL2/BAX ratio, CAT, SOD1,SOD2, HO-1,NQO1,Sirt1 | One-way ANOVA followed by post-hoc pairwise comparisons using Tukey’s test. | [128] |
| H9C2 | Resveratrol (20 µM) Doxorubicin (1 µM) | ⬆HO-1, Nrf2 (effects were canceled by Sirt1-shRNA | |||
| C57BL/6J male mice (8 weeks) | Rutin (100 mg/kg) for 11 weeks Doxorubicin (3 mg/kg) every other day for 2 weeks starting after one week administration of Rutin | ⬇LC3 II, ATG5, P62, ⬇Caspase-3 | ⬆Akt, Bcl-2 | One-way ANOVA | [145] |
| C57BL/10 mice (10 weeks) | Fluvastatin (100 mg/kg) 4 days before Doxorubicin application Doxorubicin (20 mg/kg) for 5 days | ⬇TNF-α, ⬇Bax | ⬆SOD2.Bcl-2 | Kruskal–Wallis test in conjunction with the Mann–Whitney U post-hoc test | [123] |
| Male Swiss mice and male Sprague Dawley rats | Ginsenoside Rh2 (5 mg/kg, 10 mg/kg,20 mg/kg) total 8 doses Doxorubicin (3 mg/kg or 2 mg/kg) cumulative dose 18 mg/kg or 8 mg/kg | ⬇AST,CK,LDH, MDA | ⬆SOD, CAT, GSH | One-way ANOVA; Student t-test was performed | [131] |
| C57BL/6 mice (6–8 weeks) | Resolvin D1 (2.5 µg/kg) 30 min before Doxorubicin and every day thereafter for the duration of the experiment Doxorubicin (20 mg/kg) once | ⬇LDH,CK-MB, cTnI ⬇IL-1β,IL-6, NF-kB ⬇MDA, NOX2, NOX4, GRP78, CHOP, caspase-12,Bax, c-caspase3 | ⬆SOD,GSH, Nrf-2, OH-1,Bcl-2 | One-way ANOVA followed by Tukey’s test | [136] |
| C57BL/6J mice (8–10 weeks) | Selenium (0.2 mg/kg) 2 weeks Doxorubicin (15 mg/kg) 2 weeks | ⬇CTnI, CK, LDH ⬇IL-1β,TNF-α, IL-18, NLP3,ASC, Caspase-1 | ⬆SOD, GSH ⬆mRNA level of Nrf-2, HO-1,NQO-1, GCLM | One-way ANOVA with Tukey’s post-hoc test | [146] |
| Male Wistar rats (250–300 g) | Panax ginseng (5 g/kg) for 30 days Doxorubicin (2.5 mg/kg) for 2 weeks | ⬇MDA | ⬆GSHPx, SOD | One-way ANOVA followed by Scheffe’s multiple range test | [142] |
| Kunming mice | Apigenin (125 or 250 mg/kg) for 17 days Doxorubicin (3 mg/kg) cumulative dose 24 mg/kg | ⬇LDH, CK,AST ⬇Bax/Bcl-2 ratio ⬇Beclin1, LC3B II/I | ⬆PI3K/AKT/mTOR pathway | One-way ANOVA with LSD post-hoc test | [144] |
| Male Sprague Dawley rats (6–8 weeks) | Rosuvastatin (1 mg/kg) for 6 weeks Doxorubicin (1 mg/kg) for 2 weeks | ⬇AST, CK-MB ⬇HMGB1, RAGE ⬇TNF-α, IFN-γ | ⬆IL-10, IL-4 | One-way ANOVA followed by Tukey’s post-hoc test | [125] |
| BALB/c female mice | Ginsenoside Rh2 (20 mg/kg and 30 mg/kg) every day Doxorubicin (2 mg/kg) cumulative dose of 22 mg/kg | ⬇Caspase-3,Capsase-7, caspase-9 ⬇IL-1β, TNF-α, IL-6 ⬇α-SMA, Smad2, Smad3 | ⬆p 21 | One-way ANOVA followed by Tukey’s multiple comparison test | [132] |
| H9C2 HCF HUVEC | Ginsenoside Rh2 (2.5, 5 and 10 µg/mL) 7 days after Doxorubicin treatment Doxorubicin (100 nM) for 7 days | ⬇α- SMA, Vimentin ⬇MMP2, MMP4, MMP9, MMP14, MMP17, MMP19,MMP23, MMP27, MMP28 | |||
| H9C2 | Neferine (10 µM) Doxorubicin (1 µM) | ⬇NOX2, ROS, p-ERK, p-p38 ⬇Ca2+ intracellular accumulation ⬇COX2, TNF-α, Cytochrome c, Bax | ⬆Cyclin D1 ⬆Bcl-2 | One-way ANOVA followed by Tukey’s multiple comparison test | [133] |
| C57Bl/6 (6 weeks) | Empagliflozin (10 mg/kg) daily for 3 days alone and then in combination with Doxorubicin Doxorubicin (2.17 mg/kg) daily for 7 days | ⬇Ferroptosis, MDA ⬇IL-1β, IL-6, IL-8, MyD88, NLRP3 ⬇MMP-9, Caspase-3 | Non-parametric test. ANOVA test. | [139] | |
| HL-1 | Empagliflozin (50, 100 and 500 nM) Doxorubicin (0.1 to 50 µM) | ⬇intracellular Ca2+, MDA, 4-HNA, NO ⬇IL-1β, IL-6, IL-8 ⬇MyD88, NLRP2 | |||
5. Conclusions
Funding
Conflicts of Interest
References
- Curigliano, G.; Cardinale, D.; Dent, S.; Criscitiello, C.; Aseyev, O.; Lenihan, D.; Cipolla, C.M. Cardiotoxicity of anticancer treatments: Epidemiology, detection, and management. CA Cancer J. Clin. 2016, 66, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Hershman, D.L.; Neugut, A.I. Anthracycline cardiotoxicity: One size does not fit all! J. Natl. Cancer Inst. 2008, 100, 1046–1047. [Google Scholar] [CrossRef]
- Angsutararux, P.; Luanpitpong, S.; Issaragrisil, S. Chemotherapy-Induced Cardiotoxicity: Overview of the Roles of Oxidative Stress. Oxid. Med. Cell Longev. 2015, 2015, 795602. [Google Scholar] [CrossRef]
- Timm, K.N.; Tyler, D.J. The Role of AMPK Activation for Cardioprotection in Doxorubicin-Induced Cardiotoxicity. Cardiovasc. Drugs Ther. 2020, 34, 255–269. [Google Scholar] [CrossRef]
- Ewer, M.S.; Ali, M.K.; Mackay, B.; Wallace, S.; Valdivieso, M.; Legha, S.S.; Benjamin, R.S.; Haynie, T.P. A comparison of cardiac biopsy grades and ejection fraction estimations in patients receiving Adriamycin. J. Clin. Oncol. 1984, 2, 112–117. [Google Scholar] [CrossRef]
- Legha, S.S.; Benjamin, R.S.; Mackay, B.; Ever, M.; Wallace, S.; Valdivieso, M.; Rasmussen, S.L.; Blumenshein, G.R.; Freireich, E.J. Reduction of Doxorubicin Cardiotoxicity by Prolonged Continuous Intravenous Infusion. Ann. Intern. Med. 1982, 96, 133–139. [Google Scholar] [CrossRef]
- Dalen, E.C.; Pal, H.J.; Kremer, L.C. Different dosage schedules for reducing cardiotoxicity in people with cancer receiving anthracycline chemotherapy. Cochrane Database Syst. Rev. 2016, 3, CD005008. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Layard, M.W.; Basa, P.; Davis, H.L.; Von Hoff, A.L., Jr.; Rozencweig, M.; Muggia, F.M. Risk factors for doxorubicin-induced congestive heart failure. Ann. Intern. Med. 1979, 91, 710–717. [Google Scholar] [CrossRef]
- Schirone, L.; D’Ambrosio, L.; Forte, M.; Genovese, R.; Schiavon, S.; Spinosa, G.; Iacovone, G.; Valenti, V.; Frati, G.; Sciarretta, S. Mitochondria and Doxorubicin-Induced Cardiomyopathy: A Complex Interplay. Cells 2022, 11, 2000. [Google Scholar] [CrossRef]
- Fabiani, I.; Aimo, A.; Grigoratos, C.; Castiglione, V.; Gentile, F.; Saccaro, L.F.; Arzilli, C.; Cardinale, D.; Passino, C.; Emdin, M. Oxidative stress and inflammation: Determinants of anthracycline cardiotoxicity and possible therapeutic targets. Heart Fail. Rev. 2021, 26, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Zamorano, J.L.; Lancellotti, P.; Rodriguez Muñoz, D.; Aboyans, V.; Asteggiano, R.; Galderisi, M.; Habib, G.; Lenihan, D.J.; Lip, G.Y.H.; Lyon, A.R.; et al. ESC Scientific Document Group 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur. Heart J 2016, 37, 2768–2801. [Google Scholar] [CrossRef] [PubMed]
- Linders, A.N.; Dias, I.B.; López Fernández, T.; Tocchetti, C.G.; Bomer, N.; Van der Meer, P. A review of the pathophysiological mechanisms of doxorubicin-induced cardiotoxicity and aging. NPJ Aging 2024, 23, 9. [Google Scholar] [CrossRef] [PubMed]
- Belger, C.; Abrahams, C.; Imamdin, A.; Lecour, S. Doxorubicin-induced cardiotoxicity and risk factors. Int. J. Cardiol. Heart Vasc. 2024, 50, 101332. [Google Scholar] [CrossRef] [PubMed]
- Plana, J.C.; Galderisi, M.; Barac, A.; Ewer, M.S.; Ky, B.; Scherrer-Crosbie, M.; Ganame, J.; Sebag, I.A.; Agler, D.A.; Badano, L.P.; et al. Expert consensus for multimodality imaging evaluation of adult patients during and after cancer therapy: A report from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. 2014, 27, 911–939. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Chung, W.B.; Lee, J.E.; Park, C.S.; Park, W.C.; Song, B.J.; Youn, H.J. Candesartan and carvedilol for primary prevention of subclinical cardiotoxicity in breast cancer patients without a cardiovascular risk treated with doxorubicin. Cancer Med. 2021, 10, 3964–3973. [Google Scholar] [CrossRef] [PubMed]
- Stěrba, M.; Popelová, O.; Vávrová, A.; Jirkovský, E.; Kovaříková, P.; Geršl, V.; Simůnek, T. Oxidative stress, redox signaling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxid. Redox Signal 2013, 18, 899–929. [Google Scholar] [CrossRef] [PubMed]
- Hrdina, R.; Gersl, V.; Klimtová, I.; Simůnek, T.; Machácková, J.; Adamcová, M. Anthracycline-induced cardiotoxicity. Acta Medica (Hradec Kral.) 2000, 43, 75–82. [Google Scholar] [CrossRef]
- Kong, C.Y.; Guo, Z.; Song, P.; Zhang, X.; Yuan, Y.P.; Teng, T.; Yan, L.; Tang, Q.Z. Underlying the Mechanisms of Doxorubicin-Induced Acute Cardiotoxicity: Oxidative Stress and Cell Death. Int. J. Biol. Sci. 2022, 18, 760–770. [Google Scholar] [CrossRef]
- Octavia, Y.; Tocchetti, C.G.; Gabrielson, K.L.; Janssens, S.; Crijns, H.J.; Moens, A.L. Doxorubicin-induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. J. Mol. Cell Cardiol. 2012, 52, 1213–1225. [Google Scholar] [CrossRef]
- Milczarek, A.; Starzyński, R.R.; Styś, A.; Jończy, A.; Staroń, R.; Grzelak, A.; Lipiński, P. A drastic superoxide-dependent oxidative stress is prerequisite for the down-regulation of IRP1: Insights from studies on SOD1-deficient mice and macrophages treated with paraquat. PLoS ONE 2017, 12, e0176800. [Google Scholar] [CrossRef] [PubMed]
- Kciuk, M.; Gieleci’nska, A.; Mujwar, S.; Kołat, D.; Kałuzi´nska-Kołat, Z.; Celik, I.; Kontek, R. Doxorubicin—An Agent with Multiple Mechanisms of Anticancer Activity. Cells 2023, 12, 659. [Google Scholar] [CrossRef] [PubMed]
- Bhagat, A.; Shrestha, P.; Kleinerman, E.S. The Innate Immune System in Cardiovascular Diseases and Its Role in Doxorubicin-Induced Cardiotoxicity. Int. J. Mol. Sci. 2022, 23, 14649. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.H.; Fefelova, N.; Pamarthi, S.H.; Gwathmey, J.K. Molecular Mechanisms of Ferroptosis and Relevance to Cardiovascular Disease. Cells 2022, 11, 112726. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhong, T.; Ma, Y.; Wan, X.; Qin, A.; Yao, B.; Zou, H.; Song, Y.; Yin, D. Bnip3 mediates doxorubicin induced cardiomyocyte pyroptosis via caspase-3/GSDME. Life Sci. 2020, 242, 117186. [Google Scholar] [CrossRef] [PubMed]
- Zilinyi, R.; Czompa, A.; Czegledi, A.; Gajtko, A.; Pituk, D.; Lekli, I.; Tosaki, A. The Cardioprotective Effect of Metformin in Doxorubicin-Induced Cardiotoxicity: The Role of Autophagy. Molecules 2018, 23, 1184. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Chen, Y.; Luo, Z.; Nie, G.; Dai, Y. Role of oxidative stress and inflammation-related signaling pathways in doxorubicin-induced cardiomyopathy. Cell Commun. Signal 2023, 21, 61. [Google Scholar] [CrossRef] [PubMed]
- Aryal, B.; Rao, V.A. Deficiency in Cardiolipin Reduces Doxorubicin-Induced Oxidative Stress and Mitochondrial Damage in Human B-Lymphocytes. PLoS ONE 2016, 11, e0158376. [Google Scholar] [CrossRef] [PubMed]
- Christidi, E.; Brunham, L.R. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2021, 12, 339. [Google Scholar] [CrossRef]
- Ruggeri, C.; Gioffré, S.; Achilli, F.; Colombo, G.I.; D’Alessandra, Y. Role of microRNAs in doxorubicin-induced cardiotoxicity: An overview of preclinical models and cancer patients. Heart Fail. Rev. 2018, 23, 109–122. [Google Scholar] [CrossRef]
- Panpan, T.; Yuchen, D.; Xianyong, S.; Meng, L.; Ruijuan, H.; Ranran, D.; Pengyan, Z.; Mingxi, L.; Rongrong, X. Cardiac Remodelling Following Cancer Therapy: A Review. Cardiovasc. Toxicol. 2022, 22, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Perfettini, J.L.; Roumier, T.; Kroemer, G. Mitochondrial fusion and fission in the control of apoptosis. Trends Cell Biol. 2005, 15, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Corrado, M.; Scorrano, L.; Campello, S. Mitochondrial dynamics in cancer and neurodegenerative and neuroinflammatory diseases. Int. J. Cell Biol. 2012, 2012, 729290. [Google Scholar] [CrossRef]
- Aung, L.H.H.; Li, R.; Prabhakar, B.S.; Li, P. Knockdown of Mtfp1 can minimize doxorubicin cardiotoxicity by inhibiting Dnm1l-mediated mitochondrial fission. J. Cell Mol. Med. 2017, 21, 3394–3404. [Google Scholar] [CrossRef] [PubMed]
- Antonny, B.; Burd, C.; De Camilli, P.; Chen, E.; Daumke, O.; Faelber, K.; Ford, M.; Frolov, V.A.; Frost, A.; Hinshaw, J.E.; et al. Membrane fission by dynamin: What we know and what we need to know. EMBO J. 2016, 35, 2270–2284. [Google Scholar] [CrossRef]
- Xia, Y.; Chen, Z.; Chen, A.; Fu, M.; Dong, Z.; Hu, K.; Yang, X.; Zou, Y.; Sun, A.; Qian, J.; et al. LCZ696 improves cardiac function via alleviating Drp1-mediated mitochondrial dysfunction in mice with doxorubicin-induced dilated cardiomyopathy. J. Mol. Cell. Cardiol. 2017, 108, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, T.; Nakamura, K.; Amioka, N.; Hatipoglu, O.F.; Yonezawa, T.; Saito, Y.; Yoshida, M.; Akagi, S.; Ito, H. LCZ696 ameliorates doxorubicin-induced cardiomyocyte toxicity in rats. Sci. Rep. 2022, 12, 4930. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Niu, M.; Hu, X.; He, Y. Targeting mitochondrial dynamics proteins for the treatment of doxorubicin-induced cardiotoxicity. Front. Mol. Biosci. 2023, 10, 1241225. [Google Scholar] [CrossRef]
- Bell, E.L.; Guarente, L. The SirT3 divining rod points to oxidative stress. Mol. Cell 2011, 42, 561–568. [Google Scholar] [CrossRef]
- He, L.; Liu, F.; Li, J. Mitochondrial Sirtuins and Doxorubicin-induced Cardiotoxicity. Cardiovasc. Toxicol. 2021, 21, 179–191. [Google Scholar] [CrossRef]
- Wallace, R. Fractured Symmetries: Information and Control Theory Perspectives on Mitochondrial Dysfunction. Acta Biotheor. 2021, 69, 277–301. [Google Scholar] [CrossRef]
- Wallace, K.B.; Sardão, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Marechal, X.; Montaigne, D.; Marciniak, C.; Marchetti, P.; Hassoun, S.M.; Beauvillain, J.C.; Lancel, S.; Neviere, R. Doxorubicin-induced cardiac dysfunction is attenuated by ciclosporin treatment in mice through improvements in mitochondrial bioenergetics. Clin. Sci. 2011, 121, 405–413. [Google Scholar] [CrossRef]
- Gilleron, M.; Marechal, X.; Montaigne, D.; Franczak, J.; Neviere, R.; Lancel, S. NADPH oxidases participate to doxorubicin-induced cardiac myocyte apoptosis. Biochem. Biophys. Res. Commun. 2009, 388, 727–731. [Google Scholar] [CrossRef]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases in mechano-transduction: Mechanisms and consequences. Antioxid. Redox Signal 2014, 20, 887–898. [Google Scholar] [CrossRef]
- Efentakis, P.; Doerschmann, H.; Witzler, C.; Siemer, S.; Nikolaou, P.E.; Kastritis, E.; Stauber, R.; Dimopoulos, M.A.; Wenzel, P.; Andreadou, I.; et al. Investigating the Vascular Toxicity Outcomes of the Irreversible Proteasome Inhibitor Carfilzomib. Int. J. Mol. Sci. 2020, 21, 5185. [Google Scholar] [CrossRef]
- Sirker, A.; Zhang, M.; Shah, A.M. NADPH oxidases in cardiovascular disease: Insights from in vivo models and clinical studies. Basic. Res. Cardiol. 2011, 106, 735–747. [Google Scholar] [CrossRef]
- Tayeh, Z.; Ofir, R. Asteriscus graveolens Extract in Combination with Cisplatin/Etoposide/Doxorubicin Suppresses Lymphoma Cell Growth through Induction of Caspase-3 Dependent Apoptosis. Int. J. Mol. Sci. 2018, 19, 2219. [Google Scholar] [CrossRef]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed. Pharmacother. 2021, 139, 111708. [Google Scholar] [CrossRef]
- Zhao, Y.; McLaughlin, D.; Robinson, E.; Harvey, A.P.; Hookham, M.B.; Shah, A.M.; McDermott, B.J.; Grieve, D.J. Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with Doxorubicin chemotherapy. Cancer Res. 2010, 70, 9287–9297. [Google Scholar] [CrossRef]
- Ma, Z.G.; Kong, C.Y.; Wu, H.M.; Song, P.; Zhang, X.; Yuan, Y.P.; Deng, W.; Tang, Q.Z. Toll-like receptor 5 deficiency diminishes doxorubicin-induced acute cardiotoxicity in mice. Theranostics 2020, 10, 11013–11025. [Google Scholar] [CrossRef]
- Graham, K.A.; Kulawiec, M.; Owens, K.M.; Li, X.; Desouki, M.M.; Chandra, D.; Singh, K.K. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol. Ther. 2010, 10, 223–231. [Google Scholar] [CrossRef]
- Zeng, C.; Duan, F.; Hu, J.; Luo, B.; Huang, B.; Lou, X.; Sun, X.; Li, H.; Zhang, X.; Yin, S.; et al. NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol. 2020, 34, 101523. [Google Scholar] [CrossRef]
- Wang, Y.; Liao, J.; Luo, Y.; Li, M.; Su, X.; Yu, B.; Teng, J.; Wang, H.; Lv, X. Berberine Alleviates Doxorubicin-Induced Myocardial Injury and Fibrosis by Eliminating Oxidative Stress and Mitochondrial Damage via Promoting Nrf-2 Pathway Activation. Int. J. Mol. Sci. 2023, 24, 3257. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef]
- Zhang, W.; Feng, C.; Jiang, H. Novel target for treating Alzheimer’s Diseases: Crosstalk between the Nrf2 pathway and autophagy. Ageing Res. Rev. 2021, 65, 101207. [Google Scholar] [CrossRef]
- Pi, J.; Bai, Y.; Reece, J.M.; Williams, J.; Liu, D.; Freeman, M.L.; Waalkes, M.P. Molecular mechanism of human Nrf2 activation and degradation: Role of sequential phosphorylation by protein kinase CK2. Free. Radic. Biol. Med. 2007, 42, 1797–1806. [Google Scholar] [CrossRef]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21Cip1/WAF1 upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef]
- Milani, P.; Ambrosi, G.; Gammoh, O.; Blandini, F.; Cereda, C. SOD1 and DJ-1 converge at Nrf2 pathway: A clue for antioxidant therapeutic potential in Neurodegeneration. Oxidative Med. Cell. Longev. 2013, 2013, 836760. [Google Scholar] [CrossRef]
- Su, S.; Li, Q.; Xiong, C.; Li, J.; Zhang, R.; Niu, Y.; Zhao, L.; Wang, Y.; Guo, H. Sesamin ameliorates doxorubicin-induced cardiotoxicity: Involvement of Sirt1 and Mn-SOD pathway. Toxicol. Lett. 2014, 224, 257–263. [Google Scholar] [CrossRef]
- Songbo, M.; Lang, H.; Xinyong, C.; Bin, X.; Ping, Z.; Liang, S. Oxidative stress injury in doxorubicin-induced cardiotoxicity. Toxicol. Lett. 2019, 307, 41–48. [Google Scholar] [CrossRef]
- Deng, J.; Huang, M.; Wu, H. Protective effect of limonin against doxorubicin-induced cardiotoxicity via activating nuclear factor—like 2 and Sirtuin 2 signaling pathways. Bioengineered 2021, 12, 7975–7984. [Google Scholar] [CrossRef]
- Shi, K.N.; Li, P.B.; Su, H.X.; Gao, J.; Li, H.H. MK-886 protects against cardiac ischaemia/reperfusion injury by activating proteasome-Keap1-NRF2 signalling. Redox Biol. 2023, 62, 102706. [Google Scholar] [CrossRef]
- Zhang, Y.; Ahmad, K.A.; Khan, F.U.; Yan, S.; Ihsan, A.U.; Ding, Q. Chitosan oligo saccharides prevent doxorubicin-induced oxidative stress and cardiac apoptosis through activating p38 and JNK MAPK mediated Nrf2/ARE pathway. Chem. Biol. Interact. 2019, 305, 54–65. [Google Scholar] [CrossRef]
- Li, N.; Jiang, W.; Wang, W.; Xiong, R.; Wu, X.; Geng, Q. Ferroptosis and its emerging roles in cardiovascular diseases. Pharm. Res. 2021, 166, 105466. [Google Scholar] [CrossRef]
- Pecoraro, M.; Pala, B.; Di Marcantonio, M.C.; Muraro, R.; Marzocco, S.; Pinto, A.; Mincione, G.; Popolo, A. Doxorubicin induced oxidative and nitrosative stress: Mitochondrial connexin 43 is at the crossroads. Int. J. Mol. Med. 2020, 46, 1197–1209. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Rajesh, M.; Bátkai, S.; Kashiwaya, Y.; Haskó, G.; Liaudet, L.; Szabó, C.; Pacher, P. Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1466–H1483. [Google Scholar] [CrossRef]
- Ohshima, H.; Sawa, T.; Akaike, T. 8-nitroguanine, a product of nitrative DNA damage caused by reactive nitrogen species: Formation, occurrence, and implications in inflammation and carcinogenesis. Antioxid. Redox Signal 2006, 8, 1033–1045. [Google Scholar] [CrossRef]
- Moini Jazani, A.; Arabzadeh, A.; Haghi-Aminjan, H.; Nasimi Doost Azgomi, R. The role of ginseng derivatives against chemotherapy-induced cardiotoxicity: A systematic review of non-clinical studies. Front. Cardiovasc. Med. 2023, 10, 1022360. [Google Scholar] [CrossRef]
- Li, T.; Singal, P.K. Adriamycin-induced early changes in myocardial antioxidant enzymes and their modulation by probucol. Circulation 2000, 102, 2105–2110. [Google Scholar] [CrossRef]
- Neilan, T.G.; Blake, S.L.; Ichinose, F.; Raher, M.J.; Buys, E.S.; Jassal, D.S.; Furutani, E.; Perez-Sanz, T.M.; Graveline, A.; Janssens, S.P.; et al. Disruption of nitric oxide synthase 3 protects against the cardiac injury, dysfunction, and mortality induced by doxorubicin. Circulation 2007, 116, 506–514. [Google Scholar] [CrossRef]
- Rawat, D.K.; Hecker, P.; Watanabe, M.; Chettimada, S.; Levy, R.J.; Okada, T.; Edwards, J.G.; Gupte, S.A. Glucose-6-phosphate dehydrogenase and NADPH redox regulates cardiac myocyte L-type calcium channel activity and myocardial contractile function. PLoS ONE 2012, 7, e45365. [Google Scholar] [CrossRef]
- Vásquez-Vivar, J.; Martasek, P.; Hogg, N.; Masters, B.S.; Pritchard, K.A., Jr.; Kalyanaraman, B. Endothelial nitric oxide synthase-dependent superoxide generation from adriamycin. Biochemistry 1997, 36, 11293–11297. [Google Scholar] [CrossRef]
- Cappetta, D.; De Angelis, A.; Sapio, L.; Prezioso, L.; Illiano, M.; Quaini, F.; Rossi, F.; Berrino, L.; Naviglio, S.; Urbanek, K. Oxidative Stress and Cellular Response to Doxorubicin: A Common Factor in the Complex Milieu of Anthracycline Cardiotoxicity. Oxid. Med. Cell Longev. 2017, 2017, 1521020. [Google Scholar] [CrossRef]
- Russo, M.; Guida, F.; Paparo, L.; Trinchese, G.; Aitoro, R.; Avagliano, C.; Fiordelisi, A.; Napolitano, F.; Mercurio, V.; Sala, V.; et al. The novel butyrate derivative phenylalanine-butyramide protects from doxorubicin-induced cardiotoxicity. Eur. J. Heart Fail. 2019, 21, 519–528. [Google Scholar] [CrossRef]
- Bartesaghi, S.; Radi, R. Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol. 2018, 14, 618–625. [Google Scholar] [CrossRef]
- Milano, G.; Biemmi, V.; Lazzarini, E.; Balbi, C.; Ciullo, A.; Bolis, S.; Ameri, P.; Di Silvestre, D.; Mauri, P.; Barile, L.; et al. Intravenous administration of cardiac progenitor cell-derived exosomes protects against doxorubicin/trastuzumab-induced cardiac toxicity. Cardiovasc. Res. 2020, 116, 383–392. [Google Scholar] [CrossRef]
- Akolkar, G.; Bagchi, A.K.; Ayyappan, P.; Jassal, D.S.; Singal, P.K. Doxorubicin-induced nitrosative stress is mitigated by vitamin C via the modulation of nitric oxide synthases. Am. J. Physiol. Cell Physiol. 2017, 312, C418–C427. [Google Scholar] [CrossRef]
- Liu, B.; Li, H.; Qu, H.; Sun, B. Nitric oxide synthase expressions in ADR-induced cardiomyopathy in rats. J. Biochem. Mol. Biol. 2006, 39, 759–765. [Google Scholar] [CrossRef]
- Xu, Z.; Lin, S.; Wu, W.; Tan, h.; Wang, Z.; Cheng, C.; Lu, L.; Zhang, X. Ghrelin prevents doxorubicin-induced cardiotoxicity through TNF-alpha/NF-kappaB pathways and mitochondrial protective mechanisms. Toxicology 2008, 247, 133–138. [Google Scholar] [CrossRef]
- Pecoraro, M.; Del Pizzo, M.; Marzocco, S.; Sorrentino, R.; Ciccarelli, M.; Iaccarino, G.; Pinto, A.; Popolo, A. Inflammatory mediators in a short-time mouse model of doxorubicin-induced cardiotoxicity. Toxicol. Appl. Pharmacol. 2016, 293, 44–52. [Google Scholar] [CrossRef]
- Nagy, A.; Börzsei, D.; Hoffmann, A.; Törökl, S.; Veszelka, M.; Almási, N.; Varga, C.; Szabó, R. A Comprehensive Overview on Chemotherapy-Induced Cardiotoxicity: Insights into the Underlying Inflammatory and Oxidative Mechanisms. Cardiovasc. Drugs Ther. 2024. [Google Scholar] [CrossRef]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Andersson, U. Targeting Inflammation Driven by HMGB1. Front. Immunol. 2020, 11, 484. [Google Scholar] [CrossRef]
- Shirazi, L.F.; Bissett, J.; Romeo, F.; Mehta, J.L. Role of Inflammation in Heart Failure. Curr. Atheroscler. Rep. 2017, 19, 27. [Google Scholar] [CrossRef]
- Bartekova, M.; Radosinska, J.; Jelemensky, M.; Dhalla, N.S. Role of cytokines and inflammation in heart function during health and disease. Heart Failure Rev. 2018, 23, 733–758. [Google Scholar] [CrossRef]
- Tavakoli, D.Z.; Singla, D.K. Embryonic stem cell-derived exosomes inhibit doxorubicin-induced TLR4-NLRP3-mediated cell death-pyroptosis. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H460–H471. [Google Scholar] [CrossRef]
- Singla, D.K.; Johnson, T.A.; Tavakoli, D.Z. Exosome Treatment Enhances Anti-Inflammatory M2 Macrophages and Reduces Inflammation-Induced Pyroptosis in Doxorubicin-Induced Cardiomyopathy. Cells 2019, 8, 1224. [Google Scholar] [CrossRef]
- Ren, G.; Zhang, X.; Xiamo, Y.; Zhang, W.; Wang, Y.; Ma, W.; Wang, X.; Song, P.; Lai, L.; Chen, H.; et al. ABRO1 promotes NLRP3 inflammasome activation through regulation of NLRP3 deuibiquitination. EMBO J. 2019, 38, e100376. [Google Scholar] [CrossRef]
- Li, L.L.; Wei, L.; Zhang, N.; Wei, W.Y.; Hu, C.; Deng, W.; Tang, Q.Z. Levosimendan Protects against Doxorubicin-Induced Cardiotoxicity by Regulating the PTEN/Akt Pathway. Biomed. Res. Int. 2020, 2020, 8593617. [Google Scholar] [CrossRef]
- Ni, C.; Ma, P.; Wang, R.; Lou, X.; Liu, X.; Qin, Y.; Xue, R.; Blasig, I.; Erben, U.; Qin, Z. Doxorubicin-induced cardiotoxicity involves IFNγ-mediated metabolic reprogramming in cardiomyocytes. J. Pathol. 2019, 247, 320–332. [Google Scholar] [CrossRef]
- Ye, B.; Shi, X.; Xu, J.; Dai, S.; Xu, J.; Fan, X.; Han, B.; Han, J. Gasdermin D mediates doxorubicin-induced cardiomyocyte pyroptosis and cardiotoxicity via directly binding to doxorubicin and changes in mitochondrial damage. Transl. Res. 2022, 248, 36–50. [Google Scholar] [CrossRef]
- Lan, Y.; Wang, Y.; Huang, K.; Zeng, Q. Heat shock protein 22 attenuates doxorubicin induced cardiotoxicity via regulating inflammation and apoptosis. Front. Pharm. 2020, 11, 257. [Google Scholar] [CrossRef]
- Ye, S.; Su, L.; Shan, P.; Ye, B.; Wu, S.; Liang, G.; Huang, W. LCZ696 Attenuated Doxorubicin-Induced Chronic Cardiomyopathy Through the TLR2-MyD88 Complex Formation. Front. Cell Dev. Biol. 2021, 13, 654051. [Google Scholar] [CrossRef]
- Quagliariello, V.; Passariello, M.; Di Mauro, A.; Cipullo, C.; Paccone, A.; Barbieri, A.; Palma, G.; Luciano, A.; Buccolo, S.; Bisceglia, I.; et al. Immune checkpoint inhibitor therapy increases systemic SDF-1, cardiac DAMPs Fibronectin-EDA, S100/Calgranulin, galectine-3, and NLRP3-MyD88-chemokine pathways. Front. Cardiovasc. Med. 2022, 8, 930797, Erratum in Front. Cardiovasc. Med. 2023, 10, 1129873. [Google Scholar] [CrossRef]
- D’Angelo, N.A.; Noronha, M.A.; Câmara, M.C.C.; Kurnik, I.S.; Feng, C.; Araujo, V.H.S.; Santos, J.H.P.M.; Feitosa, V.; Molino, J.V.D.; Rangel-Yagui, C.O.; et al. Doxorubicin nanoformulations on therapy against cancer: An overview from the last 10 years. Biomater. Adv. 2022, 133, 112623. [Google Scholar] [CrossRef]
- Li, X.R.; Cheng, X.H.; Zhang, G.N.; Wang, X.X.; Huang, J.M. Cardiac safety analysis of first-line chemotherapy drug pegylated liposomal doxorubicin in ovarian cancer. J. Ovarian Res. 2022, 15, 96. [Google Scholar] [CrossRef]
- Dempke, W.C.M.; Zielinski, R.; Winkler, C.; Silberman, S.; Reuther, S.; Priebe, W. Anthracycline-induced cardiotoxicity—Are we about to clear this hurdle? Eur. J. Cancer 2023, 185, 94–104. [Google Scholar] [CrossRef]
- Pendlebury, A.; De Bernardo, R.; Rose, P.G. Long-term use of pegylated liposomal doxorubicin to a cumulative dose of 4600 mg/m2 in recurrent ovarian cancer. Anti-Cancer Drugs 2017, 28, 815–817. [Google Scholar] [CrossRef]
- Misra, R.; Das, M.; Sahoo, B.S.; Sahoo, S.K. Reversal of multidrug resistance in vitro by co-delivery of MDR1 targeting siRNA and doxorubicin using a novel cationic poly(lactide-co-glycolide) nanoformulation. Int. J. Pharm. 2014, 475, 372–384. [Google Scholar] [CrossRef]
- Xing, M.; Yan, F.; Yu, S.; Shen, P. Efficacy and Cardiotoxicity of Liposomal Doxorubicin Based Chemotherapy in Advanced Breast Cancer: A Meta-Analysis of Ten Randomized Controlled Trials. PLoS ONE 2015, 10, e0133569. [Google Scholar] [CrossRef]
- Cagel, M.; Grotz, E.; Bernabeu, E.; Moretton, M.A.; Chiappetta, D.A. Doxorubicin: Nanotechnological overviews from bench to bedside. Drug Discov. Today 2017, 22, 270–281. [Google Scholar] [CrossRef]
- Gallo, E.; Diaferia, C.; Rosa, E.; Smaldone, G.; Morelli, G.; Accardo, A. Peptide-Based Hydrogels and Nanogels for Delivery of Doxorubicin. Int. J. Nanomed. 2021, 16, 1617–1630. [Google Scholar] [CrossRef]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071. [Google Scholar] [CrossRef]
- Norouzi, M.; Yathindranath, V.; Thliveris, J.A.; Kopec, B.M.; Siahaan, T.J.; Miller, D.W. Doxorubicin-loaded iron oxide nanoparticles for glioblastoma therapy: A combinational approach for enhanced delivery of nanoparticles. Sci. Rep. 2020, 10, 11292. [Google Scholar] [CrossRef]
- Mu, Q.; Kievit, F.M.; Kant, R.J.; Lin, G.; Jeon, M.; Zhang, M. Anti-HER2/neu peptide-conjugated iron oxide nanoparticles for targeted delivery of paclitaxel to breast cancer cells. Nanoscale 2015, 7, 18010–18014. [Google Scholar] [CrossRef]
- Norouzi, M.; Nazari, B.; Miller, D.W. Injectable hydrogel-based drug delivery systems for local cancer therapy. Drug Discov. Today 2016, 1, 1835–1849. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, D.; Wei, X.; Zhu, X.; Li, J.; Tu, C.; Su, Y.; Wu, J.; Zhu, B.; Yan, D. Multifunctional pH-sensitive superparamagnetic iron-oxide nanocomposites for targeted drug delivery and MR imaging. J. Control Release 2013, 169, 228–238. [Google Scholar] [CrossRef]
- Chang, Y.; Li, Y.; Meng, X.; Liu, N.; Sun, D.; Wang, J. Dendrimer functionalized water soluble magnetic iron oxide conjugates as dual imaging probe for tumor targeting and drug delivery. Polym. Chem. 2013, 4, 789–794. [Google Scholar] [CrossRef]
- Shen, B.; Ma, Y.; Yu, S.; Ji, C. Smart multifunctional magnetic nanoparticle-based drug delivery system for cancer thermo chemotherapy and intracellular imaging. ACS Appl. Mater. Interfaces 2016, 8, 24502–24508. [Google Scholar] [CrossRef]
- Weiss, G.; Loyevsky, M.; Gordeuk, V.R. Dexrazoxane (ICRF-187). Gen. Pharmacol. 1999, 32, 155–158. [Google Scholar] [CrossRef]
- Hutchins, K.K.; Siddeek, H.; Franco, V.I.; Lipshultz, S.E. Prevention of cardiotoxicity among survivors of childhood cancer. Br. J. Clin. Pharmacol. 2017, 83, 455–465. [Google Scholar] [CrossRef]
- Kourek, C.; Touloupaki, M.; Rempakos, A.; Loritis, K.; Tsougkos, E.; Paraskevaidis, I.; Briasoulis, A. Cardioprotective Strategies from Cardiotoxicity in Cancer Patients: A Comprehensive Review. J. Cardiovasc. Dev. Dis. 2022, 9, 9259. [Google Scholar] [CrossRef]
- Yue, T.L.; Cheng, H.Y.; Lysko, P.G.; McKenna, P.J.; Feuerstein, R.; Gu, J.L.; Lysko, K.A.; Davis, L.L.; Feuerstein, G. Carvedilol, a new vasodilator and beta adrenoceptor antagonist, is an antioxidant and free radical scavenger. J. Pharmacol. Exp. Ther. 1992, 263, 92–98. [Google Scholar] [PubMed]
- Spallarossa, P.; Garibaldi, S.; Altieri, P.; Fabbi, P.; Manca, V.; Nasti, S.; Rossettin, P.; Ghigliotti, G.; Ballestrero, A.; Patrone, F.; et al. Carvedilol prevents doxorubicin-induced free radical release and apoptosis in cardiomyocytes in vitro. J. Mol. Cell Cardiol. 2004, 37, 837–846. [Google Scholar] [CrossRef]
- Tashakori Beheshti, A.; Mostafavi Toroghi, H.; Hosseini, G.; Zarifian, A.; Homaei Shandiz, F.; Fazlinezhad, A. Carvedilol Administration Can Prevent Doxorubicin-Induced Cardiotoxicity: A Double-Blind Randomized Trial. Cardiology 2016, 134, 47–53. [Google Scholar] [CrossRef]
- Abuosa, A.M.; Elshiekh, A.H.; Qureshi, K.; Abrar, M.B.; Kholeif, M.A.; Kinsara, A.J.; Andejani, A.; Ahmed, A.H.; Cleland, J.G.F. Prophylactic use of carvedilol to prevent ventricular dysfunction in patients with cancer treated with doxorubicin. Indian Heart J. 2018, 70 (Suppl. S3), S96–S100. [Google Scholar] [CrossRef]
- Carrasco, R.; Ramirez, M.C.; Nes, K.; Schuster, A.; Aguayo, R.; Morales, M.; Ramos, C.; Hasson, D.; Sotomayor, C.G.; Henriquez, P.; et al. Prevention of doxorubicin-induced Cardiotoxicity by pharmacological non-hypoxic myocardial preconditioning based on Docosahexaenoic Acid (DHA) and carvedilol direct antioxidant effects: Study protocol for a pilot, randomized, double-blind, controlled trial (CarDHA trial). Trials 2020, 21, 137. [Google Scholar] [CrossRef]
- Nabati, M.; Janbabai, G.; Baghyari, S.; Esmaili, K.; Yazdani, J. Cardioprotective Effects of Carvedilol in Inhibiting Doxorubicin-induced Cardiotoxicity. J. Cardiovasc. Pharmacol. 2017, 69, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, I.; Connolly, C.; Anis, M.; Mustafa, H.; Oosthuizen, F.; Viljoen, M. A prospective study to evaluate the efficacy and safety of vitamin E and levocarnitine prophylaxis against doxorubicin-induced cardiotoxicity in adult breast cancer patients. J. Oncol. Pharm. Pract. 2024, 30, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Riad, A.; Bien, S.; Westermann, D.; Becher, P.M.; Loya, K.; Landmesser, U.; Kroemer, H.K.; Schultheiss, H.P.; Tschöpe, C. Pretreatment with statin attenuates the cardiotoxicity of Doxorubicin in mice. Cancer Res. 2009, 69, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Dadson, K.; Thavendiranathan, P.; Hauck, L.; Grothe, D.; Azam, M.A.; Stanley-Hasnain, S.; Mahiny-Shahmohammady, D.; Si, D.; Bokhari, M.; Lai, P.F.H.; et al. Statins Protect Against Early Stages of Doxorubicin-induced Cardiotoxicity Through the Regulation of Akt Signaling and SERCA2. CJC Open 2022, 4, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lu, X.; Liu, Z.; Du, K. Rosuvastatin reduces the pro-inflammatory effects of adriamycin on the expression of HMGB1 and RAGE in rats. Int. J. Mol. Med. 2018, 42, 3415–3423. [Google Scholar] [CrossRef] [PubMed]
- Neilan, T.G.; Quinaglia, T.; Onoue, T.; Mahmood, S.S.; Drobni, Z.D.; Gilman, H.K.; Smith, A.; Heemelaar, J.C.; Brahmbhatt, P.; Ho, J.S.; et al. Atorvastatin for Anthracycline-Associated Cardiac Dysfunction: The STOP-CA Randomized Clinical Trial. JAMA 2023, 330, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Makhlin, I.; Demissei, B.G.; D’Agostino, R.; Hundley, W.G.; Baleanu-Gogonea, C.; Wilcox, N.S.; Chen, A.; Smith, A.M.; O’Connell, N.S.; Januzzi, J.; et al. Statins Do Not Significantly Affect Oxidative Nitrosative Stress Biomarkers in the PREVENT Randomized Clinical Trial. Clin. Cancer Res. 2024, 30, 2370–2376. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Liu, Q.; Gao, T.; Li, J.; Zhang, J.; Chen, O.; Cao, C.; Mao, M.; Xiao, M.; Zhang, X.; et al. Resveratrol and FGF1 Synergistically Ameliorates Doxorubicin-Induced Cardiotoxicity via Activation of SIRT1-NRF2 Pathway. Nutrients 2022, 14, 4017. [Google Scholar] [CrossRef]
- Chen, L.; Sun, X.; Wang, Z.; Chen, M.; He, Y.; Zhang, H.; Han, D.; Zheng, L. Resveratrol protects against doxorubicin-induced cardiotoxicity by attenuating ferroptosis through modulating the MAPK signaling pathway. Toxicol. Appl. Pharmacol. 2024, 482, 116794. [Google Scholar] [CrossRef]
- Abdel-Daim, M.M.; Kilany, O.E.; Khalifa, H.A.; Ahmed, A.A.M. Allicin ameliorates doxorubicin-induced cardiotoxicity in rats via suppression of oxidative stress, inflammation and apoptosis. Cancer Chemother. Pharmacol. 2017, 80, 745–753. [Google Scholar] [CrossRef]
- Wang, H.; Yu, P.; Gou, H.; Zhang, J.; Zhu, M.; Wang, Z.H.; Tian, J.W.; Jiang, Y.T.; Fu, F.H. Cardioprotective Effects of 20(S)-Ginsenoside Rh2 against Doxorubicin-Induced Cardiotoxicity In Vitro and In Vivo. Evid. Based Complement. Alternat Med. 2012, 2012, 506214. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Yun, Y.; Cui, C.; Kim, S. Ginsenoside Rh2 mitigates doxorubicin-induced cardiotoxicity by inhibiting apoptotic and inflammatory damage and weakening pathological remodelling in breast cancer-bearing mice. Cell Prolif. 2022, 55, e13246. [Google Scholar] [CrossRef] [PubMed]
- Priya, L.B.; Baskaran, R.; Huang, C.Y.; Padma, V.V. Neferine ameliorates cardiomyoblast apoptosis induced by doxorubicin: Possible role in modulating NADPH oxidase/ROS-mediated NFκB redox signaling cascade. Sci. Rep. 2017, 7, 12283. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Wang, Q.; Sun, S.; Xu, G.; Wu, Q.; Qi, M.; Bai, F.; Yu, J. Astragaloside IV promotes the eNOS/NO/cGMP pathway and improves left ventricular diastolic function in rats with metabolic syndrome. J. Int. Med. Res. 2019, 48, 0300060519826848. [Google Scholar] [CrossRef] [PubMed]
- Afsar, T.; Razak, S.; Batoo, K.M.; Khan, M.R. Acacia hydaspica R. Parker prevents doxorubicin-induced cardiac injury by attenuation of oxidative stress and structural Cardiomyocyte alterations in rats. BMC Complement. Altern. Med. 2017, 17, 554. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, J.; Zhao, M.; Liu, J.; Ye, J.; Xu, Y.; Wang, Z.; Ye, D.; Li, D.; Wan, J. Resolvin D1 Attenuates Doxorubicin-Induced Cardiotoxicity by Inhibiting Inflammation, Oxidative and Endoplasmic Reticulum Stress. Front. Pharmacol. 2022, 12, 749899. [Google Scholar] [CrossRef]
- Cheng, D.; Chen, L.; Tu, W.; Wang, H.; Wang, Q.; Meng, L.; Li, Z.; Yu, Q. Protective effects of valsartan administration on doxorubicin induced myocardial injury in rats and the role of oxidative stress and NOX2/NOX4 signaling. Mol. Med. Rep. 2020, 22, 4151–4162. [Google Scholar] [CrossRef]
- Hsieh, P.L.; Chu, P.M.; Cheng, H.C.; Huang, Y.T.; Chou, W.C.; Tsai, K.L.; Chan, S.H. Dapagliflozin Mitigates Doxorubicin-Caused Myocardium Damage by Regulating AKT-Mediated Oxidative Stress, Cardiac Remodeling, and Inflammation. Int. J. Mol. Sci. 2022, 23, 10146. [Google Scholar] [CrossRef] [PubMed]
- Quagliariello, V.; De Laurentiis, M.; Rea, D.; Barbieri, A.; Monti, M.G.; Carbone, A.; Paccone, A.; Altucci, L.; Conte, M.; Canale, M.L.; et al. The SGLT-2 inhibitor empagliflozin improves myocardial strain, reduces cardiac fibrosis and pro-inflammatory cytokines in non-diabetic mice treated with doxorubicin. Cardiovasc. Diabetol. 2021, 23, 150. [Google Scholar] [CrossRef]
- Draginic, N.; Jakovljevic, V.; Andjic, M.; Jeremic, J.; Srejovic, I.; Rankovic, M.; Tomovic, M.; Nikolic Turnic, T.; Svistunov, A.; Bolevich, S.; et al. Melissa officinalis L. as a Nutritional Strategy for Cardioprotection. Front. Physiol. 2021, 12, 661778. [Google Scholar] [CrossRef]
- Wan, Y.; Wang, J.; Xu, J.F.; Tang, F.; Chen, L.; Tan, Y.Z.; Rao, C.L.; Ao, H.; Peng, C. Panax ginseng and its ginsenosides: Potential candidates for the prevention and treatment of chemotherapy-induced side effects. J. Ginseng Res. 2021, 45, 617–630. [Google Scholar] [CrossRef] [PubMed]
- You, J.S.; Huang, H.F.; Chang, Y.L. Panax ginseng reduces adriamycin-induced heart failure in rats. Phytother. Res. 2005, 19, 1018–1022. [Google Scholar] [CrossRef] [PubMed]
- Syahputra, R.A.; Harahap, U.; Dalimunthe, A.; Nasution, M.P.; Satria, D. The Role of Flavonoids as a Cardioprotective Strategy against Doxorubicin-Induced Cardiotoxicity: A Review. Molecules 2022, 27, 1320. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Sun, H.; Zha, W.; Cui, W.; Xu, L.; Min, Q.; Wu, J. Apigenin Attenuates Adriamycin-Induced Cardiomyocyte Apoptosis via the PI3K/AKT/mTOR Pathway. Evid. Based Complement. Alternat Med. 2017, 2017, 2590676. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yang, L.; Ma, J.; Lu, L.; Wang, X.; Ren, J.; Yang, J. Rutin attenuates doxorubicin-induced cardiotoxicity via regulating autophagy and apoptosis. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1904–1911. [Google Scholar] [CrossRef]
- Yang, H.B.; Lu, Z.Y.; Yuan, W.; Li, W.D.; Mao, S. Selenium Attenuates Doxorubicin-Induced Cardiotoxicity Through Nrf2-NLRP3 Pathway. Biol. Trace Elem. Res. 2022, 200, 2848–2856. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitale, R.; Marzocco, S.; Popolo, A. Role of Oxidative Stress and Inflammation in Doxorubicin-Induced Cardiotoxicity: A Brief Account. Int. J. Mol. Sci. 2024, 25, 7477. https://doi.org/10.3390/ijms25137477
Vitale R, Marzocco S, Popolo A. Role of Oxidative Stress and Inflammation in Doxorubicin-Induced Cardiotoxicity: A Brief Account. International Journal of Molecular Sciences. 2024; 25(13):7477. https://doi.org/10.3390/ijms25137477
Chicago/Turabian StyleVitale, Roberta, Stefania Marzocco, and Ada Popolo. 2024. "Role of Oxidative Stress and Inflammation in Doxorubicin-Induced Cardiotoxicity: A Brief Account" International Journal of Molecular Sciences 25, no. 13: 7477. https://doi.org/10.3390/ijms25137477
APA StyleVitale, R., Marzocco, S., & Popolo, A. (2024). Role of Oxidative Stress and Inflammation in Doxorubicin-Induced Cardiotoxicity: A Brief Account. International Journal of Molecular Sciences, 25(13), 7477. https://doi.org/10.3390/ijms25137477