
Genome-Wide Investigation of the CRF Gene Family in Maize and Functional Analysis of ZmCRF9 in Response to Multiple Abiotic Stresses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Identification of CRF Genes in Maize and Sequence Analysis
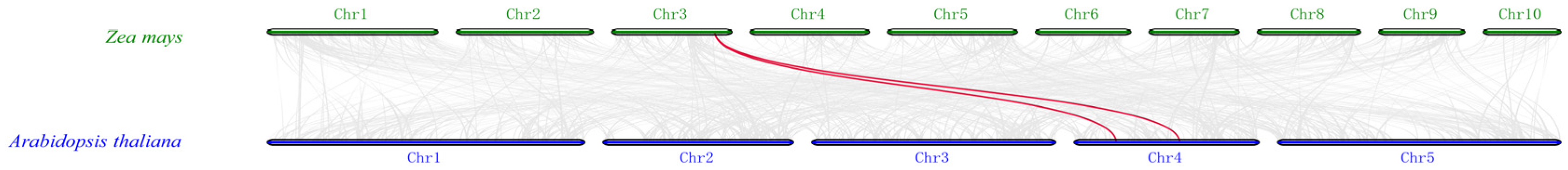
2.2. Chromosomal Localization and Syntenic Analysis
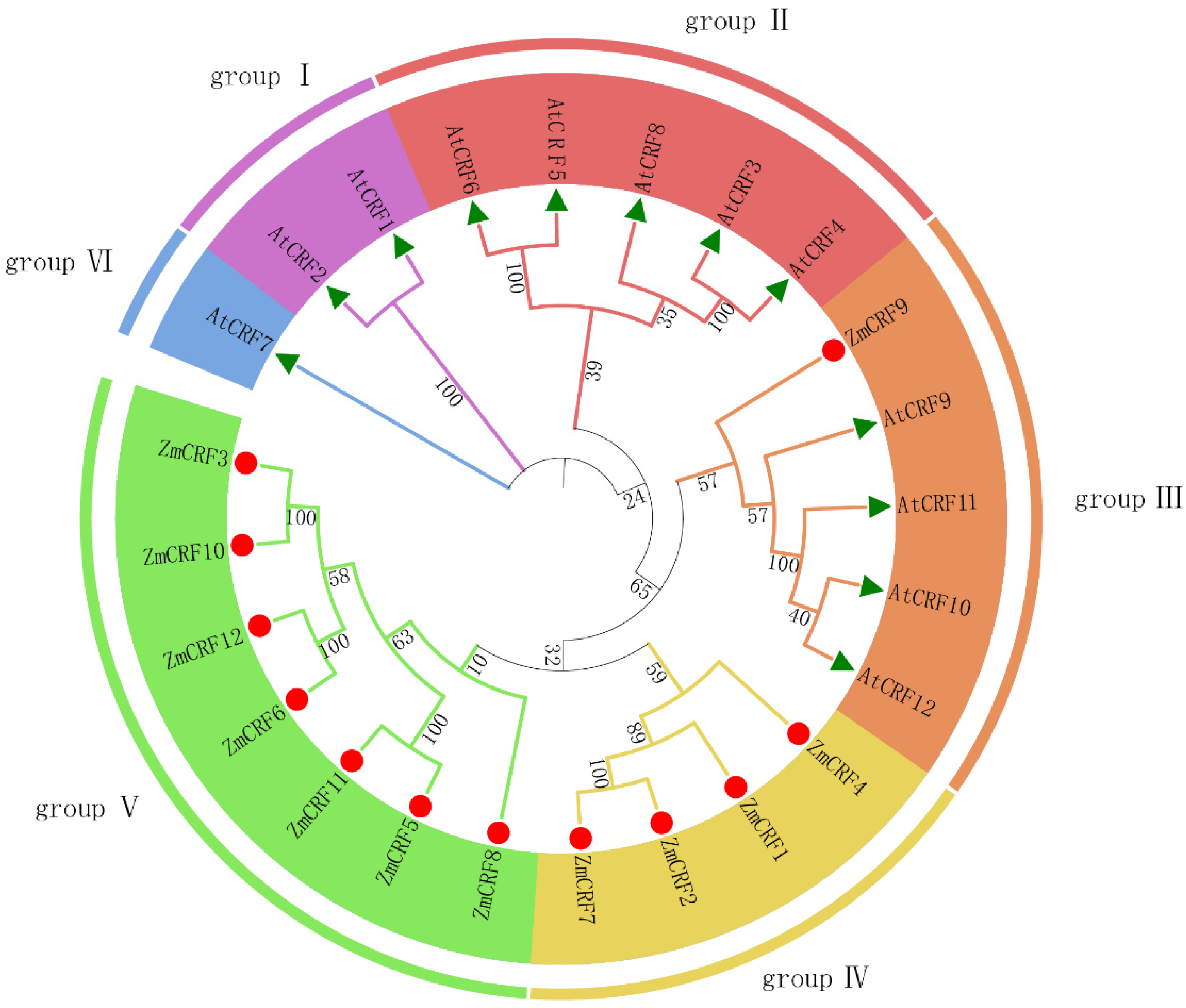
2.3. Phylogenetic Classification of ZmCRF Genes
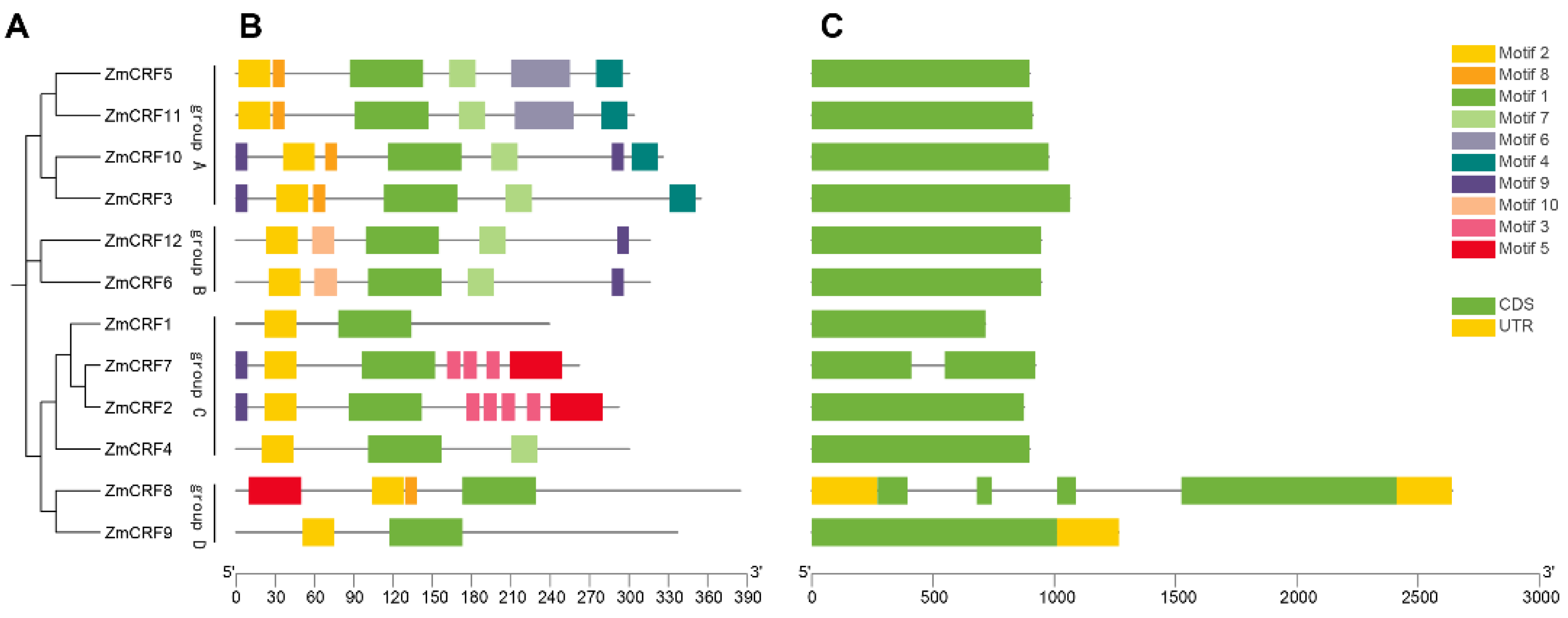
2.4. Conserved Motifs and Gene Structures of the ZmCRFs
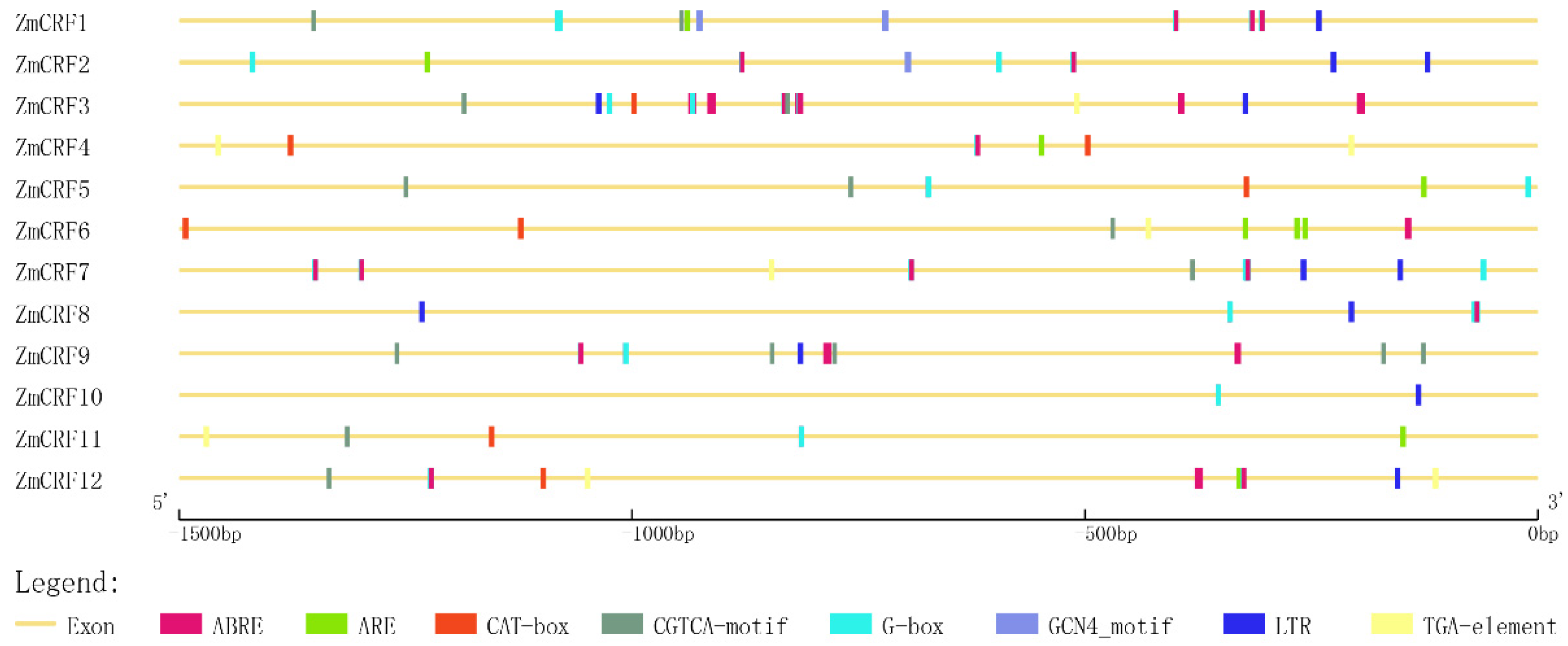
2.5. Cis-Element Analysis in the Promoter Regions of Maize CRF Genes
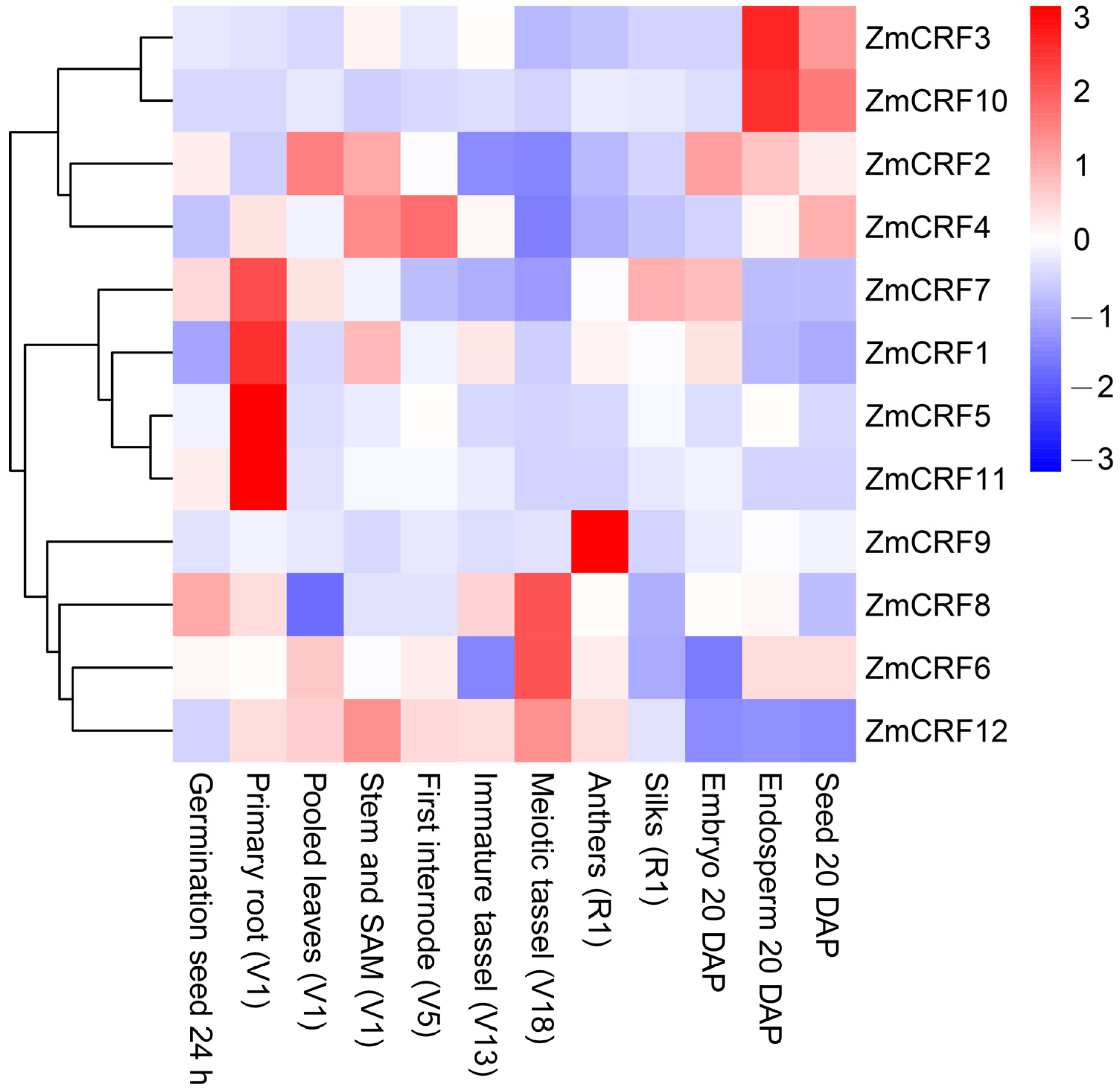
2.6. Expression Profiles of Maize CRFs in Diverse Tissues and Developmental Stages
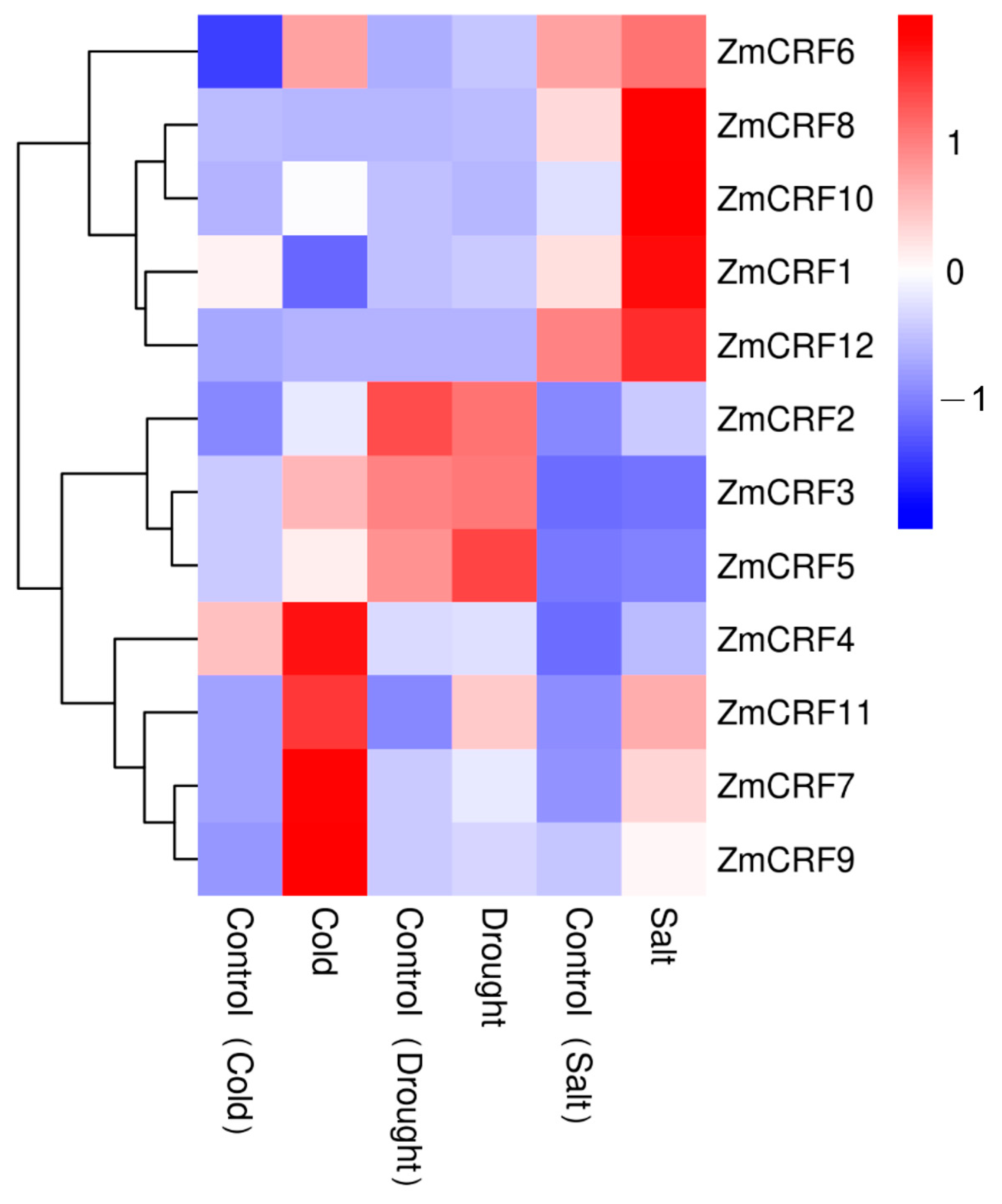
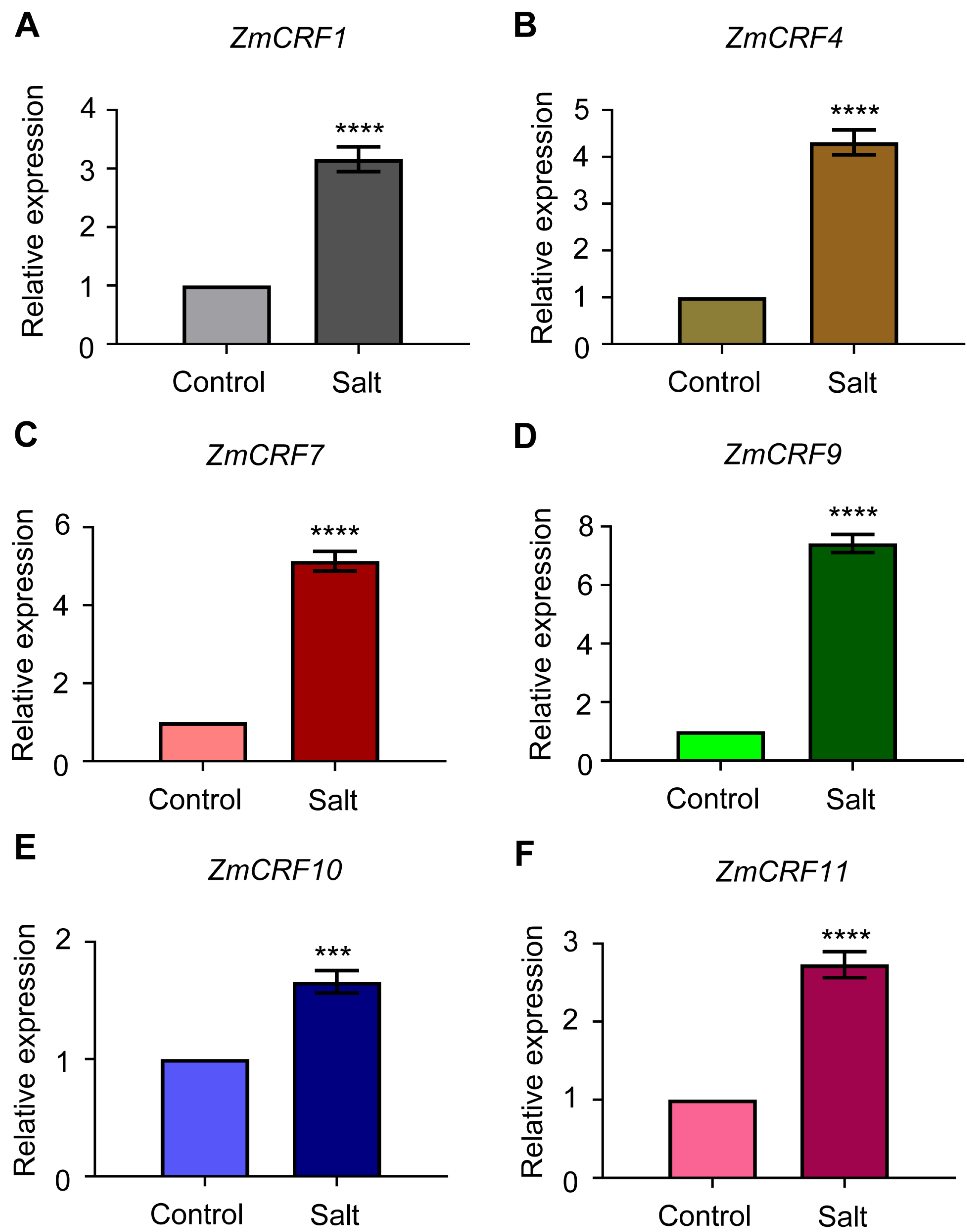
2.7. Expression Profiles of ZmCRFs under Multiple Abiotic Stresses
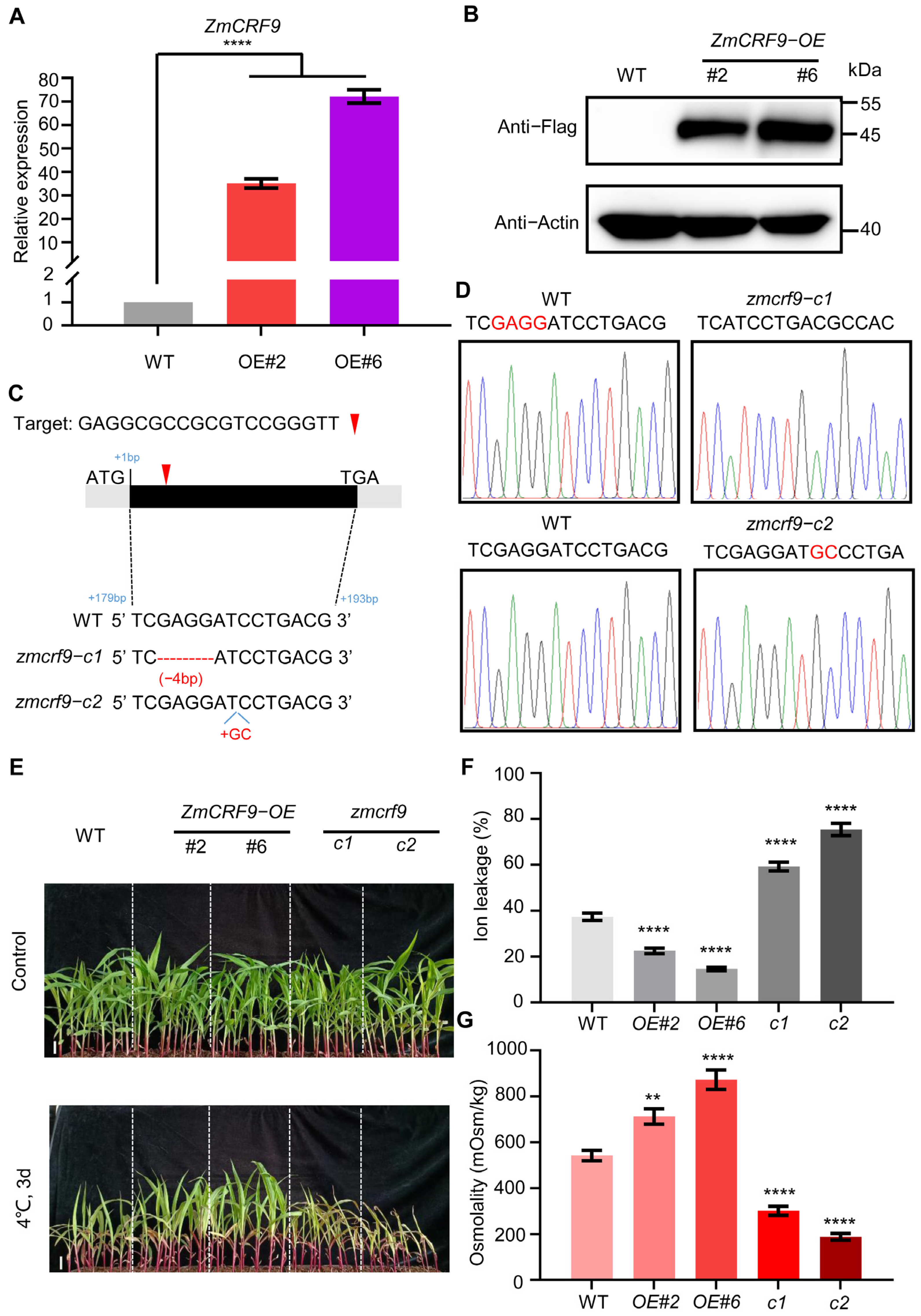
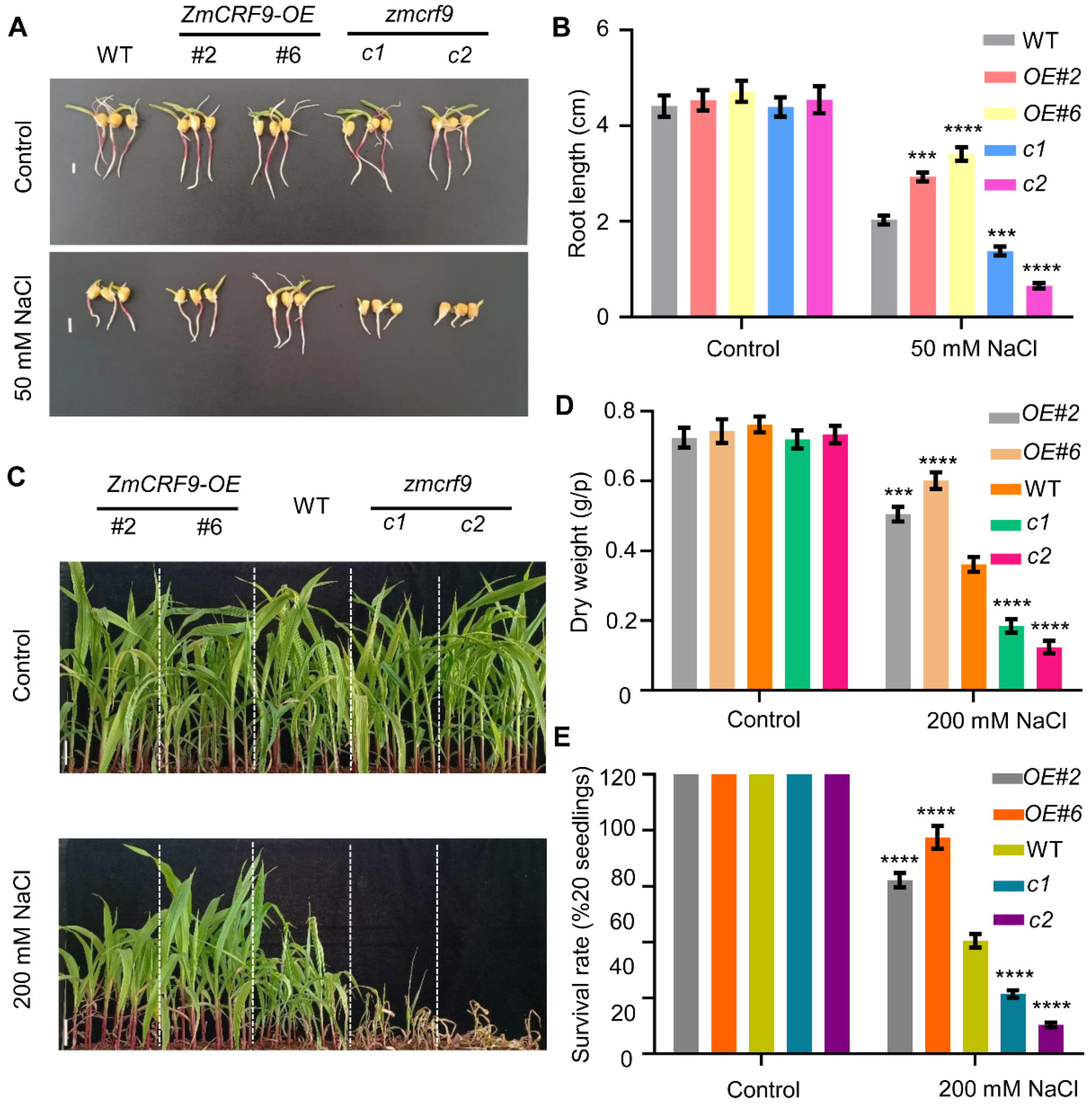
2.8. ZmCRF9 Functions as a Positive Regulator of Cold and Salt Tolerance in Maize
2.9. Interaction Network Analysis of ZmCRF Proteins
3. Discussion
4. Material and Methods
4.1. Identification and Bioinformatics Analysis of the ZmCRF Genes
4.2. Characterization of Chromosomal Locations
4.3. ZmCRF Gene Duplication and Synteny Analyses
4.4. Comparative Sequence Alignment and Phylogenetic Assessment
4.5. Conserved Motifs and Gene Structure Analysis
4.6. Cis-Acting Regulatory Element Analysis of ZmCRFs
4.7. Expression Profiles of ZmCRFs in Maize
4.8. Plant Materials and Growth Conditions
4.9. RNA Isolation and qRT-PCR
4.10. Plant Transformation
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cortleven, A.; Leuendorf, J.E.; Frank, M.; Pezzetta, D.; Bolt, S.; Schmülling, T. Cytokinin action in response to abiotic and biotic stresses in plants. Plant Cell Environ. 2019, 42, 998–1018. [Google Scholar] [CrossRef] [PubMed]
- Werner, T.; Motyka, V.; Strnad, M.; Schmülling, T. Regulation of plant growth by cytokinin. Proc. Nat. Acad. Sci. USA 2001, 98, 10487–10492. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, M.; Meng, Z.; Wang, B.; Chen, M. Research Progress on the Roles of Cytokinin in Plant Response to Stress. Int. J. Mol. Sci. 2020, 21, 6574. [Google Scholar] [CrossRef]
- Schaller, G.E.; Bishopp, A.; Kieber, J.J. The Yin-Yang of Hormones: Cytokinin and Auxin Interactions in Plant Development. Plant Cell 2015, 27, 44–63. [Google Scholar] [CrossRef] [PubMed]
- El-Showk, S.; Ruonala, R.; Helariutta, Y. Crossing paths: Cytokinin signalling and crosstalk. Development 2013, 140, 1373–1383. [Google Scholar] [CrossRef] [PubMed]
- Kieber, J.J.; Schaller, G.E. Cytokinin signaling in plant development. Development 2018, 145, 149344. [Google Scholar] [CrossRef] [PubMed]
- Hallmark, H.T.; Rashotte, A.M. Review—Cytokinin Response Factors: Responding to more than cytokinin. Plant Sci. 2019, 289, 110251. [Google Scholar] [CrossRef] [PubMed]
- Rashotte, A.M.; Mason, M.G.; Hutchison, C.E.; Ferreira, F.J.; Schaller, G.E.; Kieber, J.J. A subset of Arabidopsis AP2 transcription factors mediates cytokinin responses in concert with a two-component pathway. Proc. Nat. Acad. Sci. USA 2006, 103, 11081–11085. [Google Scholar] [CrossRef] [PubMed]
- Rashotte, A.M.; Goertzen, L.R. The CRF domain defines Cytokinin Response Factor proteins in plants. BMC Plant Biol. 2010, 10, 74. [Google Scholar] [CrossRef]
- Cutcliffe, J.W.; Hellmann, E.; Heyl, A.; Rashotte, A.M. CRFs form protein–protein interactions with each other and with members of the cytokinin signalling pathway in Arabidopsis via the CRF domain. J. Exp. Bot. 2011, 62, 4995–5002. [Google Scholar] [CrossRef]
- Zwack, P.J.; Shi, X.; Robinson, B.R.; Gupta, S.; Compton, M.A.; Gerken, D.M.; Goertzen, L.R.; Rashotte, A.M. Vascular Expression and C-Terminal Sequence Divergence of Cytokinin Response Factors in Flowering Plants. Plant Cell Physiol. 2012, 53, 1683–1695. [Google Scholar] [CrossRef] [PubMed]
- Kim, J. Cytokinin Response Factors Gating Environmental Signals and Hormones. Trends Plant Sci. 2016, 21, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; Cho, C.; Lee, M.R.; Van Binh, N.; Kim, J. Cytokinin Response Factor2 (CRF2) and CRF3 Regulate Lateral Root Development in Response to Cold Stress in Arabidopsis. Plant Cell 2016, 28, 1828–1843. [Google Scholar] [CrossRef] [PubMed]
- Zwack, P.J.; Compton, M.A.; Adams, C.I.; Rashotte, A.M. Cytokinin response factor 4 (CRF4) is induced by cold and involved in freezing tolerance. Plant Cell Rep. 2015, 35, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Wang, L.; Guo, Y.; Li, Y.; Ümüt, H.; Wang, Y. An ERF transcription factor from Tamarix hispida, ThCRF1, can adjust osmotic potential and reactive oxygen species scavenging capability to improve salt tolerance. Plant Sci. 2017, 265, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, H.; Shi, L.; Xu, F.; Ding, G. Genome-Wide Dissection of the CRF Gene Family in Brassica napus Indicates that BnaCRF8s Specifically Regulate Root Architecture and Phosphate Homeostasis against Phosphate Fluctuation in Plants. Int. J. Mol. Sci. 2020, 21, 3660. [Google Scholar] [CrossRef] [PubMed]
- Zwack, P.J.; Robinson, B.R.; Risley, M.G.; Rashotte, A.M. Cytokinin Response Factor 6 Negatively Regulates Leaf Senescence and is Induced in Response to Cytokinin and Numerous Abiotic Stresses. Plant Cell Physiol. 2013, 54, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Gupta, S.; Rashotte, A.M. Characterization of two tomato AP2/ERF genes, SlCRF1 and SlCRF2 in hormone and stress responses. Plant Cell Rep. 2013, 33, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Rashotte, A.M. Expression patterns and regulation of SlCRF3 and SlCRF5 in response to cytokinin and abiotic stresses in tomato (Solanum lycopersicum). J Plant Physiol. 2014, 171, 349–358. [Google Scholar] [CrossRef]
- Zwack, P.J.; De Clercq, I.; Howton, T.C.; Hallmark, H.T.; Hurny, A.; Keshishian, E.A.; Parish, A.M.; Benkova, E.; Mukhtar, M.S.; Van Breusegem, F.; et al. Cytokinin Response Factor 6 Represses Cytokinin-Associated Genes during Oxidative Stress. Plant Physiol. 2016, 172, 1249–1258. [Google Scholar] [CrossRef]
- Shi, X.; Gupta, S.; Rashotte, A.M. Solanum lycopersicum cytokinin response factor (SlCRF) genes: Characterization of CRF domain-containing ERF genes in tomato. J. Exp. Bot. 2012, 63, 973–982. [Google Scholar] [CrossRef]
- Wu, S.-B.; Liu, Z.; Kong, L.; Zhang, M.; Lv, Y.; Liu, Y.; Zou, M.; Lu, G.; Cao, J.; Yu, X. Genome-Wide Identification, Phylogeny, Evolution and Expression Patterns of AP2/ERF Genes and Cytokinin Response Factors in Brassica rapa ssp. pekinensis. PLoS ONE 2013, 8, e83444. [Google Scholar]
- Duan, X.; Zhang, K.; Duanmu, H.; Yu, Y. Genome-Wide Identification and Expression Characteristics of Cytokinin Response Factors in Soybean. J. Plant Growth Regul. 2023, 42, 4484–4496. [Google Scholar] [CrossRef]
- Schnable, J.C. Genome Evolution in Maize: From Genomes Back to Genes. Annu. Rev. Plant Biol. 2015, 66, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Shiferaw, B.; Prasanna, B.M.; Hellin, J.; Bänziger, M. Crops that feed the world 6. past successes and future challenges to the role played by maize in global food security. Food Sec. 2011, 3, 307–327. [Google Scholar] [CrossRef]
- Bellon, M.R.; Mastretta-Yanes, A.; Ponce-Mendoza, A.; Ortiz-Santamaría, D.; Oliveros-Galindo, O.; Perales, H.; Acevedo, F.; Sarukhán, J. Evolutionary and food supply implications of ongoing maize domestication by Mexican campesinos. Proc. R. Soc. B 2018, 285, 1049. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef]
- Kong, L.; Zhao, K.; Gao, Y.; Miao, L.; Chen, C.; Deng, H.; Liu, Z.; Yu, X. Comparative analysis of cytokinin response factors in Brassica diploids and amphidiploids and insights into the evolution of Brassica species. BMC Genom. 2018, 19, 728. [Google Scholar] [CrossRef]
- Hurley, I.; Hale, M.E.; Prince, V.E. Duplication events and the evolution of segmental identity. Evol. Dev. 2010, 7, 556–567. [Google Scholar] [CrossRef]
- Prince, V.E.; Pickett, F.B. Splitting pairs: The diverging fates of duplicated genes. Nat. Rev. Genet. 2002, 3, 827–837. [Google Scholar] [CrossRef]
- Hu, T.; Banzhaf, W. Nonsynonymous to Synonymous Substitution Ratio ka/ks: Measurement for Rate of Evolution in Evolutionary Computation. In Parallel Problem Solving from Nature—PPSN X, 10th International Conference, Dortmund, Germany, 13–17 September 2008; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Travers, A.; Muskhelishvili, G. DNA structure and function. FEBS J. 2015, 282, 2279–2295. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Peng, D.; Bai, L.-P.; Ma, H.; Chen, L.-J.; Zhao, M.-H.; Xu, Z.-J.; Guo, Z.-F. Molecular switch for cold acclimation—Anatomy of the cold-inducible promoter in plants. Biochemistry 2013, 78, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.Z.; Harper, L.C.; Schaeffer, M.L.; Andorf, C.M.; Seigfried, T.E.; Campbell, D.A.; Lawrence, C.J. Choosing a genome browser for a Model Organism Database: Surveying the Maize community. Database 2010, 2010, baq007. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.J.; Jian, L.; Xu, J.; Zhang, Q.; Zhang, M.; Jin, M.; Peng, Y.; Yan, J.; Han, B.; Liu, J.; et al. High-Throughput CRISPR/Cas9 Mutagenesis Streamlines Trait Gene Identification in Maize. Plant Cell 2020, 32, 1397–1413. [Google Scholar] [CrossRef] [PubMed]
- Salomé, P.A. A Roadmap toward Large-Scale Genome Editing in Crops. Plant Cell 2020, 32, 1340–1341. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-h.; Zhu, J.-K. Phenotypic Analysis of Arabidopsis Mutants: Electrolyte Leakage after Freezing Stress. Cold Spring Harb. Protoc. 2010, 2010, prot4970. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, Y.; Lu, J.; Shao, H. Roles of plant soluble sugars and their responses to plant cold stress. Afr. J. Biotechnol. 2009, 8, 2004–2010. [Google Scholar]
- An, H.; Roussot, C.; Suárez-López, P.; Corbesier, L.; Vincent, C.; Piñeiro, M.; Hepworth, S.; Mouradov, A.; Justin, S.; Turnbull, C.; et al. CONSTANS acts in the phloem to regulate a systemic signal that induces photoperiodic flowering of Arabidopsis. Development 2004, 131, 3615–3626. [Google Scholar] [CrossRef]
- Jaenicke, U.H.R.; Franks, F.; Chapman, D.; Griffin, M.C.A.; Hvidt, A.; Cowan, D.A. Protein structure and function at low temperatures. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1997, 326, 535–553. [Google Scholar]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.J.S. The B73 Maize Genome: Complexity, Diversity, and Dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef]
- Li, G.; Wang, J.; Liao, Y.; Jiang, D.; Chen, C.; Zheng, Y. A high-continuity genome assembly of Chinese flowering cabbage (Brassica rapa var. parachinensis) provides new insights into Brassica genome structure evolution. Plant 2023, 12, 2498. [Google Scholar] [CrossRef]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef]
- Barone, A.; Matteo, A.; Carputo, D.; Frusciante, L. High-Throughput Genomics Enhances Tomato Breeding Efficiency. Curr. Genom. 2009, 10, 1–9. [Google Scholar] [CrossRef]
- Bennett, M.D. Comparisons with Caenorhabditis (100 Mb) and Drosophila (175 Mb) Using Flow Cytometry Show Genome Size in Arabidopsis to be 157 Mb and thus 25% Larger than the Arabidopsis Genome Initiative Estimate of 125 Mb. Ann. Bot. 2003, 91, 547–557. [Google Scholar] [CrossRef]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.P.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef]
- Liu, L.; White, M.J.; MacRae, T.H. Transcription factors and their genes in higher plants. Eur. J. Biochem. 2001, 262, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Nott, A.; Meislin, S.; Moore, M. A quantitative analysis of intron effects on mammalian gene expression. RNA 2003, 9, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Marand, A.P.; Eveland, A.L.; Kaufmann, K.; Springer, N.M. Cis-Regulatory Elements in Plant Development, Adaptation, and Evolution. Annu. Rev. Plant Biol. 2023, 74, 111–137. [Google Scholar] [CrossRef]
- Brady, S.M.; Long, T.A.; Benfey, P.N. Unraveling the Dynamic Transcriptome. Plant Cell 2006, 18, 2101–2111. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Guo, Y. Elucidating the molecular mechanisms mediating plant salt-stress responses. New Phytol. 2018, 217, 523–539. [Google Scholar] [CrossRef]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [PubMed]
- Voorrips, R.E. MapChart: Software for the Graphical Presentation of Linkage Maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, İ.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wang, J.; Lin, W.; Li, S.; Li, H.; Zhou, J.; Ni, P.; Dong, W.; Hu, S.; Zeng, C.; et al. The Genomes of Oryza sativa: A history of duplications. PLoS Biol. 2005, 3, e38. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, 50, W276–W279. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Peterson, D.; Tamura, K. MEGA-CC: Computing core of molecular evolutionary genetics analysis program for automated and iterative data analysis. Bioinformatics 2012, 28, 2685–2686. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L. Discovering novel sequence motifs with MEME. Curr. Protoc. Bioinform. 2002, 2, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jin, J.; Guo, A.-Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef]
- Rombauts, S.; Déhais, P.; Van Montagu, M.; Rouzé, P. PlantCARE, a plant cis-acting regulatory element database. Nucleic Acids Res. 1999, 27, 295–296. [Google Scholar] [CrossRef]
- Andorf, C.M.; Cannon, E.K.; Portwood, J.L.; Gardiner, J.M.; Harper, L.C.; Schaeffer, M.L.; Braun, B.L.; Campbell, D.A.; Vinnakota, A.G.; Sribalusu, V.V.; et al. MaizeGDB update: New tools, data and interface for the maize model organism database. Nucleic Acids Res. 2016, 44, D1195–D1201. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Fu, D.; Wang, X.; Zeng, R.; Zhang, X.; Tian, J.; Zhang, S.; Yang, X.; Tian, F.; Lai, J.; et al. The transcription factor bZIP68 negatively regulates cold tolerance in maize. Plant Cell 2022, 34, 2833–2851. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Liu, J.; Zhai, L.; Gan, Z.; Zhang, G.; Yang, S.; Wang, Y.; Wu, T.; Zhang, X.; Xu, X.; et al. Natural variation in cytokinin maintenance improves salt tolerance in apple rootstocks. Plant Cell Environ. 2018, 42, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Li, K.; Li, Y.; Wang, W.; Leng, B.; Yao, G.; Zhang, F.; Mu, C.; Liu, X. The ZmbHLH32-ZmIAA9-ZmARF1 module regulates salt tolerance in maize. Int. J. Biol. Macromol. 2023, 253, 126978. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Sun, X.; Bian, X.; Wei, T.; Han, T.; Yan, J.; Zhang, A.; Zhang, J. The transcription factor ZmNAC49 reduces stomatal density and improves drought tolerance in maize. J. Exp. Bot. 2021, 72, 1399–1410. [Google Scholar] [CrossRef]
- Le Pioufle, O.; Ganoudi, M.; Calonne-Salmon, M.; Ben Dhaou, F.; Declerck, S. Rhizophagus irregularis MUCL 41833 Improves Phosphorus Uptake and Water Use Efficiency in Maize Plants During Recovery From Drought Stress. Front. Plant Sci. 2019, 10, 897. [Google Scholar] [CrossRef] [PubMed]
- Hodges, D.M.; DeLong, J.M.; Forney, C.F. Improving the thiobarbituric acid-reactive-substances assay for estimating lipid peroxidation in plant tissues containing anthocyanin and other interfering compounds. Planta 1999, 207, 604–611. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, J. Role of Abscisic Acid in Water Stress-induced Antioxidant Defense in Leaves of Maize Seedlings. Free Radic. Res. 2009, 36, 1001–1015. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, Z.; Hou, J.; Leng, B.; Yao, G.; Ma, C.; Sun, Y.; Zhang, F.; Mu, C.; Liu, X. Genome-Wide Investigation of the CRF Gene Family in Maize and Functional Analysis of ZmCRF9 in Response to Multiple Abiotic Stresses. Int. J. Mol. Sci. 2024, 25, 7650. https://doi.org/10.3390/ijms25147650
Yan Z, Hou J, Leng B, Yao G, Ma C, Sun Y, Zhang F, Mu C, Liu X. Genome-Wide Investigation of the CRF Gene Family in Maize and Functional Analysis of ZmCRF9 in Response to Multiple Abiotic Stresses. International Journal of Molecular Sciences. 2024; 25(14):7650. https://doi.org/10.3390/ijms25147650
Chicago/Turabian StyleYan, Zhenwei, Jing Hou, Bingying Leng, Guoqi Yao, Changle Ma, Yue Sun, Fajun Zhang, Chunhua Mu, and Xia Liu. 2024. "Genome-Wide Investigation of the CRF Gene Family in Maize and Functional Analysis of ZmCRF9 in Response to Multiple Abiotic Stresses" International Journal of Molecular Sciences 25, no. 14: 7650. https://doi.org/10.3390/ijms25147650
APA StyleYan, Z., Hou, J., Leng, B., Yao, G., Ma, C., Sun, Y., Zhang, F., Mu, C., & Liu, X. (2024). Genome-Wide Investigation of the CRF Gene Family in Maize and Functional Analysis of ZmCRF9 in Response to Multiple Abiotic Stresses. International Journal of Molecular Sciences, 25(14), 7650. https://doi.org/10.3390/ijms25147650