
Pathogenesis of Endometriosis and Endometriosis-Associated Cancers


{kind=link}
{kind=link}
Abstract
:1. Introduction
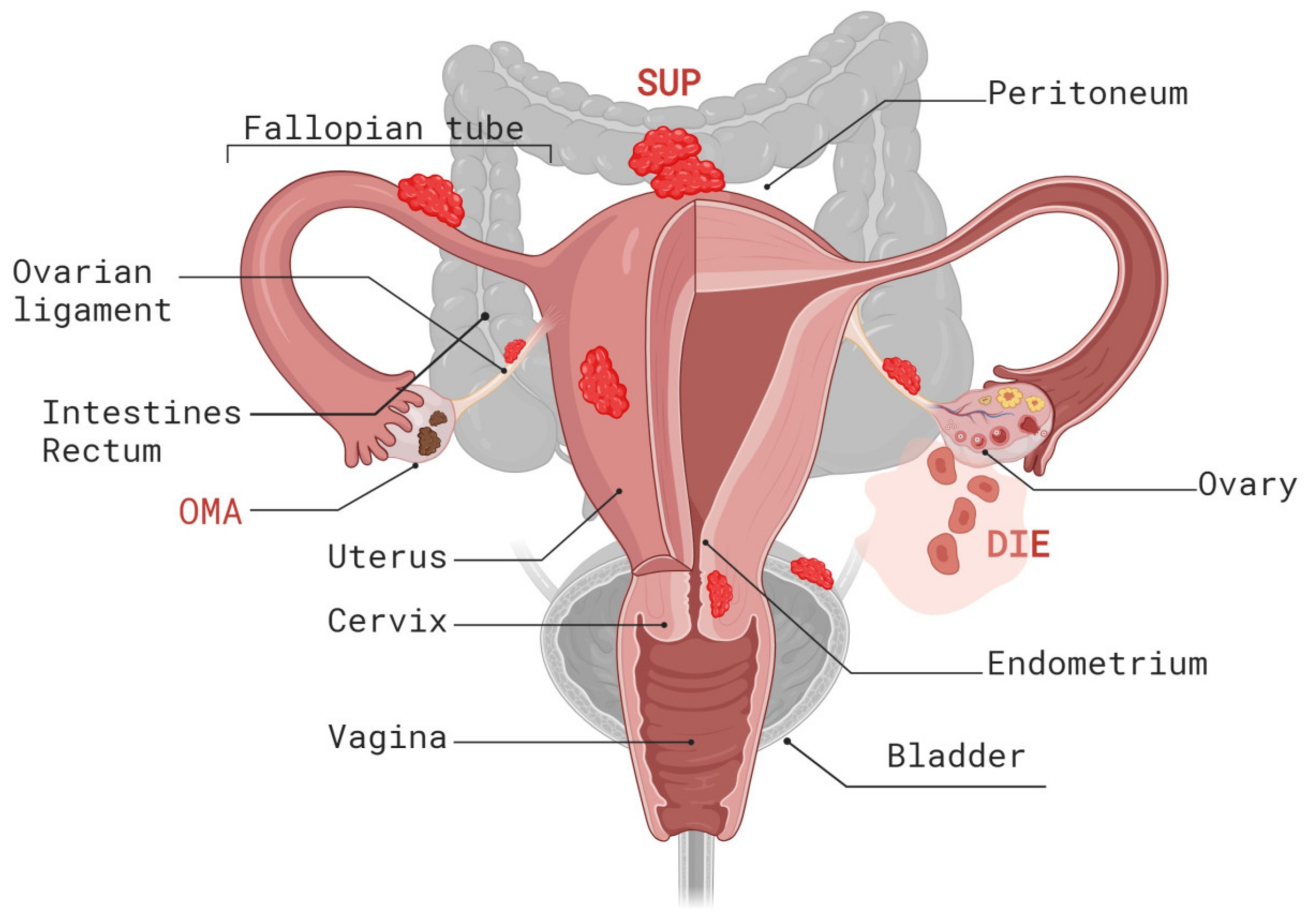
2. Histopathogenesis
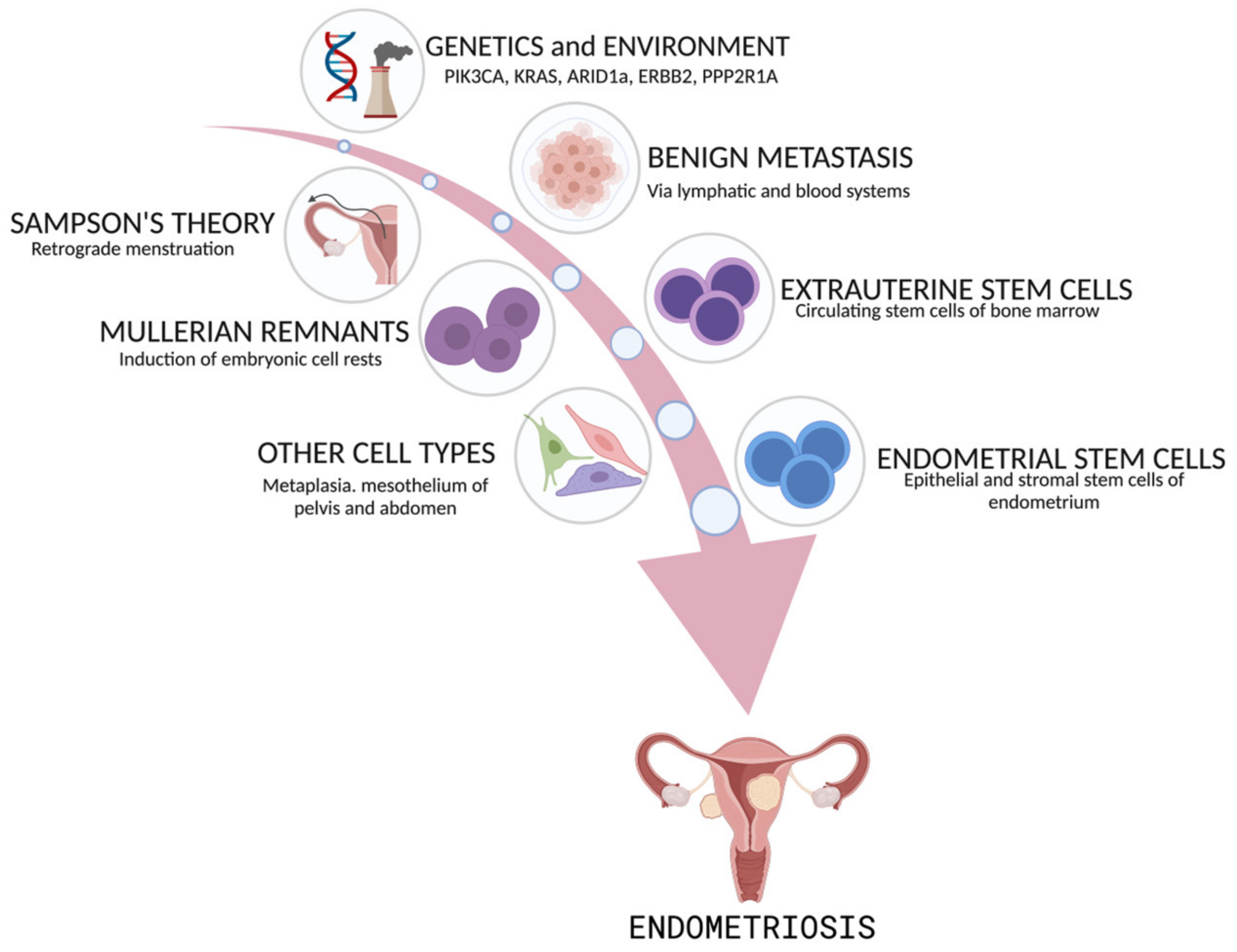
3. Origin of Endometriosis
4. Risk Factors for Endometriosis
4.1. Genetic Risk Factors
4.2. Modifiable Risk Factors
5. The Genomic Landscape of Endometriosis
5.1. Risk Loci and Epigenetic Events Associated with Steroid Hormone Imbalance
5.2. Identification of Additional Endometriosis Risk Loci
6. Endometriosis as a Neoplastic Disease
6.1. Somatic Mutations in Cancer Driver Genes
6.2. The Role of Cancer Driver Mutations in Endometriosis Development
6.3. Endometriosis Development: Clonal Expansion and Heterogeneity
7. Endometriosis-Associated Cancers
Genetic Basis of Endometriosis-Associated Cancers
8. Novel Insights into Disease Pathology: 3D Organoids, Single-Cell Omics, and Imaging Studies
8.1. Organoid Models of Endometriosis
8.2. Single-Cell Omics Approaches
9. Crosstalk between Endometriotic Lesions and the Microenvironment
10. Conclusions and Future Directions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Correction Statement
References
- Giudice, L.C. Clinical practice. Endometriosis. N. Engl. J. Med. 2010, 362, 2389–2398. [Google Scholar] [CrossRef] [PubMed]
- Bulun, S.E.; Yilmaz, B.D.; Sison, C.; Miyazaki, K.; Bernardi, L.; Liu, S.; Kohlmeier, A.; Yin, P.; Milad, M.; Wei, J. Endometriosis. Endocr. Rev. 2019, 40, 1048–1079. [Google Scholar] [CrossRef] [PubMed]
- Zondervan, K.T.; Becker, C.M.; Koga, K.; Missmer, S.A.; Taylor, R.N.; Vigano, P. Endometriosis. Nat. Rev. Dis. Primers 2018, 4, 9. [Google Scholar] [CrossRef]
- Zondervan, K.T.; Becker, C.M.; Missmer, S.A. Endometriosis. N. Engl. J. Med. 2020, 382, 1244–1256. [Google Scholar] [CrossRef]
- Maddern, J.; Grundy, L.; Castro, J.; Brierley, S.M. Pain in Endometriosis. Front. Cell. Neurosci. 2020, 14, 590823. [Google Scholar] [CrossRef]
- Anaf, V.; El Nakadi, I.; Simon, P.; Van de Stadt, J.; Fayt, I.; Simonart, T.; Noel, J.C. Preferential infiltration of large bowel endometriosis along the nerves of the colon. Hum. Reprod. 2004, 19, 996–1002. [Google Scholar] [CrossRef]
- Anaf, V.; Simon, P.; El Nakadi, I.; Fayt, I.; Simonart, T.; Buxant, F.; Noel, J.C. Hyperalgesia, nerve infiltration and nerve growth factor expression in deep adenomyotic nodules, peritoneal and ovarian endometriosis. Hum. Reprod. 2002, 17, 1895–1900. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, R.; Mardon, H.; Barlow, D.; Kennedy, S. Delay in the diagnosis of endometriosis: A survey of women from the USA and the UK. Hum. Reprod. 1996, 11, 878–880. [Google Scholar] [CrossRef]
- D’Hooghe, T.M.; Debrock, S. Endometriosis, retrograde menstruation and peritoneal inflammation in women and in baboons. Hum. Reprod. Update 2002, 8, 84–88. [Google Scholar] [CrossRef]
- Guerriero, S.; Condous, G.; van den Bosch, T.; Valentin, L.; Leone, F.P.; Van Schoubroeck, D.; Exacoustos, C.; Installe, A.J.; Martins, W.P.; Abrao, M.S.; et al. Systematic approach to sonographic evaluation of the pelvis in women with suspected endometriosis, including terms, definitions and measurements: A consensus opinion from the International Deep Endometriosis Analysis (IDEA) group. Ultrasound Obstet. Gynecol. 2016, 48, 318–332. [Google Scholar] [CrossRef]
- Bazot, M.; Bharwani, N.; Huchon, C.; Kinkel, K.; Cunha, T.M.; Guerra, A.; Manganaro, L.; Bunesch, L.; Kido, A.; Togashi, K.; et al. European society of urogenital radiology (ESUR) guidelines: MR imaging of pelvic endometriosis. Eur. Radiol. 2017, 27, 2765–2775. [Google Scholar] [CrossRef]
- Keckstein, J.; Hoopmann, M.; Merz, E.; Grab, D.; Weichert, J.; Helmy-Bader, S.; Wolfler, M.; Bajka, M.; Mechsner, S.; Schafer, S.; et al. Expert opinion on the use of transvaginal sonography for presurgical staging and classification of endometriosis. Arch. Gynecol. Obstet. 2023, 307, 5–19. [Google Scholar] [CrossRef]
- Nirgianakis, K.; Ma, L.; McKinnon, B.; Mueller, M.D. Recurrence Patterns after Surgery in Patients with Different Endometriosis Subtypes: A Long-Term Hospital-Based Cohort Study. J. Clin. Med. 2020, 9, 496. [Google Scholar] [CrossRef] [PubMed]
- Vercellini, P.; Vigano, P.; Somigliana, E.; Fedele, L. Endometriosis: Pathogenesis and treatment. Nat. Rev. Endocrinol. 2014, 10, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Canis, M.; Donnez, J.G.; Guzick, D.S.; Halme, J.K.; Rock, J.A.; Schenken, R.S.; Vernon, M.W. Revised American Society for Reproductive Medicine classification of endometriosis: 1996. Fertil. Steril. 1997, 67, 817–821. [Google Scholar] [CrossRef]
- Johnson, N.P.; Hummelshoj, L.; Adamson, G.D.; Keckstein, J.; Taylor, H.S.; Abrao, M.S.; Bush, D.; Kiesel, L.; Tamimi, R.; Sharpe-Timms, K.L.; et al. World Endometriosis Society consensus on the classification of endometriosis. Hum. Reprod. 2017, 32, 315–324. [Google Scholar] [CrossRef]
- Keckstein, J.; Saridogan, E.; Ulrich, U.A.; Sillem, M.; Oppelt, P.; Schweppe, K.W.; Krentel, H.; Janschek, E.; Exacoustos, C.; Malzoni, M.; et al. The #Enzian classification: A comprehensive non-invasive and surgical description system for endometriosis. Acta Obstet. Gynecol. Scand. 2021, 100, 1165–1175. [Google Scholar] [CrossRef]
- Chapron, C.; Santulli, P.; de Ziegler, D.; Noel, J.C.; Anaf, V.; Streuli, I.; Foulot, H.; Souza, C.; Borghese, B. Ovarian endometrioma: Severe pelvic pain is associated with deeply infiltrating endometriosis. Hum. Reprod. 2012, 27, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Saavalainen, L.; Lassus, H.; But, A.; Tiitinen, A.; Harkki, P.; Gissler, M.; Pukkala, E.; Heikinheimo, O. Risk of Gynecologic Cancer according to the Type of Endometriosis. Obstet. Gynecol. 2018, 131, 1095–1102. [Google Scholar] [CrossRef]
- Sampson, J.A. Peritoneal endometriosis due to the menstrual dissemination of endometrial tissue into the peritoneal cavity. Am. J. Obstet. Gynecol. 1927, 14, 422–469. [Google Scholar] [CrossRef]
- Signorile, P.G.; Baldi, A. New evidence in endometriosis. Int. J. Biochem. Cell Biol. 2015, 60, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Taylor, H.S. Endometrial cells derived from donor stem cells in bone marrow transplant recipients. JAMA 2004, 292, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Padykula, H.A.; Coles, L.G.; Okulicz, W.C.; Rapaport, S.I.; McCracken, J.A.; King, N.W., Jr.; Longcope, C.; Kaiserman-Abramof, I.R. The basalis of the primate endometrium: A bifunctional germinal compartment. Biol. Reprod. 1989, 40, 681–690. [Google Scholar] [CrossRef] [PubMed]
- de Miguel-Gomez, L.; Lopez-Martinez, S.; Frances-Herrero, E.; Rodriguez-Eguren, A.; Pellicer, A.; Cervello, I. Stem Cells and the Endometrium: From the Discovery of Adult Stem Cells to Pre-Clinical Models. Cells 2021, 10, 595. [Google Scholar] [CrossRef] [PubMed]
- Sasson, I.E.; Taylor, H.S. Stem cells and the pathogenesis of endometriosis. Ann. N. Y. Acad. Sci. 2008, 1127, 106–115. [Google Scholar] [CrossRef]
- Leyendecker, G.; Herbertz, M.; Kunz, G.; Mall, G. Endometriosis results from the dislocation of basal endometrium. Hum. Reprod. 2002, 17, 2725–2736. [Google Scholar] [CrossRef]
- Du, H.; Taylor, H.S. Contribution of bone marrow-derived stem cells to endometrium and endometriosis. Stem Cells 2007, 25, 2082–2086. [Google Scholar] [CrossRef] [PubMed]
- Sakr, S.; Naqvi, H.; Komm, B.; Taylor, H.S. Endometriosis impairs bone marrow-derived stem cell recruitment to the uterus whereas bazedoxifene treatment leads to endometriosis regression and improved uterine stem cell engraftment. Endocrinology 2014, 155, 1489–1497. [Google Scholar] [CrossRef]
- Mok-Lin, E.Y.; Wolfberg, A.; Hollinquist, H.; Laufer, M.R. Endometriosis in a patient with Mayer-Rokitansky-Kuster-Hauser syndrome and complete uterine agenesis: Evidence to support the theory of coelomic metaplasia. J. Pediatr. Adolesc. Gynecol. 2010, 23, e35–e37. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Nakaoka, H.; Suda, K.; Yoshihara, K.; Ishiguro, T.; Yachida, N.; Saito, K.; Ueda, H.; Sugino, K.; Mori, Y.; et al. Spatiotemporal dynamics of clonal selection and diversification in normal endometrial epithelium. Nat. Commun. 2022, 13, 943. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Yoshihara, K.; Suda, K.; Nakaoka, H.; Yachida, N.; Ueda, H.; Sugino, K.; Mori, Y.; Yamawaki, K.; Tamura, R.; et al. Three-dimensional understanding of the morphological complexity of the human uterine endometrium. iScience 2021, 24, 102258. [Google Scholar] [CrossRef]
- Maruyama, T. A Revised Stem Cell Theory for the Pathogenesis of Endometriosis. J. Pers. Med. 2022, 12, 216. [Google Scholar] [CrossRef]
- Cousins, F.L.; Pandoy, R.; Jin, S.; Gargett, C.E. The Elusive Endometrial Epithelial Stem/Progenitor Cells. Front. Cell Dev. Biol. 2021, 9, 640319. [Google Scholar] [CrossRef]
- Nguyen, H.P.T.; Xiao, L.; Deane, J.A.; Tan, K.S.; Cousins, F.L.; Masuda, H.; Sprung, C.N.; Rosamilia, A.; Gargett, C.E. N-cadherin identifies human endometrial epithelial progenitor cells by in vitro stem cell assays. Hum. Reprod. 2017, 32, 2254–2268. [Google Scholar] [CrossRef] [PubMed]
- Valentijn, A.J.; Palial, K.; Al-Lamee, H.; Tempest, N.; Drury, J.; Von Zglinicki, T.; Saretzki, G.; Murray, P.; Gargett, C.E.; Hapangama, D.K. SSEA-1 isolates human endometrial basal glandular epithelial cells: Phenotypic and functional characterization and implications in the pathogenesis of endometriosis. Hum. Reprod. 2013, 28, 2695–2708. [Google Scholar] [CrossRef]
- Tan, Y.; Flynn, W.F.; Sivajothi, S.; Luo, D.; Bozal, S.B.; Dave, M.; Luciano, A.A.; Robson, P.; Luciano, D.E.; Courtois, E.T. Single-cell analysis of endometriosis reveals a coordinated transcriptional programme driving immunotolerance and angiogenesis across eutopic and ectopic tissues. Nat. Cell Biol. 2022, 24, 1306–1318. [Google Scholar] [CrossRef]
- Masuda, H.; Anwar, S.S.; Buhring, H.J.; Rao, J.R.; Gargett, C.E. A novel marker of human endometrial mesenchymal stem-like cells. Cell Transplant. 2012, 21, 2201–2214. [Google Scholar] [CrossRef] [PubMed]
- Darzi, S.; Werkmeister, J.A.; Deane, J.A.; Gargett, C.E. Identification and Characterization of Human Endometrial Mesenchymal Stem/Stromal Cells and Their Potential for Cellular Therapy. Stem Cells Transl. Med. 2016, 5, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Laschke, M.W.; Giebels, C.; Nickels, R.M.; Scheuer, C.; Menger, M.D. Endothelial progenitor cells contribute to the vascularization of endometriotic lesions. Am. J. Pathol. 2011, 178, 442–450. [Google Scholar] [CrossRef]
- Becker, C.M.; Beaudry, P.; Funakoshi, T.; Benny, O.; Zaslavsky, A.; Zurakowski, D.; Folkman, J.; D’Amato, R.J.; Ryeom, S. Circulating endothelial progenitor cells are up-regulated in a mouse model of endometriosis. Am. J. Pathol. 2011, 178, 1782–1791. [Google Scholar] [CrossRef]
- Ono, M.; Maruyama, T.; Masuda, H.; Kajitani, T.; Nagashima, T.; Arase, T.; Ito, M.; Ohta, K.; Uchida, H.; Asada, H.; et al. Side population in human uterine myometrium displays phenotypic and functional characteristics of myometrial stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 18700–18705. [Google Scholar] [CrossRef]
- Cervello, I.; Gil-Sanchis, C.; Mas, A.; Delgado-Rosas, F.; Martinez-Conejero, J.A.; Galan, A.; Martinez-Romero, A.; Martinez, S.; Navarro, I.; Ferro, J.; et al. Human endometrial side population cells exhibit genotypic, phenotypic and functional features of somatic stem cells. PLoS ONE 2010, 5, e10964. [Google Scholar] [CrossRef]
- Masuda, H.; Matsuzaki, Y.; Hiratsu, E.; Ono, M.; Nagashima, T.; Kajitani, T.; Arase, T.; Oda, H.; Uchida, H.; Asada, H.; et al. Stem cell-like properties of the endometrial side population: Implication in endometrial regeneration. PLoS ONE 2010, 5, e10387. [Google Scholar] [CrossRef]
- Miyazaki, K.; Maruyama, T.; Masuda, H.; Yamasaki, A.; Uchida, S.; Oda, H.; Uchida, H.; Yoshimura, Y. Stem cell-like differentiation potentials of endometrial side population cells as revealed by a newly developed in vivo endometrial stem cell assay. PLoS ONE 2012, 7, e50749. [Google Scholar] [CrossRef] [PubMed]
- Cervello, I.; Mas, A.; Gil-Sanchis, C.; Peris, L.; Faus, A.; Saunders, P.T.; Critchley, H.O.; Simon, C. Reconstruction of endometrium from human endometrial side population cell lines. PLoS ONE 2011, 6, e21221. [Google Scholar] [CrossRef]
- Gardner, G.H.; Greene, R.R.; Ranney, B. The histogenesis of endometriosis; recent contributions. Obstet. Gynecol. 1953, 1, 615–637. [Google Scholar] [PubMed]
- Simpson, J.L.; Elias, S.; Malinak, L.R.; Buttram, V.C., Jr. Heritable aspects of endometriosis. I. Genetic studies. Am. J. Obstet. Gynecol. 1980, 137, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Parasar, P.; Ozcan, P.; Terry, K.L. Endometriosis: Epidemiology, Diagnosis and Clinical Management. Curr. Obstet. Gynecol. Rep. 2017, 6, 34–41. [Google Scholar] [CrossRef]
- Frey, G.H. The familial occurrence of endometriosis; report of five instances and review of the literature. Am. J. Obstet. Gynecol. 1957, 73, 418–421. [Google Scholar] [CrossRef]
- Kennedy, S.; Hadfield, R.; Mardon, H.; Barlow, D. Age of onset of pain symptoms in non-twin sisters concordant for endometriosis. Hum. Reprod. 1996, 11, 403–405. [Google Scholar] [CrossRef]
- Treloar, S.A.; O’Connor, D.T.; O’Connor, V.M.; Martin, N.G. Genetic influences on endometriosis in an Australian twin sample. Fertil. Steril. 1999, 71, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Bulun, S.E.; Cheng, Y.H.; Pavone, M.E.; Xue, Q.; Attar, E.; Trukhacheva, E.; Tokunaga, H.; Utsunomiya, H.; Yin, P.; Luo, X.; et al. Estrogen receptor-β, estrogen receptor-α, and progesterone resistance in endometriosis. Semin. Reprod. Med. 2010, 28, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Nnoaham, K.E.; Webster, P.; Kumbang, J.; Kennedy, S.H.; Zondervan, K.T. Is early age at menarche a risk factor for endometriosis? A systematic review and meta-analysis of case-control studies. Fertil. Steril. 2012, 98, 702–712.e706. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, H.; Geirsson, R.T.; Steinthorsdottir, V.; Jonsson, H.; Manolescu, A.; Kong, A.; Ingadottir, G.; Gulcher, J.; Stefansson, K. Genetic factors contribute to the risk of developing endometriosis. Hum. Reprod. 2002, 17, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Hemmert, R.; Schliep, K.C.; Willis, S.; Peterson, C.M.; Louis, G.B.; Allen-Brady, K.; Simonsen, S.E.; Stanford, J.B.; Byun, J.; Smith, K.R. Modifiable life style factors and risk for incident endometriosis. Paediatr. Perinat. Epidemiol. 2019, 33, 19–25. [Google Scholar] [CrossRef]
- Jurkiewicz-Przondziono, J.; Lemm, M.; Kwiatkowska-Pamula, A.; Ziolko, E.; Wojtowicz, M.K. Influence of diet on the risk of developing endometriosis. Ginekol. Pol. 2017, 88, 96–102. [Google Scholar] [CrossRef]
- Parazzini, F.; Vigano, P.; Candiani, M.; Fedele, L. Diet and endometriosis risk: A literature review. Reprod. Biomed. Online 2013, 26, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Fiuza-Luces, C.; Garatachea, N.; Berger, N.A.; Lucia, A. Exercise is the real polypill. Physiology 2013, 28, 330–358. [Google Scholar] [CrossRef]
- Sofo, V.; Gotte, M.; Lagana, A.S.; Salmeri, F.M.; Triolo, O.; Sturlese, E.; Retto, G.; Alfa, M.; Granese, R.; Abrao, M.S. Correlation between dioxin and endometriosis: An epigenetic route to unravel the pathogenesis of the disease. Arch. Gynecol. Obstet. 2015, 292, 973–986. [Google Scholar] [CrossRef]
- Zhou, B.; Yang, L.; Sun, Q.; Cong, R.; Gu, H.; Tang, N.; Zhu, H.; Wang, B. Cigarette smoking and the risk of endometrial cancer: A meta-analysis. Am. J. Med. 2008, 121, 501–508.e503. [Google Scholar] [CrossRef]
- Aban, M.; Ertunc, D.; Tok, E.C.; Tamer, L.; Arslan, M.; Dilek, S. Modulating interaction of glutathione-S-transferase polymorphisms with smoking in endometriosis. J. Reprod. Med. 2007, 52, 715–721. [Google Scholar] [PubMed]
- Calhaz-Jorge, C.; Mol, B.W.; Nunes, J.; Costa, A.P. Clinical predictive factors for endometriosis in a Portuguese infertile population. Hum. Reprod. 2004, 19, 2126–2131. [Google Scholar] [CrossRef] [PubMed]
- Cramer, D.W.; Wilson, E.; Stillman, R.J.; Berger, M.J.; Belisle, S.; Schiff, I.; Albrecht, B.; Gibson, M.; Stadel, B.V.; Schoenbaum, S.C. The relation of endometriosis to menstrual characteristics, smoking, and exercise. JAMA 1986, 255, 1904–1908. [Google Scholar] [CrossRef]
- Bravi, F.; Parazzini, F.; Cipriani, S.; Chiaffarino, F.; Ricci, E.; Chiantera, V.; Vigano, P.; La Vecchia, C. Tobacco smoking and risk of endometriosis: A systematic review and meta-analysis. BMJ Open 2014, 4, e006325. [Google Scholar] [CrossRef] [PubMed]
- Missmer, S.A.; Hankinson, S.E.; Spiegelman, D.; Barbieri, R.L.; Marshall, L.M.; Hunter, D.J. Incidence of laparoscopically confirmed endometriosis by demographic, anthropometric, and lifestyle factors. Am. J. Epidemiol. 2004, 160, 784–796. [Google Scholar] [CrossRef]
- Vidal, J.D.; VandeVoort, C.A.; Marcus, C.B.; Lazarewicz, N.R.; Conley, A.J. In vitro exposure to environmental tobacco smoke induces CYP1B1 expression in human luteinized granulosa cells. Reprod. Toxicol. 2006, 22, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Matorras, R.; Rodiquez, F.; Pijoan, J.I.; Ramon, O.; Gutierrez de Teran, G.; Rodriguez-Escudero, F. Epidemiology of endometriosis in infertile women. Fertil. Steril. 1995, 63, 34–38. [Google Scholar] [CrossRef]
- Berube, S.; Marcoux, S.; Maheux, R. Characteristics related to the prevalence of minimal or mild endometriosis in infertile women. Canadian Collaborative Group on Endometriosis. Epidemiology 1998, 9, 504–510. [Google Scholar] [CrossRef]
- Chapron, C.; Souza, C.; de Ziegler, D.; Lafay-Pillet, M.C.; Ngo, C.; Bijaoui, G.; Goffinet, F.; Borghese, B. Smoking habits of 411 women with histologically proven endometriosis and 567 unaffected women. Fertil. Steril. 2010, 94, 2353–2355. [Google Scholar] [CrossRef]
- Signorello, L.B.; Harlow, B.L.; Cramer, D.W.; Spiegelman, D.; Hill, J.A. Epidemiologic determinants of endometriosis: A hospital-based case-control study. Ann. Epidemiol. 1997, 7, 267–274. [Google Scholar] [CrossRef]
- Li Piani, L.; Chiaffarino, F.; Cipriani, S.; Vigano, P.; Somigliana, E.; Parazzini, F. A systematic review and meta-analysis on alcohol consumption and risk of endometriosis: An update from 2012. Sci. Rep. 2022, 12, 19122. [Google Scholar] [CrossRef] [PubMed]
- Parazzini, F.; Cipriani, S.; Bravi, F.; Pelucchi, C.; Chiaffarino, F.; Ricci, E.; Vigano, P. A metaanalysis on alcohol consumption and risk of endometriosis. Am. J. Obstet. Gynecol. 2013, 209, 106.e1–106.e10. [Google Scholar] [CrossRef] [PubMed]
- Salliss, M.E.; Farland, L.V.; Mahnert, N.D.; Herbst-Kralovetz, M.M. The role of gut and genital microbiota and the estrobolome in endometriosis, infertility and chronic pelvic pain. Hum. Reprod. Update 2021, 28, 92–131. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, A.; Suzuki, M.; Hamaguchi, T.; Watanabe, S.; Iijima, K.; Murofushi, Y.; Shinjo, K.; Osuka, S.; Hariyama, Y.; Ito, M.; et al. Fusobacterium infection facilitates the development of endometriosis through the phenotypic transition of endometrial fibroblasts. Sci. Transl. Med. 2023, 15, eadd1531. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Gong, P.; Chen, Y.; Nwachukwu, J.C.; Srinivasan, S.; Ko, C.; Bagchi, M.K.; Taylor, R.N.; Korach, K.S.; Nettles, K.W.; et al. Dual suppression of estrogenic and inflammatory activities for targeting of endometriosis. Sci. Transl. Med. 2015, 7, 271ra279. [Google Scholar] [CrossRef]
- Chantalat, E.; Valera, M.C.; Vaysse, C.; Noirrit, E.; Rusidze, M.; Weyl, A.; Vergriete, K.; Buscail, E.; Lluel, P.; Fontaine, C.; et al. Estrogen Receptors and Endometriosis. Int. J. Mol. Sci. 2020, 21, 2815. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, Y.; Fassbender, A.; Bowdler, L.; Fung, J.N.; Peterse, D.; Dorien, O.; Montgomery, G.W.; Nyholt, D.R.; D’Hooghe, T.M. Independent Replication and Meta-Analysis for Endometriosis Risk Loci. Twin Res. Hum. Genet. 2015, 18, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, Y.; Steinthorsdottir, V.; Morris, A.P.; Fassbender, A.; Rahmioglu, N.; De Vivo, I.; Buring, J.E.; Zhang, F.; Edwards, T.L.; Jones, S.; et al. Meta-analysis identifies five novel loci associated with endometriosis highlighting key genes involved in hormone metabolism. Nat. Commun. 2017, 8, 15539. [Google Scholar] [CrossRef]
- Sapkota, Y.; Vivo, I.; Steinthorsdottir, V.; Fassbender, A.; Bowdler, L.; Buring, J.E.; Edwards, T.L.; Jones, S.; Dorien, O.; Peterse, D.; et al. Analysis of potential protein-modifying variants in 9000 endometriosis patients and 150,000 controls of European ancestry. Sci. Rep. 2017, 7, 11380. [Google Scholar] [CrossRef]
- Burns, K.A.; Rodriguez, K.F.; Hewitt, S.C.; Janardhan, K.S.; Young, S.L.; Korach, K.S. Role of estrogen receptor signaling required for endometriosis-like lesion establishment in a mouse model. Endocrinology 2012, 153, 3960–3971. [Google Scholar] [CrossRef]
- Gougeon, A. Human ovarian follicular development: From activation of resting follicles to preovulatory maturation. Ann. Endocrinol. 2010, 71, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Nyholt, D.R.; Low, S.K.; Anderson, C.A.; Painter, J.N.; Uno, S.; Morris, A.P.; MacGregor, S.; Gordon, S.D.; Henders, A.K.; Martin, N.G.; et al. Genome-wide association meta-analysis identifies new endometriosis risk loci. Nat. Genet. 2012, 44, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Rahmioglu, N.; Nyholt, D.R.; Morris, A.P.; Missmer, S.A.; Montgomery, G.W.; Zondervan, K.T. Genetic variants underlying risk of endometriosis: Insights from meta-analysis of eight genome-wide association and replication datasets. Hum. Reprod. Update 2014, 20, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Chadchan, S.B.; Popli, P.; Liao, Z.; Andreas, E.; Dias, M.; Wang, T.; Gunderson, S.J.; Jimenez, P.T.; Lanza, D.G.; Lanz, R.B.; et al. A GREB1-steroid receptor feedforward mechanism governs differential GREB1 action in endometrial function and endometriosis. Nat. Commun. 2024, 15, 1947. [Google Scholar] [CrossRef]
- Mohammed, H.; D’Santos, C.; Serandour, A.A.; Ali, H.R.; Brown, G.D.; Atkins, A.; Rueda, O.M.; Holmes, K.A.; Theodorou, V.; Robinson, J.L.; et al. Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor. Cell Rep. 2013, 3, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Matalliotaki, C.; Matalliotakis, M.; Rahmioglu, N.; Mavromatidis, G.; Matalliotakis, I.; Koumantakis, G.; Zondervan, K.; Spandidos, D.A.; Goulielmos, G.N.; Zervou, M.I. Role of FN1 and GREB1 gene polymorphisms in endometriosis. Mol. Med. Rep. 2019, 20, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Fung, J.N.; Holdsworth-Carson, S.J.; Sapkota, Y.; Zhao, Z.Z.; Jones, L.; Girling, J.E.; Paiva, P.; Healey, M.; Nyholt, D.R.; Rogers, P.A.; et al. Functional evaluation of genetic variants associated with endometriosis near GREB1. Hum. Reprod. 2015, 30, 1263–1275. [Google Scholar] [CrossRef]
- Christofolini, D.M.; Amaro, A.; Mafra, F.; Sonnewend, A.; Bianco, B.; Barbosa, C.P. CYP2C19 polymorphism increases the risk of endometriosis. J. Assist. Reprod. Genet. 2015, 32, 91–94. [Google Scholar] [CrossRef]
- Perini, J.A.; Machado, D.E.; Cardoso, J.V.; Fernandes, V.C.; Struchiner, C.J.; Suarez-Kurtz, G. CYP2C19 metabolic estrogen phenotypes and endometriosis risk in Brazilian women. Clinics 2023, 78, 100176. [Google Scholar] [CrossRef]
- Hodgkinson, K.; Forrest, L.A.; Vuong, N.; Garson, K.; Djordjevic, B.; Vanderhyden, B.C. GREB1 is an estrogen receptor-regulated tumour promoter that is frequently expressed in ovarian cancer. Oncogene 2018, 37, 5873–5886. [Google Scholar] [CrossRef]
- Veeraraghavan, J.; Tan, Y.; Cao, X.X.; Kim, J.A.; Wang, X.; Chamness, G.C.; Maiti, S.N.; Cooper, L.J.; Edwards, D.P.; Contreras, A.; et al. Recurrent ESR1-CCDC170 rearrangements in an aggressive subset of oestrogen receptor-positive breast cancers. Nat. Commun. 2014, 5, 4577. [Google Scholar] [CrossRef] [PubMed]
- Dunning, A.M.; Michailidou, K.; Kuchenbaecker, K.B.; Thompson, D.; French, J.D.; Beesley, J.; Healey, C.S.; Kar, S.; Pooley, K.A.; Lopez-Knowles, E.; et al. Breast cancer risk variants at 6q25 display different phenotype associations and regulate ESR1, RMND1 and CCDC170. Nat. Genet. 2016, 48, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Malentacchi, F.; Fambrini, M.; Harrath, A.H.; Huang, H.; Petraglia, F. Epigenetics of Estrogen and Progesterone Receptors in Endometriosis. Reprod. Sci. 2020, 27, 1967–1974. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Seidita, I.; Vannuccini, S.; Prisinzano, M.; Donati, C.; Petraglia, F. Epigenetics, endometriosis and sex steroid receptors: An update on the epigenetic regulatory mechanisms of estrogen and progesterone receptors in patients with endometriosis. Vitam. Horm. 2023, 122, 171–191. [Google Scholar] [CrossRef] [PubMed]
- Houshdaran, S.; Nezhat, C.R.; Vo, K.C.; Zelenko, Z.; Irwin, J.C.; Giudice, L.C. Aberrant Endometrial DNA Methylome and Associated Gene Expression in Women with Endometriosis. Biol. Reprod. 2016, 95, 93. [Google Scholar] [CrossRef] [PubMed]
- Ochoa Bernal, M.A.; Fazleabas, A.T. The Known, the Unknown and the Future of the Pathophysiology of Endometriosis. Int. J. Mol. Sci. 2024, 25, 5815. [Google Scholar] [CrossRef] [PubMed]
- Treloar, S.A.; Wicks, J.; Nyholt, D.R.; Montgomery, G.W.; Bahlo, M.; Smith, V.; Dawson, G.; Mackay, I.J.; Weeks, D.E.; Bennett, S.T.; et al. Genomewide linkage study in 1176 affected sister pair families identifies a significant susceptibility locus for endometriosis on chromosome 10q26. Am. J. Hum. Genet. 2005, 77, 365–376. [Google Scholar] [CrossRef]
- Zondervan, K.T.; Treloar, S.A.; Lin, J.; Weeks, D.E.; Nyholt, D.R.; Mangion, J.; MacKay, I.J.; Cardon, L.R.; Martin, N.G.; Kennedy, S.H.; et al. Significant evidence of one or more susceptibility loci for endometriosis with near-Mendelian inheritance on chromosome 7p13–15. Hum. Reprod. 2007, 22, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Painter, J.N.; Anderson, C.A.; Nyholt, D.R.; Macgregor, S.; Lin, J.; Lee, S.H.; Lambert, A.; Zhao, Z.Z.; Roseman, F.; Guo, Q.; et al. Genome-wide association study identifies a locus at 7p15.2 associated with endometriosis. Nat. Genet. 2011, 43, 51–54. [Google Scholar] [CrossRef]
- Uno, S.; Zembutsu, H.; Hirasawa, A.; Takahashi, A.; Kubo, M.; Akahane, T.; Aoki, D.; Kamatani, N.; Hirata, K.; Nakamura, Y. A genome-wide association study identifies genetic variants in the CDKN2BAS locus associated with endometriosis in Japanese. Nat. Genet. 2010, 42, 707–710. [Google Scholar] [CrossRef]
- Tapmeier, T.T.; Rahmioglu, N.; Lin, J.; De Leo, B.; Obendorf, M.; Raveendran, M.; Fischer, O.M.; Bafligil, C.; Guo, M.; Harris, R.A.; et al. Neuropeptide S receptor 1 is a nonhormonal treatment target in endometriosis. Sci. Transl. Med. 2021, 13, eabd6469. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, M.; Bruce, S.; Bresso, F.; Zucchelli, M.; Ezer, S.; Pulkkinen, V.; Lindgren, C.; Astegiano, M.; Rizzetto, M.; Gionchetti, P.; et al. Neuropeptide s receptor 1 gene polymorphism is associated with susceptibility to inflammatory bowel disease. Gastroenterology 2007, 133, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, T.; Polvi, A.; Rydman, P.; Vendelin, J.; Pulkkinen, V.; Salmikangas, P.; Makela, S.; Rehn, M.; Pirskanen, A.; Rautanen, A.; et al. Characterization of a common susceptibility locus for asthma-related traits. Science 2004, 304, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Pulkkinen, V.; Majuri, M.L.; Wang, G.; Holopainen, P.; Obase, Y.; Vendelin, J.; Wolff, H.; Rytila, P.; Laitinen, L.A.; Haahtela, T.; et al. Neuropeptide S and G protein-coupled receptor 154 modulate macrophage immune responses. Hum. Mol. Genet. 2006, 15, 1667–1679. [Google Scholar] [CrossRef] [PubMed]
- Rahmioglu, N.; Mortlock, S.; Ghiasi, M.; Moller, P.L.; Stefansdottir, L.; Galarneau, G.; Turman, C.; Danning, R.; Law, M.H.; Sapkota, Y.; et al. The genetic basis of endometriosis and comorbidity with other pain and inflammatory conditions. Nat. Genet. 2023, 55, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Adachi, S.; Tajima, A.; Quan, J.; Haino, K.; Yoshihara, K.; Masuzaki, H.; Katabuchi, H.; Ikuma, K.; Suginami, H.; Nishida, N.; et al. Meta-analysis of genome-wide association scans for genetic susceptibility to endometriosis in Japanese population. J. Hum. Genet. 2010, 55, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Pavlicev, M.; McDonough-Goldstein, C.E.; Zupan, A.M.; Muglia, L.; Hu, Y.C.; Kong, F.; Monangi, N.; Dagdas, G.; Zupancic, N.; Maziarz, J.; et al. A common allele increases endometrial Wnt4 expression, with antagonistic implications for pregnancy, reproductive cancers, and endometriosis. Nat. Commun. 2024, 15, 1152. [Google Scholar] [CrossRef] [PubMed]
- Albertsen, H.M.; Chettier, R.; Farrington, P.; Ward, K. Genome-wide association study link novel loci to endometriosis. PLoS ONE 2013, 8, e58257. [Google Scholar] [CrossRef]
- Uimari, O.; Rahmioglu, N.; Nyholt, D.R.; Vincent, K.; Missmer, S.A.; Becker, C.; Morris, A.P.; Montgomery, G.W.; Zondervan, K.T. Genome-wide genetic analyses highlight mitogen-activated protein kinase (MAPK) signaling in the pathogenesis of endometriosis. Hum. Reprod. 2017, 32, 780–793. [Google Scholar] [CrossRef]
- Siufi Neto, J.; Kho, R.M.; Siufi, D.F.; Baracat, E.C.; Anderson, K.S.; Abrao, M.S. Cellular, histologic, and molecular changes associated with endometriosis and ovarian cancer. J. Minim. Invasive Gynecol. 2014, 21, 55–63. [Google Scholar] [CrossRef]
- Sato, N.; Tsunoda, H.; Nishida, M.; Morishita, Y.; Takimoto, Y.; Kubo, T.; Noguchi, M. Loss of heterozygosity on 10q23.3 and mutation of the tumor suppressor gene PTEN in benign endometrial cyst of the ovary: Possible sequence progression from benign endometrial cyst to endometrioid carcinoma and clear cell carcinoma of the ovary. Cancer Res. 2000, 60, 7052–7056. [Google Scholar] [PubMed]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Anglesio, M.S.; Papadopoulos, N.; Ayhan, A.; Nazeran, T.M.; Noe, M.; Horlings, H.M.; Lum, A.; Jones, S.; Senz, J.; Seckin, T.; et al. Cancer-Associated Mutations in Endometriosis without Cancer. N. Engl. J. Med. 2017, 376, 1835–1848. [Google Scholar] [CrossRef] [PubMed]
- Suda, K.; Nakaoka, H.; Yoshihara, K.; Ishiguro, T.; Tamura, R.; Mori, Y.; Yamawaki, K.; Adachi, S.; Takahashi, T.; Kase, H.; et al. Clonal Expansion and Diversification of Cancer-Associated Mutations in Endometriosis and Normal Endometrium. Cell Rep. 2018, 24, 1777–1789. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.; Leongamornlert, D.; Coorens, T.H.H.; Sanders, M.A.; Ellis, P.; Dentro, S.C.; Dawson, K.J.; Butler, T.; Rahbari, R.; Mitchell, T.J.; et al. The mutational landscape of normal human endometrial epithelium. Nature 2020, 580, 640–646. [Google Scholar] [CrossRef]
- Moore, L.; Cagan, A.; Coorens, T.H.H.; Neville, M.D.C.; Sanghvi, R.; Sanders, M.A.; Oliver, T.R.W.; Leongamornlert, D.; Ellis, P.; Noorani, A.; et al. The mutational landscape of human somatic and germline cells. Nature 2021, 597, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Noe, M.; Ayhan, A.; Wang, T.L.; Shih, I.M. Independent development of endometrial epithelium and stroma within the same endometriosis. J. Pathol. 2018, 245, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Lac, V.; Verhoef, L.; Aguirre-Hernandez, R.; Nazeran, T.M.; Tessier-Cloutier, B.; Praetorius, T.; Orr, N.L.; Noga, H.; Lum, A.; Khattra, J.; et al. Iatrogenic endometriosis harbors somatic cancer-driver mutations. Hum. Reprod. 2019, 34, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Yachida, N.; Yoshihara, K.; Suda, K.; Nakaoka, H.; Ueda, H.; Sugino, K.; Yamaguchi, M.; Mori, Y.; Yamawaki, K.; Tamura, R.; et al. ARID1A protein expression is retained in ovarian endometriosis with ARID1A loss-of-function mutations: Implication for the two-hit hypothesis. Sci. Rep. 2020, 10, 14260. [Google Scholar] [CrossRef]
- Yamamoto, S.; Tsuda, H.; Takano, M.; Tamai, S.; Matsubara, O. Loss of ARID1A protein expression occurs as an early event in ovarian clear-cell carcinoma development and frequently coexists with PIK3CA mutations. Mod. Pathol. 2012, 25, 615–624. [Google Scholar] [CrossRef]
- Preatorius, T.H.; Lac, V.; Tessier-Cloutier, B.; Senz, J.; Nazeran, T.; Grube, M.; Koebel, M.; Kraemer, B.; Yong, P.; Kommoss, S.; et al. Is endometriosis metastasizing? Shared somatic alterations suggest common origins across endometriotic lesions. Geburtshilfe Frauenheilkd 2020, 80, e160–e161. [Google Scholar]
- Revathidevi, S.; Nakaoka, H.; Suda, K.; Fujito, N.; Munirajan, A.K.; Yoshihara, K.; Enomoto, T.; Inoue, I. APOBEC mediated mutagenesis drives genomic heterogeneity in endometriosis. J. Hum. Genet. 2022, 67, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Durfee, C.; Temiz, N.A.; Levin-Klein, R.; Argyris, P.P.; Alsoe, L.; Carracedo, S.; Alonso de la Vega, A.; Proehl, J.; Holzhauer, A.M.; Seeman, Z.J.; et al. Human APOBEC3B promotes tumor development in vivo including signature mutations and metastases. Cell Rep. Med. 2023, 4, 101211. [Google Scholar] [CrossRef]
- Long, X.; Lu, H.; Cai, M.C.; Zang, J.; Zhang, Z.; Wu, J.; Liu, X.; Cheng, L.; Cheng, J.; Cheung, L.W.T.; et al. APOBEC3B stratifies ovarian clear cell carcinoma with distinct immunophenotype and prognosis. Br. J. Cancer 2023, 128, 2054–2062. [Google Scholar] [CrossRef] [PubMed]
- Mostoufizadeh, M.; Scully, R.E. Malignant tumors arising in endometriosis. Clin. Obstet. Gynecol. 1980, 23, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Anglesio, M.S.; Yong, P.J. Endometriosis-associated Ovarian Cancers. Clin. Obstet. Gynecol. 2017, 60, 711–727. [Google Scholar] [CrossRef] [PubMed]
- Melin, A.; Sparen, P.; Bergqvist, A. The risk of cancer and the role of parity among women with endometriosis. Hum. Reprod. 2007, 22, 3021–3026. [Google Scholar] [CrossRef] [PubMed]
- Melin, A.; Sparen, P.; Persson, I.; Bergqvist, A. Endometriosis and the risk of cancer with special emphasis on ovarian cancer. Hum. Reprod. 2006, 21, 1237–1242. [Google Scholar] [CrossRef] [PubMed]
- Somigliana, E.; Vigano, P.; Parazzini, F.; Stoppelli, S.; Giambattista, E.; Vercellini, P. Association between endometriosis and cancer: A comprehensive review and a critical analysis of clinical and epidemiological evidence. Gynecol. Oncol. 2006, 101, 331–341. [Google Scholar] [CrossRef]
- Kvaskoff, M.; Mahamat-Saleh, Y.; Farland, L.V.; Shigesi, N.; Terry, K.L.; Harris, H.R.; Roman, H.; Becker, C.M.; As-Sanie, S.; Zondervan, K.T.; et al. Endometriosis and cancer: A systematic review and meta-analysis. Hum. Reprod. Update 2021, 27, 393–420. [Google Scholar] [CrossRef]
- Hollis, R.L.; Gourley, C. Genetic and molecular changes in ovarian cancer. Cancer Biol. Med. 2016, 13, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, S.; Coward, J.I.; Bast, R.C., Jr.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef]
- Prat, J.; D’Angelo, E.; Espinosa, I. Ovarian carcinomas: At least five different diseases with distinct histological features and molecular genetics. Hum. Pathol. 2018, 80, 11–27. [Google Scholar] [CrossRef] [PubMed]
- McCluggage, W.G. Endometriosis-related pathology: A discussion of selected uncommon benign, premalignant and malignant lesions. Histopathology 2020, 76, 76–92. [Google Scholar] [CrossRef]
- Worley, M.J.; Welch, W.R.; Berkowitz, R.S.; Ng, S.W. Endometriosis-associated ovarian cancer: A review of pathogenesis. Int. J. Mol. Sci. 2013, 14, 5367–5379. [Google Scholar] [CrossRef]
- Wang, Y.; Nicholes, K.; Shih, I.M. The Origin and Pathogenesis of Endometriosis. Annu. Rev. Pathol. 2020, 15, 71–95. [Google Scholar] [CrossRef]
- Kobayashi, H.; Sumimoto, K.; Moniwa, N.; Imai, M.; Takakura, K.; Kuromaki, T.; Morioka, E.; Arisawa, K.; Terao, T. Risk of developing ovarian cancer among women with ovarian endometrioma: A cohort study in Shizuoka, Japan. Int. J. Gynecol. Cancer 2007, 17, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Nagase, S.; Ohta, T.; Takahashi, F.; Enomoto, T.; The 2017 Committee on Gynecologic Oncology of the Japan Society of Obstetrics and Gynecology. Annual report of the committee on gynecologic oncology, the Japan Society of Obstetrics and Gynecology: Annual patients report for 2015 and annual treatment report for 2010. J. Obstet. Gynaecol. Res. 2019, 45, 289–298. [Google Scholar] [CrossRef]
- Brinton, L.A.; Sakoda, L.C.; Sherman, M.E.; Frederiksen, K.; Kjaer, S.K.; Graubard, B.I.; Olsen, J.H.; Mellemkjaer, L. Relationship of benign gynecologic diseases to subsequent risk of ovarian and uterine tumors. Cancer Epidemiol. Biomark. Prev. 2005, 14, 2929–2935. [Google Scholar] [CrossRef]
- Rossing, M.A.; Cushing-Haugen, K.L.; Wicklund, K.G.; Doherty, J.A.; Weiss, N.S. Risk of epithelial ovarian cancer in relation to benign ovarian conditions and ovarian surgery. Cancer Causes Control 2008, 19, 1357–1364. [Google Scholar] [CrossRef]
- Pearce, C.L.; Templeman, C.; Rossing, M.A.; Lee, A.; Near, A.M.; Webb, P.M.; Nagle, C.M.; Doherty, J.A.; Cushing-Haugen, K.L.; Wicklund, K.G.; et al. Association between endometriosis and risk of histological subtypes of ovarian cancer: A pooled analysis of case-control studies. Lancet Oncol. 2012, 13, 385–394. [Google Scholar] [CrossRef]
- Mortlock, S.; Corona, R.I.; Kho, P.F.; Pharoah, P.; Seo, J.H.; Freedman, M.L.; Gayther, S.A.; Siedhoff, M.T.; Rogers, P.A.W.; Leuchter, R.; et al. A multi-level investigation of the genetic relationship between endometriosis and ovarian cancer histotypes. Cell Rep. Med. 2022, 3, 100542. [Google Scholar] [CrossRef]
- Stamp, J.P.; Gilks, C.B.; Wesseling, M.; Eshragh, S.; Ceballos, K.; Anglesio, M.S.; Kwon, J.S.; Tone, A.; Huntsman, D.G.; Carey, M.S. BAF250a Expression in Atypical Endometriosis and Endometriosis-Associated Ovarian Cancer. Int. J. Gynecol. Cancer 2016, 26, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Vercellini, P.; Parazzini, F.; Bolis, G.; Carinelli, S.; Dindelli, M.; Vendola, N.; Luchini, L.; Crosignani, P.G. Endometriosis and ovarian cancer. Am. J. Obstet. Gynecol. 1993, 169, 181–182. [Google Scholar] [CrossRef] [PubMed]
- Jimbo, H.; Yoshikawa, H.; Onda, T.; Yasugi, T.; Sakamoto, A.; Taketani, Y. Prevalence of ovarian endometriosis in epithelial ovarian cancer. Int. J. Gynaecol. Obstet. 1997, 59, 245–250. [Google Scholar] [CrossRef]
- Hashiguchi, Y.; Tsuda, H.; Inoue, T.; Berkowitz, R.S.; Mok, S.C. PTEN expression in clear cell adenocarcinoma of the ovary. Gynecol. Oncol. 2006, 101, 71–75. [Google Scholar] [CrossRef]
- Huang, H.N.; Lin, M.C.; Huang, W.C.; Chiang, Y.C.; Kuo, K.T. Loss of ARID1A expression and its relationship with PI3K-Akt pathway alterations and ZNF217 amplification in ovarian clear cell carcinoma. Mod. Pathol. 2014, 27, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.C.; Couturier, D.L.; Paterson, A.; Karnezis, A.N.; Chow, C.; Nazeran, T.M.; Odunsi, A.; Gentry-Maharaj, A.; Vrvilo, A.; Hein, A.; et al. Clinical and pathological associations of PTEN expression in ovarian cancer: A multicentre study from the Ovarian Tumour Tissue Analysis Consortium. Br. J. Cancer 2020, 123, 793–802. [Google Scholar] [CrossRef]
- Martini, M.; Ciccarone, M.; Garganese, G.; Maggiore, C.; Evangelista, A.; Rahimi, S.; Zannoni, G.; Vittori, G.; Larocca, L.M. Possible involvement of hMLH1, p16INK4a and PTEN in the malignant transformation of endometriosis. Int. J. Cancer 2002, 102, 398–406. [Google Scholar] [CrossRef]
- Murakami, R.; Matsumura, N.; Brown, J.B.; Higasa, K.; Tsutsumi, T.; Kamada, M.; Abou-Taleb, H.; Hosoe, Y.; Kitamura, S.; Yamaguchi, K.; et al. Exome Sequencing Landscape Analysis in Ovarian Clear Cell Carcinoma Shed Light on Key Chromosomal Regions and Mutation Gene Networks. Am. J. Pathol. 2017, 187, 2246–2258. [Google Scholar] [CrossRef]
- Shibuya, Y.; Tokunaga, H.; Saito, S.; Shimokawa, K.; Katsuoka, F.; Bin, L.; Kojima, K.; Nagasaki, M.; Yamamoto, M.; Yaegashi, N.; et al. Identification of somatic genetic alterations in ovarian clear cell carcinoma with next generation sequencing. Genes Chromosomes Cancer 2018, 57, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.I.; Lee, J.W.; Lee, M.; Kim, H.S.; Chung, H.H.; Kim, J.W.; Park, N.H.; Song, Y.S.; Seo, J.S. Genomic landscape of ovarian clear cell carcinoma via whole exome sequencing. Gynecol. Oncol. 2018, 148, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Guida, M.; Sanguedolce, F.; Bufo, P.; Di Spiezio Sardo, A.; Bifulco, G.; Nappi, C.; Pannone, G. Aberrant DNA hypermethylation of hMLH-1 and CDKN2A/p16 genes in benign, premalignant and malignant endometrial lesions. Eur. J. Gynaecol. Oncol. 2009, 30, 267–270. [Google Scholar] [PubMed]
- Boretto, M.; Cox, B.; Noben, M.; Hendriks, N.; Fassbender, A.; Roose, H.; Amant, F.; Timmerman, D.; Tomassetti, C.; Vanhie, A.; et al. Development of organoids from mouse and human endometrium showing endometrial epithelium physiology and long-term expandability. Development 2017, 144, 1775–1786. [Google Scholar] [CrossRef]
- Boretto, M.; Maenhoudt, N.; Luo, X.; Hennes, A.; Boeckx, B.; Bui, B.; Heremans, R.; Perneel, L.; Kobayashi, H.; Van Zundert, I.; et al. Patient-derived organoids from endometrial disease capture clinical heterogeneity and are amenable to drug screening. Nat. Cell Biol. 2019, 21, 1041–1051. [Google Scholar] [CrossRef]
- Cindrova-Davies, T.; Zhao, X.; Elder, K.; Jones, C.J.P.; Moffett, A.; Burton, G.J.; Turco, M.Y. Menstrual flow as a non-invasive source of endometrial organoids. Commun. Biol. 2021, 4, 651. [Google Scholar] [CrossRef]
- De Vriendt, S.; Casares, C.M.; Rocha, S.; Vankelecom, H. Matrix scaffolds for endometrium-derived organoid models. Front. Endocrinol. 2023, 14, 1240064. [Google Scholar] [CrossRef]
- Kopper, O.; de Witte, C.J.; Lohmussaar, K.; Valle-Inclan, J.E.; Hami, N.; Kester, L.; Balgobind, A.V.; Korving, J.; Proost, N.; Begthel, H.; et al. An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat. Med. 2019, 25, 838–849. [Google Scholar] [CrossRef]
- Wang, W.; Vilella, F.; Alama, P.; Moreno, I.; Mignardi, M.; Isakova, A.; Pan, W.; Simon, C.; Quake, S.R. Single-cell transcriptomic atlas of the human endometrium during the menstrual cycle. Nat. Med. 2020, 26, 1644–1653. [Google Scholar] [CrossRef]
- Garcia-Alonso, L.; Handfield, L.F.; Roberts, K.; Nikolakopoulou, K.; Fernando, R.C.; Gardner, L.; Woodhams, B.; Arutyunyan, A.; Polanski, K.; Hoo, R.; et al. Mapping the temporal and spatial dynamics of the human endometrium in vivo and in vitro. Nat. Genet. 2021, 53, 1698–1711. [Google Scholar] [CrossRef]
- Vento-Tormo, R.; Efremova, M.; Botting, R.A.; Turco, M.Y.; Vento-Tormo, M.; Meyer, K.B.; Park, J.E.; Stephenson, E.; Polanski, K.; Goncalves, A.; et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 2018, 563, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.J.; Adelson, R.P.; Vashistha, H.; Khalili, H.; Nayyar, A.; Puran, R.; Herrera, R.; Chatterjee, P.K.; Lee, A.T.; Truskinovsky, A.M.; et al. Single-cell analysis of menstrual endometrial tissues defines phenotypes associated with endometriosis. BMC Med. 2022, 20, 315. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Chung, Y.J.; Moon, S.W.; Choi, E.J.; Kim, M.R.; Chung, Y.J.; Lee, S.H. Single-cell profiling identifies distinct hormonal, immunologic, and inflammatory signatures of endometriosis-constituting cells. J. Pathol. 2023, 261, 323–334. [Google Scholar] [CrossRef]
- Yan, J.; Zhou, L.; Liu, M.; Zhu, H.; Zhang, X.; Cai, E.; Xu, X.; Chen, T.; Cheng, H.; Liu, J.; et al. Single-cell analysis reveals insights into epithelial abnormalities in ovarian endometriosis. Cell Rep. 2024, 43, 113716. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, M.A.S.; Haro, M.; Wright, K.N.; Lin, X.; Abbasi, F.; Sun, J.; Hernandez, L.; Orr, N.L.; Hong, J.; Choi-Kuaea, Y.; et al. Single-cell transcriptomic analysis of endometriosis. Nat. Genet. 2023, 55, 255–267. [Google Scholar] [CrossRef]
- Zou, G.; Wang, J.; Xu, X.; Xu, P.; Zhu, L.; Yu, Q.; Peng, Y.; Guo, X.; Li, T.; Zhang, X. Cell subtypes and immune dysfunction in peritoneal fluid of endometriosis revealed by single-cell RNA-sequencing. Cell Biosci. 2021, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, L.; Zhan, H.; Mo, Y.; Ren, Z.; Shao, A.; Lin, J. Single-cell transcriptomic analysis of endometriosis provides insights into fibroblast fates and immune cell heterogeneity. Cell Biosci. 2021, 11, 125. [Google Scholar] [CrossRef]
- Guo, M.; Bafligil, C.; Tapmeier, T.; Hubbard, C.; Manek, S.; Shang, C.; Martinez, F.O.; Schmidt, N.; Obendorf, M.; Hess-Stumpp, H.; et al. Mass cytometry analysis reveals a distinct immune environment in peritoneal fluid in endometriosis: A characterisation study. BMC Med. 2020, 18, 3. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.E.; Li, Y.; Cai, B.; He, Q.; Chen, G.; Wang, M.; Wang, K.; Wan, X.; Yan, Q. Phenotyping of immune and endometrial epithelial cells in endometrial carcinomas revealed by single-cell RNA sequencing. Aging 2021, 13, 6565–6591. [Google Scholar] [CrossRef]
- Herington, J.L.; Bruner-Tran, K.L.; Lucas, J.A.; Osteen, K.G. Immune interactions in endometriosis. Expert Rev. Clin. Immunol. 2011, 7, 611–626. [Google Scholar] [CrossRef]
- Marečková, M.; Garcia-Alonso, L.; Moullet, M.; Lorenzi, V.; Petryszak, R.; Sancho-Serra, C.; Oszlanczi, A.; Mazzeo, C.I.; Hoffman, S.; Krassowski, M.; et al. An integrated single-cell reference atlas of the human endometrium. bioRxiv 2023. [Google Scholar] [CrossRef]
- Oskotsky, T.T.; Bhoja, A.; Bunis, D.; Le, B.L.; Tang, A.S.; Kosti, I.; Li, C.; Houshdaran, S.; Sen, S.; Vallve-Juanico, J.; et al. Identifying therapeutic candidates for endometriosis through a transcriptomics-based drug repositioning approach. iScience 2024, 27, 109388. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, B.D.; Bulun, S.E. Endometriosis and nuclear receptors. Hum. Reprod. Update 2019, 25, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Cao, S.; Wang, X.; Feng, Y.; Billig, H. The elusive and controversial roles of estrogen and progesterone receptors in human endometriosis. Am. J. Transl. Res. 2014, 6, 104–113. [Google Scholar] [PubMed]
- Patel, B.G.; Lenk, E.E.; Lebovic, D.I.; Shu, Y.; Yu, J.; Taylor, R.N. Pathogenesis of endometriosis: Interaction between Endocrine and inflammatory pathways. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 50, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Ansariniya, H.; Yavari, A.; Javaheri, A.; Zare, F. Oxidative stress-related effects on various aspects of endometriosis. Am. J. Reprod. Immunol. 2022, 88, e13593. [Google Scholar] [CrossRef] [PubMed]
- Scutiero, G.; Iannone, P.; Bernardi, G.; Bonaccorsi, G.; Spadaro, S.; Volta, C.A.; Greco, P.; Nappi, L. Oxidative Stress and Endometriosis: A Systematic Review of the Literature. Oxid. Med. Cell Longev. 2017, 2017, 7265238. [Google Scholar] [CrossRef]
- Chung, M.S.; Han, S.J. Endometriosis-Associated Angiogenesis and Anti-angiogenic Therapy for Endometriosis. Front. Glob. Women’s Health 2022, 3, 856316. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adilbayeva, A.; Kunz, J. Pathogenesis of Endometriosis and Endometriosis-Associated Cancers. Int. J. Mol. Sci. 2024, 25, 7624. https://doi.org/10.3390/ijms25147624
Adilbayeva A, Kunz J. Pathogenesis of Endometriosis and Endometriosis-Associated Cancers. International Journal of Molecular Sciences. 2024; 25(14):7624. https://doi.org/10.3390/ijms25147624
Chicago/Turabian StyleAdilbayeva, Altynay, and Jeannette Kunz. 2024. "Pathogenesis of Endometriosis and Endometriosis-Associated Cancers" International Journal of Molecular Sciences 25, no. 14: 7624. https://doi.org/10.3390/ijms25147624
APA StyleAdilbayeva, A., & Kunz, J. (2024). Pathogenesis of Endometriosis and Endometriosis-Associated Cancers. International Journal of Molecular Sciences, 25(14), 7624. https://doi.org/10.3390/ijms25147624