Abstract
Colon cancer, one of the most common and fatal cancers worldwide, is characterized by stepwise accumulation of specific genetic alterations in tumor suppressor genes or oncogenes, leading to tumor growth and metastasis. HIPK2 (homeodomain-interacting protein kinase 2) is a serine/threonine protein kinase and a “bona fide” oncosuppressor protein. Its activation inhibits tumor growth mainly by promoting apoptosis, while its inactivation increases tumorigenicity and resistance to therapies of many different cancer types, including colon cancer. HIPK2 interacts with many molecular pathways by means of its kinase activity or transcriptional co-repressor function modulating cell growth and apoptosis, invasion, angiogenesis, inflammation and hypoxia. HIPK2 has been shown to participate in several molecular pathways involved in colon cancer including p53, Wnt/β-catenin and the newly identified nuclear factor erythroid 2 (NF-E2) p45-related factor 2 (NRF2). HIPK2 also plays a role in tumor–host interaction in the tumor microenvironment (TME) by inducing angiogenesis and cancer-associated fibroblast (CAF) differentiation. The aim of this review is to assess the role of HIPK2 in colon cancer and the underlying molecular pathways for a better understanding of its involvement in colon cancer carcinogenesis and response to therapies, which will likely pave the way for novel colon cancer therapies.
Keywords:
HIPK2; p53; colorectal cancer; colon cancer; hypoxia; hyperglycemia; cancer therapy; Wnt/β-catenin; NRF2; microRNA; angiogenesis; tumor inflammation 1. Introduction
Colorectal cancer (herein, colon cancer), which comprises colon and/or rectum cancer, is one of the most common cancers in women and men: it is the third most frequently diagnosed type of cancer (11% of all diagnosed cancer cases) and the second leading cause of cancer-related mortality worldwide [1,2]. The 5-year survival rate depends on the tumor stage, ranging from 90% for the localized stage to 71% for regional and 14% for patients with distant metastasis [3]. Thus, early detection of the disease is a priority. In addition, a worrying rise in patients presenting with colon cancer younger than 50 years has been observed [4]. There are three principal types of colon cancer: sporadic, hereditary and colitis-associated. Global incidence and mortality are likely to be increased in the coming decades and especially in highly developed countries, mostly depending on dietary factors and lifestyle, family history and chronic inflammation [5,6]. Risk factors comprise a high-calorie diet rich in animal-derived proteins, especially red and processed meat, wheat products and high sugar consumption, combined with poor physical activity. In addition, other risk factors are family history of colon cancer (stronger association for first-degree relatives), inflammatory bowel disease (IBD), Crohn’s disease, smoking, excessive alcohol consumption, obesity and diabetes [7,8] (Figure 1).

Figure 1.
Schematic representation of the risk factors of colon cancer development.
The most important preventive factor represents routine endoscopic check-ups, namely colonoscopy, with the removal of precancerous lesions (polypectomy) and, interestingly, the role of new technologies in detection of neoplasia, such as artificial intelligence, which is rapidly emerging [9]. Colon cancer development is characterized by a “multistep carcinogenesis“ process, according to the Fearon and Vogelstein genetic model [10], involving a series of histological and morphological changes beginning as a benign adenomatous intestinal polyp from epithelial tissue of the colon, which progresses to advanced adenoma with high-grade dysplasia, invasive adenocarcinoma, and metastasis to distant organs such as the liver [10] (Figure 2). These changes are triggered by a sequential accumulation of specific genomic alterations involving different oncogenes such as Kirsten rat sarcoma virus (KRAS), serine/threonine-protein kinase B-Raf (BRAF), phosphatidylinositol kinase catalytic subunit alpha (PIK3CA) and tumor suppressor genes such adenomatous polyposis coli (APC), tumor suppressor protein (TP)53, SMAD4, phosphatase and tensin homolog (PTEN) [11,12], along with chromosomal (microsatellite) instabilities (Figure 2), that deregulate key signaling pathways driving disease progression, including Wnt/β-catenin, downstream mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K), epidermal growth factor receptor (EGFR) and transforming growth factor beta (TGF-β) [13]. In the early steps of tumorigenesis, the function of the tumor suppressor p53 is inhibited by increased levels of its suppressor MDM2 (murine double minute 2) caused by a KRAS mutation actioned by the PI3K–AKT pathway [14,15]. Later stages of tumorigenesis are induced by a further loss of wild type p53 protein, by gene mutation, leading to a more malignant phenotype and resistance to therapies [16,17,18].
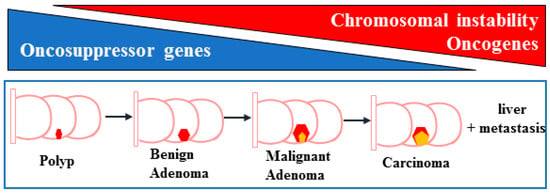
Figure 2.
Schematic representation of the steps of colon cancer development. The passages from polyp to benign adenoma, malignant adenoma, carcinoma (represented by the pink/yellow pentagons), in the colon regions, and liver metastasis, include the reduction of oncosuppressor genes (blue triangle) and the activation of oncogenes with increased chromosomal instability (red triangle).
Recently, the impact of the tumor microenvironment (TME)—composed of cancer-associated fibroblasts (CAF), endothelial cells sprouting angiogenesis, immune cells, along with the microbiota and the tumor-derived exosomes (TEXs)—has gained attention in colon cancer progression and metastasis, either by promoting or inhibiting the processes, becoming an interesting target in colon cancer therapies [19,20]. The basis of colon cancer treatment consists of surgery, targeted therapy, neoadjuvant radiotherapy and adjuvant chemotherapy along with the promising immunotherapy [21,22]. Unfortunately, drug-resistance remains one of the deadlocks for the low survival rates of colon cancer patients, as several molecular alterations confer resistance to standard chemotherapy and targeted agents [21]. A better understanding of the mechanisms leading to intrinsic and acquired resistance to therapies will be a great asset for prognostic and therapeutic purpose in colon cancer. Colon cancers carrying common genetic events have been shown to markedly vary in their biology [23]; thus, to further improve treatment opportunities, additional diagnostic parameters and molecular determinants of clinical outcome for colon cancer are required.
In this regard, the aim of this review is to assess the role of the “bona fide” oncosuppressor homeodomain interacting protein kinase-2 (HIPK2) in colon cancer progression and response to therapies. By reviewing the current literature, the review will summarize the molecular interplay that occurs between HIPK2 and molecular pathways involved in colon cancer regression and response to therapies that could likely be exploited as a potential biomarker and therapeutic target in colon cancer therapy.
2. HIPK2 in Colon Cancer
HIPK2 is a serine-threonine and an evolutionarily conserved protein kinase that interacts with many different molecules by means of its kinase or transcriptional activities [24,25]. Through these interactions, HIPK2 regulates many cellular functions including cell growth and apoptosis, fibrosis, angiogenesis and inflammation involved in cancer, fibrotic and neurodegenerative diseases [26,27,28]. Scientific consensus suggests that HIPK2 is a “bona fide” oncosuppressor protein, thus, its activation inhibits tumor growth, mainly by induction of apoptosis with p53 activation, while its inactivation increases tumorigenicity and chemoresistance through inhibition of apoptotic pathways or de-repression of pro-survival pathways [29]. The role of HIPK2 in cancer has been previously well reviewed [24,25,26,27,28,29]. Briefly, HIPK2 is activated by anticancer drugs or DNA damage and inactivated by hypoxia, hyperglycemia or micro (mi) RNAs [29,30,31]. The role of HIPK2 in colon cancer comes from studies that demonstrate that HIPK2 participates in most of the pathways affected in colon cancer: it positively modulates oncosuppressor p53 through direct phosphorylation, inducing apoptosis in response to DNA damage [32,33]; it interacts with the Wnt/β-catenin and TGF-β at multiple levels (e.g., by targeting β-catenin, by forming a multimeric complex with AXIN, as a transcriptional corepressor of SMADs and by interacting with the common Wnt/TGF-β mediator TAK1) [34,35,36,37,38,39,40,41]; and it interacts with the nuclear factor erythroid 2 (NF-E2) p45-related factor 2 (NRF2) [42,43,44]. Recently, a role for HIPK2 in the remodeling of the TME has been unveiled. In particular, HIPK2 inhibition induces a pro-inflammatory phenotype, angiogenesis and cancer-associated fibroblasts (CAF) differentiation [27,45,46,47], supporting tumor progression and chemoresistance. HIPK2 is downregulated by hypoxia and by hyperglycemia [48,49] (the latter being a common feature of diabetes mellitus), two conditions often present in solid tumors, including colon cancer, and having a role in cancer progression and resistance to therapies [5,50,51]. For these reasons, HIPK2 is becoming an appealing molecule in the colon cancer field, deserving in-depth analysis in future studies, for further understanding of colon cancer development and response to therapies. Below is a summary of the studies that, up to now, have evaluated the HIPK2 expression in colon cancer tissues, genomic data sets and cell lines, with a focus on the interaction with molecular pathways involved in cancer progression and response to therapies.
2.1. HIPK2 and p53
The TP53 protein is considered “the guardian of the genome”, as it plays a crucial role in regulation of the cell cycle and the genome stability [52]. Given its key role in drug-induced apoptosis, wild-type (WT) p53 plays a critical role in efficient tumor response to therapies [53]. Up to 60% of patients with colon cancer show somatic mutations of the TP53 gene and in the remaining percentage, p53 can be deregulated at the protein level. In both cases, inactivation of p53 oncosuppressor functions drives the transition from adenoma to adenocarcinoma and impairs the tumor response to therapies [14,16,18]. As HIPK2 is a positive regulator of p53 apoptotic function [54], the effect of HIPK2 depletion on p53 transcriptional activity was first analyzed using a microarray assay. The study highlighted that HIPK2 depletion with small interfering (si) RNA in colon cancer cells leads to the loss of WTp53 target gene activation [55]. Mechanistically, the results showed that stable HIPK2 knockdown, by modulating metallothionein and zinc, induces WTp53 protein misfolding that impairs p53–DNA binding and WTp53 target gene transcription. Interestingly, the WTp53 misfolding could be reverted by zinc supplementation, as evidenced by in vitro and in vivo studies [55,56,57]. Bioinformatics analysis of microarray data from colon cancer patients with known clinical record and p53 mutation status, using the Kaplan–Meier procedure, showed significant association of poor survival with low HIPK2 expression only in tumors expressing WTp53, in agreement with the in vitro data [55] (Table 1). These findings unveiled an unexpected mechanism of WTp53 inactivation following HIPK2 depletion and proposed a new way (that is, zinc supplementation) to modify the possible equilibrium between active and inactive WTp53 in tumors (i.e., by increasing WTp53 oncosuppressor function), which may be applied in the clinic. These findings were in line with a study showing that HIPK2 mutations in acute myeloid leukemia impair p53-mediated transcription [58]. HIPK2 protein activity was also required in vivo for efficient p300/p53 co-recruitment onto apoptotic gene promoters; thus, by balancing p53 acetylation and deacetylation [59], other than through phosphorylating p53 at serine 46 (Ser46) [53,54], HIPK2 regulates p53 apoptosis-promoting transcriptional activity [54]. These pioneering studies revealed a critical role of HIPK2 in maintaining the transactivation activity of WTp53 and further suggest that low expression of HIPK2 may impair the p53 function in tumors harboring WTp53. They also opened the way to anticancer treatments using zinc, in combination with classical chemotherapy, to restore WTp53 activity when cancers present low expression of HIPK2 [57] or in p53-functionally deficient cancer cells [60].
Looking for a prognostic role of HIPK2 in colon cancer, the authors analyzed a retrospective series of 80 primary patients’ samples at different tumor stage of colon cancer and normal mucosa samples (taken at distance from the tumor) by immunofluorescence and tissue microarray (TMA) by using multiplexed tissue cytometry capable of exploring correlative protein expression at the single tumor cell level on TMA, simultaneously monitoring HIPK2 and p53 protein expression at the single cell level. The authors found that an increase of HIPK2 protein level in the tumor, compared to the normal mucosa, in univariate analysis, was associated with a better prognosis that did not depend on WTp53 status because it was also observed in p53-mutated background, highlighting the p53-independent function of HIPK2 [61] (Table 1). The expression of HIPK2 was then evaluated in primary tumor specimens of human colon cancer, with particular regard to post-operative cancer recurrence, metastasis and malignancy grades [62]. The authors started from the finding that in China, the rate of colon cancer incidence is increasing faster nationally than all other cancers [63] and that colon tumorigenesis may depend on functional abnormalities of relevant genes. Immunohistochemistry (IHC) analysis of 100 colon tumor samples and 20 normal intestinal tissues displayed that HIPK2 expression inversely correlates with Dukes stage and depth of cancer invasion [62] (Table 1). This is in line with previous studies showing that HIPK2 protein downregulation in samples of other cancer types, such as breast [64], thyroid [65], and pancreatic [66], increases tumor progression. By using a xenograft colon cancer mouse model, the authors tested the in vivo anti-tumor effect of verbascoside (VB), an active constituent of a Chinese traditional medical plant genus that has an effect in many cancers, including colon cancer [67,68], and measured protein levels of HIPK2 and p53, and the apoptosis-related gene products Bax and Bcl-2. They showed that VB stimulates the HIPK2–p53 signaling pathway, thus inhibiting cell proliferation and promoting apoptosis in colon cancer [62] (Table 1), pointing again to the key role of a functional HIPK2–p53 axis for achieving efficient tumor cell death in response to cytotoxic therapies [26].

Table 1.
Summary of the HIPK2 expression in colon cancer tissues and cell lines along with the biological and molecular effects and the specific references. IHC: Immunohistochemistry; cPLA2: cytosolic phospholipase A2; PGE2: prostaglandin E2; COX-2: cyclooxygenase-2; VEGF: vascular endothelial growth factor; HIF-1: hypoxia inducible factor-1; TMA: tissue microarray; 5-FU: 5-Fluorouracil; OXA: oxaliplatin; lncRNA PRNT: long non-coding RNA prion protein testis specific.
Table 1.
Summary of the HIPK2 expression in colon cancer tissues and cell lines along with the biological and molecular effects and the specific references. IHC: Immunohistochemistry; cPLA2: cytosolic phospholipase A2; PGE2: prostaglandin E2; COX-2: cyclooxygenase-2; VEGF: vascular endothelial growth factor; HIF-1: hypoxia inducible factor-1; TMA: tissue microarray; 5-FU: 5-Fluorouracil; OXA: oxaliplatin; lncRNA PRNT: long non-coding RNA prion protein testis specific.
| HIPK2 Expression in Colon Cancer Tissues and/or Cell Lines | Molecular/Cellular Effects | Biological Outcome | Ref. |
|---|---|---|---|
| ↓ HIPK2 protein expression by siRNA in colon cancer cell lines in vitro; Microarray data on >300 samples from colon cancer patients with known clinical records and p53 mutation status. | ↓ p53 apoptotic activity in vitro and in vivo in tumor xenografts | ↓ Patient survival with low HIPK2 expression only in tumors expressing WTp53 | [55] |
| ↑ HIPK2 protein levels, by multiplexed tissue cytometry, in 80 colon cancer tissues | ↑ Patient prognosis | [61] | |
| HIPK2 protein expression by IHC in 100 colon cancer tissues | Verbascoside treatment stimulates HIPK2–p53 apoptotic pathway in vitro and in vivo, in tumor xenografts | Inverse correlation between HIPK2 expression and Dukes stage and invasion | [62] |
| ↓ HIPK2 mRNA levels in nine colon cancer tissues of patients with sporadic colorectal cancer | ↑ cPLA2 mRNA expression and ↑ PGE2 production; HIPK2 represses cPLA2 promoter activity in vitro. | ↓ HIPK2 expression increases the growth of tumor xenografts; Inverse correlation between HIPK2 expression and Dukes stage. | [45] |
| Inverse correlation between HIPK2 and COX-2 expression in primary colon adenocarcinomas in silico | ↓ HIPK2 expression leads to ↑ COX-2/VEGF pathway through HIF-1 in vitro; VEGF inhibition in HIPK2-depleted cells restored dendritic cell (DC) maturation. | [69] | |
| Colitis-associated colon cancer in HIPK2+/− mice | ↑ Percentage of macrophages in the tumors of HIPK2+/− mice; ↑ Serum concentration of pro-inflammatory IL-6, IL-1β and TNF-α cytokines through NF-κB; HIPK2 inhibits NF-κB activity in vitro. | ↑ Colon cancers grow in HIPK2+/− mice | [46] |
| ↑ HIPK2 expression, by IHC, in TMA of 270 colon cancer samples | ↑ Tumor progression and TNM stages; ↓ HIPK2 expression reduces ERK phosphorylation in vitro. | ↓ HIPK2 expression reduces the growth of tumors derived from KRAS mutated colon cancer cells | [70] |
| ↑ HIPK2 expression, by immunohistochemistry (IHC), in tissue microarray (TMA) of 84 stage II colon cancer samples | ↑ Response to therapy (5-FU, OXA) | [71] | |
| ↑ lncRNA PRNT in datasets of OXA-resistant cancer cells | ↓ HIPK2 mRNA | ↑ Colon cancer cell growth and migration, and resistance to OXA in vitro and in vivo | [72] |
| ↓ HIPK2 mRNA levels in colon cancer tissues | ↑ exomiR-1229, ↑ VEGFA, ↑ VEGFR1, and ↑ p-AKT; exomiR-1229 target and inhibits HIPK2 in vitro. | ↑ Angiogenesis, metastasis and poor survival | [73] |
| HIPK2 Expression in Colon Cancer Tissues and/or Cell Lines | Molecular and Biological Effects | Ref. | |
| Reduced HIPK2 protein expression by siRNA in cell lines | Reduced p53 apoptotic activity in vitro and in vivo; Poor patient survival with low HIPK2 expression only in tumors expressing WTp53. | [55] | |
| Increased HIPK2 levels in CRC tissues | Better patient prognosis that did not depend on WTp53 status | [61] | |
| HIPK2 expression by IHC in CRC | Inverse correlation between HIPK2 expression and Dukes stage and invasion; Verbascoside treatment stimulates HIPK2/p53 apoptotic pathway in vitro and in vivo. | [62] | |
| Low HIPK2 mRNA levels in CRC tissues of patients with sporadic colorectal cancer | High cytosolic phospholipase A2 (cPLA2) expression and PGE2 production; HIPK2 represses cPLA2 promoter activity; Increased in vivo tumor growth of xenografts from HIPK2-depleted colon cancer cells; Inverse correlation between HIPK2 expression and Dukes stage. | [45] | |
| Inverse correlation between HIPK2 and COX-2 expression in primary colon adenocarcinomas in silico | HIPK2 inhibition leads to upregulation of COX-2/VEGF pathway through HIF-1; VEGF inhibition in HIPK2-depleted cells restored dendritic cell (DC) maturation. | [69] | |
| Colitis-associated CRC in Hipk2+/− mice | Tumors grow more rapidly in Hipk2+/− mice; The percentage of macrophages is increased in the tumors of Hipk2+/− mice; Increased serum concentration of pro-inflammatory IL-6, IL-1β and TNF-α cytokines through NF-κB; HIPK2 inhibits NF-κB activity. | [46] | |
| Increase of HIPK2-positive cancer cells in TMAs of colon cancer samples | Correlation with tumor progression and TNM stages; HIPK2 knockdown reduces ERK phosphorylation in vitro and the growth of tumors derived from KRAS mutated cells. | [70] | |
| High HIPK2 positivity in TMAs from tumor samples | Association with improved response to therapy (5-FU, OXA), independent from p53 status | [71] | |
| Inverse correlation between HIPK2 mRNA and lncRNA PRNT in datasets of OXA-resistant cancer cells | PRNT regulates HIPK2 expression in CRC by sponging ZNF184 transcription factor | [72] | |
| HIPK2 mRNA downregulation in CRC tissues compared to the adjacent normal tissues | HIPK2 inhibition by exomiR-1229 with consequent angiogenesis by VEGFA, VEGFR1 and p-AKT upregulation | [73] | |
↑ high; ↓ low.
2.2. HIPK2 and Inflammatory Pathways (COX/PGE2/VEGF and NF-κB)
HIPK2 mRNA levels were found to be significantly lower in colon cancer tissues of patients with sporadic colorectal cancer, expressing cytosolic phospholipase A2 (cPLA2), compared to tissues of patients with familial adenomatous polyposis, which showed undetectable cPLA2 levels [45]. An interesting link between HIPK2 and cPLA has been revealed. cPLA2 is involved in the generation of inflammatory prostanoids, including prostaglandin E2 (PGE2), which is the predominant prostanoid found in most colon cancers and is known to promote colon carcinoma growth by stimulating proliferation, angiogenesis, invasiveness and by inhibiting apoptosis [74,75]. In vitro studies with colon cancer cell lines, using siRNA, showed that HIPK2 silencing is associated with increased PGE2 biosynthesis that was profoundly suppressed by the cPLA2 inhibitor. Thus, molecular studies showed that HIPK2 overexpression, in a complex with histone deacetylase 1 (HDAC1), inhibits the cPLA2-luciferase promoter activity [45] (Table 1). The tumors derived from HIPK2-silenced cells, injected in nude mice, showed noticeably increased growth compared with tumors derived from parental cells, underscoring the role of HIPK2 to restrain progression of human colon tumorigenesis, at least in part, by turning off cPLA2-dependent PGE2 generation [45]. As a proof of principle, analysis of HIPK2 mRNA expression in human colon cancers showed that it tended to correlate inversely with the Dukes staging of the tumors [45] (Table 1), in agreement with the Zhou and colleagues’ study [62].
The production of prostaglandins via ciclooxygenase-2 (COX-2) is an important signaling pathway in the pathogenesis of colon cancer [76,77]. Transcriptional control of the COX-2 gene may depend on Wnt/β-catenin [78] or hypoxia-inducible factor-1 (HIF-1) [79] signaling pathways that can both undergo activation when HIPK2 is inhibited [29]. Thus, HIPK2 knockdown, by using siRNA, in colon cancer cells has been shown to upregulate COX-2 expression and, by using the Oncomine integrated cancer database research tool (http://www.oncomine.org, accessed on 23 June 2024), the analysis of datasets, obtained from specimens of normal tissues and primary colon adenocarcinomas, revealed an inverse correlation between HIPK2 and COX-2 expression [69] (Table 1). Activation of COX-2 and HIF-1 pathways can induce vascular endothelial growth factor (VEGF) expression [80,81], which in turn potentiates HIF-1 transcriptional activity and contributes to tumor progression [82]. Inflammatory mediators such as VEGF have been shown to suppress dendritic cell (DC) maturation, permitting evasion of anti-tumor immune responses, and to induce tumor progression [83,84]. In this regard, HIPK2 knockdown, by using siRNA, has been shown to generate a proinflammatory phenotype by upregulating COX-2/VEGF pathway through HIF-1 [85], then, VEGF inhibition in HIPK2-depleted cell culture, by the use of anti-VEGF monoclonal antibody, efficiently restored DC maturation [69] (Table 1). Therefore, colon cancer progression and resistance to therapies may worsen, not only in the presence of chronic inflammatory diseases such as inflammatory bowel disease (IBD) [86], obesity [87] or hyperglycemia [51], but also by an inflammatory phenotype induced by genetic or epigenetic changes of HIPK2.
Inflammation is a hallmark of cancer and may arise from a variety of factors including infections, environmental carcinogens (e.g., smoking, alcohol, radiation), cellular senescence and obesity [83]. Chronic inflammation is an important driver to the development of many cancers, such as colon cancer and hepatocellular carcinoma (HCC) [88]. Starting from the findings that the tumor suppressor gene HIPK2 was expressed at higher levels from IL-6low HCC samples than from IL-6high HCC samples, based on the cluster analysis of protein kinases in hepatitis B virus infection-associated HCC patient samples, the authors analyzed the potential regulatory function of HIPK2 in chronic inflammation-driven cancers by generating HIPK2 knockout (KO) mice [46]. Because HIPK2 is known to be haploinsufficient [36,89], the authors mainly used HIPK2+/− mice that grow normally as wild-type (WT) mice. MC38 colon carcinoma cells were subcutaneously injected into WT and HIPK2+/− mice, and the authors found that tumors grow more rapidly in HIPK2+/− mice and that, surprisingly, the percentage of macrophages is increased in the tumors of HIPK2+/− mice [46]. In vivo studies demonstrated that HIPK2-deficient mice are more susceptible to lipopolysaccharide (LPS)-induced endotoxemia and cecal ligation and puncture (CLP)-induced sepsis, with increased serum concentration of proinflammatory IL-6, IL-1β and TNF-α cytokines [46]. In vitro studies showed that HIPK2 knockdown significantly enhances the messenger RNA (mRNA) levels of Il6, Il1b and Tnf-α, as well as the IL-6 and TNF-α protein levels [46]. Mechanistically, the authors found that, in macrophages, HIPK2 phosphorylates histone deacetylase 3 (HDAC3) at serine 374 to inhibit its deacetylase activity, thus suppressing NF-κB/p65 activation and the production of proinflammatory cytokines [46]. Induction of NF-κB-mediated transactivation is critical to induce inflammation, therefore, unveiling novel mechanisms that specifically regulate NF-κB activation will provide important therapeutic applications in inflammatory diseases. In this regard, the findings of Zhang and colleagues propose to target NF-κB through the axis HIPK2–HDAC3–NF-κB to ameliorate not only colitis-associated colorectal cancer and sepsis but also inflammation-related cancers [46] (Table 1). These findings are in line with previous ones showing that overexpression of HIPK2 suppresses the expression levels of proinflammatory TNF-α and IL-1β proinflammatory cytokines in spinal cord-injured rats [90] and attenuates sepsis-mediated liver injury [91] and that, in cancer cells, HIPK2 can block the inflammatory phenotype induced by VEGF/PGE2 production [45,69], underlining an important role for HIPK2 in suppressing inflammation-induced tumor progression.
2.3. HIPK2 and Liver Metastasis
Liver metastasis of colon cancer is an important cause of death, but its molecular mechanisms are still unclear [92]. In a pilot study aiming at investigating new markers and therapeutic targets of colon cancer liver metastasis, the authors performed high-depth whole-exome sequencing of primary tumor tissues, matched paracancerous, normal tissues, and liver metastases of four colorectal cancer patients. They found that the genes with the highest mutation frequency were titin (TTN), obscurin (OBSCN), and HIPK2 [93]. The very limited number of patient samples prevented demonstration of any significant involvement of those genes in colon cancer development and metastasis; therefore, additional studies are necessary to correlate the potential high frequency of mutations in the HIPK2 gene with colon cancer development and liver metastasis.
2.4. HIPK2 and Mutant KRAS
Opposite to the above findings, a role for HIPK2 as positive mediator of colon cancer progression has been proposed. Through the production of TMAs of cancer samples from a retrospective series of 270 stage I to IV patients with colon cancer, the HIPK2 protein expression was evaluated by IHC analyses. The authors found that the number of HIPK2-positive cancer cells increases with tumor progression and correlates with the TNM stages. Results from next generation sequencing (NGS) analysis revealed that high HIPK2 expression significantly associates with risk of colon cancer recurrence [70]. By using overexpression or silencing experiments with the CRISPR/Cas9 technology, the authors found that HIPK2 expression increases when KRAS pathway is activated and that in HIPK2-knockout cells the phosphorylation of extracellular signal regulated kinase (ERK) and its upstream activator, MEK, in basal condition and after KRAS pathway activation, is impaired compared with the HIPK2-WT cells [70]. In agreement, HIPK2 silencing reduced the growth of tumors derived from KRAS mutated cells [70] (Table 1). These data counteract the previous data showing that HIPK2 depletion increases colon cancer growth compared to control tumors, depending on activation of several tumor-promoting pathways, and inversely correlates with the stage of colon cancer [37,45,62]. They also counteract the finding that HIPK2 overexpression reduces ERK phosphorylation and pancreatic cancer proliferation, making HIPK2 a suitable target for inhibiting KRAS/ERK activation in pancreatic cancer [66]. Therefore, further studies are necessary to demonstrate the pro-tumorigenic role of HIPK2 in the context of mutant KRAS and the mechanisms that led to increased HIPK2 protein levels, as the authors did not define it [70]. In this regard, it is known that oncogenic KRAS induces nuclear factor erythroid 2 (NF-E2) p45-related factor 2 (NRF2) expression, conferring chemoresistance [94], and that NRF2 impairs the HIPK2 apoptotic activity [42] (see below). Therefore, it could be interesting to assess whether, in the cooperation between mutant KRAS and HIPK2, NRF2 might have a role and whether the HIPK2 pro-survival role may depend on posttranslational modifications [95,96] leading to HIPK2 protein stability unbalancing its pro-survival/apoptotic functions. Thus, previous findings support the existence of a “HIPK2 protein modification code” [97,98] that might fine-tune HIPK2 as a signaling hub to determine cell fate, depending on cellular context.
2.5. HIPK2 and Response to Colon Cancer Chemotherapy
Using part of the above TMA of primary human colon tumor samples [70], the authors analyzed 84 patients with stage II colon cancer, treated or not with adjuvant chemotherapy, to evaluate the contribution of HIPK2 in chemotherapy response [71]. Stage II colon cancer accounts for ~25% all colon cancer cases and it is an early-stage tumor without lymph node spread or distant sites, and with low risk of recurrence. However, the management of stage II colon cancer after surgical resection remains a clinical dilemma due to the lack of reliable criteria for identifying patients who may benefit from adjuvant chemotherapy [99]. The authors showed that TMAs from normal colon tissues exhibit a low level (≤5%) of HIPK2-positive cells. By contrast, TMAs from tumor samples exhibit a broad range of HIPK2 positivity with up to 50% positivity that associated with an improved response to therapy which was independent from p53 status [71] (Table 1). In vitro studies using the CRISPR/Cas9 technology confirmed that, compared with Ctrl-Cas9 cells, HIPK2-knockout significantly decreases cell sensitivity to 5-Fluorouracil (5-FU) and oxaliplatin (OXA) [71], drugs frequently used in the treatment of colon cancer, including stage II cases [100]. Therefore, the authors concluded that HIPK2 protein expression is associated with chemo-response in early-stage colon cancer, which represents an initial step toward defying a novel predictive marker for patients with stage II colon cancer who may benefit from adjuvant therapy [71]. In a recent study trying to identify long non-coding (lnc) RNA and mRNA associated with oxaliplatin resistance, the authors found that lncRNA prion protein testis specific (PRNT) is upregulated in colon cancer [72]. By performing in vitro and in vivo studies, the authors found that PRNT upregulation induces colon cancer cell migration, resistance to OXA and tumor growth [72]. Analysis of datasets showed PRNT upregulation in OXA-resistant colon cancer along with downregulation of HIPK2. Mechanistically, PRNT regulates HIPK2 mRNA expression in colon cancer by sponging ZNF184 transcription factor [72] (Table 1). These findings are in line with previous studies showing the positive role of HIPK2 in drug-induced apoptosis [29].
2.6. HIPK2 and NRF2
It has been recently evidenced that nuclear factor erythroid 2 (NF-E2) p45-related factor 2 (NRF2), encoded by the NFE2L2 gene [101,102], is among the molecular pathways altered in colon cancer. NRF2 is the master regulator of oxidative stress that, in normal cells, is downregulated by proteasomal degradation through binding to Kelch-like ECH-associated protein 1 (Keap1). Under oxidative stress, NRF2 is released from its inhibitor Keap1 to activate the transcription of antioxidant and cytoprotective genes [103]. Although NRF2 is a cytoprotective factor and its transient activation is linked to chemoprevention, its sustained activation has been shown to protect cancer cells against chemo- and radiotherapy and promote metabolic activities that support cell proliferation and tumor growth [104]. For this opposite behavior, NRF2 is considered a “double face” molecule [105]. In solid cancer, NRF2 is upregulated by several mechanisms (e.g., Keap1 inhibition, NFE2L2 gene mutation, oncogene-induced transcription of NRF2 via KRAS, BRAF, MYC activation), promoting cell proliferation, resistance to therapy and an unfavorable prognosis [106]. For this reason, NRF2 has recently drawn the attention of many researchers for its role as a biomarker of therapy response and prognostic factor in tumors. A role for NRF2 in colon cancer progression has been unveiled and associated with patients’ poor prognosis and resistance to chemo-and radiotherapy [101,107,108,109]. Mechanistically, overexpression of NRF2 in colon cancer tissues, compared to normal tissues, has been found to correlate with ERK1/2 and AKT signaling pathway activation [109]. Although with the limitation due to small samples size, NRF2 expression was related to pathologic stage, indicating that overexpression of NRF2 may be linked with formation and stage of colon cancer [109]. These findings were in agreement with the study analyzing NRF2 expression in 76 colon cancer patients’ tissues and paired normal tissues showing that NRF2 protein expression is significantly higher in cancer tissues and that it positively associates with larger tumor size, advanced TNM stages and metastasis [108]. In another study, by measuring the NRF2 activity in silico and performing its validation in patient samples, the authors showed that NRF2 pathway is upregulated in colon cancer and that such high NRF2 expression associates with patients’ poor prognosis [110]. Then, a correlation between NRF2 expression and reduced HIPK2 activity in colon cancer cells was found in pre-clinical studies showing that NRF2 activation correlates with the inhibition of phosphorylation of p53Ser46 and with the reduction of cisplatin-induced cell death, highlighting an interplay between NRF2 and HIPK2/p53 axis in response to anticancer chemotherapy that can be hijacked by cancer cells to bypass drug cytotoxicity [43,44]. In this regard, NRF2 has been suggested to engage with the HIPK2 protein in a pro-survival crosstalk to the detriment of the HIPK2 apoptotic activity [42]. In that setting, HIPK2 has been shown to induce some antioxidant target genes that it has in common with NRF2 (i.e., NQO1, HO-1), promoting cancer cell survival [42]. Therefore, NRF2 activation in colon cancer, as for instance in the above studies [101,108,109,110], may impair HIPK2/p53 apoptotic activity, and favor HIPK2-dependent transcriptional program of genes promoting chemoresistance and tumor progression. Further studies are necessary to demonstrate that hypothesis and the underlying molecular mechanisms of the NRF2/HIPK2 interplay in colon cancer, but also in other types of cancers, tilting the death/survival outcome. In this regard, targeting NRF2 in tumors might be a useful strategy to restore cancer chemosensitivity [111] and likely HIPK2 apoptotic activity. As a proof of principle, inhibition of NRF2 has been shown to selectively induce cell death in NRF2-addicted colorectal cancer cells by promoting ferroptosis and pyroptosis [112,113] and to increase chemotherapy response only when HIPK2 is expressed [71].
3. HIPK2 Role in the Colon Cancer Tumor Microenvironment (TME)
In the tumor–host interaction in the TME, a key role for tumor progression is played by angiogenesis, the formation of new blood capillaries taking place from preexisting functional vessels [114]. Angiogenesis contributes to tumor growth, resistance to therapies, inhibition of apoptosis, tumor invasion and metastasis and, for those reasons, is considered a hallmark of cancer progression [115]. In many solid cancers, angiogenesis is constantly activated by the “angiogenic switch” that is triggered by hypoxia, a common condition in solid cancers [116]. Hypoxia leads to the transcription of several angiogenic factors, the most important being VEGF [117], by means of the HIF-1 transcription factor [80]. HIF-1 is a heterodimeric molecule consisting of the constitutively expressed HIF-1β subunit and the hypoxia-stimulated HIF-1α subunit [118]. After dimerization with HIF-1β, HIF-1α binds to a consensus sequence called hypoxia-response element (HRE) and controls, in the nucleus, the expression of several genes involved in many aspects of cancer progression, including angiogenesis, metabolic adaptation, apoptosis resistance, invasion and metastasis [80,119]. In the extracellular milieu, important mediators of intercellular communications are the exosomes, i.e., spherical membrane-closed nanovesicles secreted by all types of cells [120]. Tumor cells produce abundant exosomes that participate in intercellular communication performing intercellular transfer of components such as microRNA (miRNA), locally and systemically interacting with the surrounding cells in the TME [121]. Growing evidence suggests that tumor-derived exosomes (TEXs) are considered new players in tumor growth and invasion, tumor-associated angiogenesis, tissue inflammation and immunologic remodeling [19]. Exosomes and their cargos may serve as cancer prognostic marker, therapeutic targets or even as anticancer drug carriers [122].
In a series of recent reports, miRNAs have been shown to bidirectionally regulate angiogenesis in colon cancer. Many miRNAs can directly act on VEGF or inhibit angiogenesis through other pathways (e.g., HIF-1α, PI3K/AKT, etc.), while some miRNAs, specifically many exosomal (exo)miRNAs, are capable of promoting colon cancer angiogenesis [123]. A study of patients with colon cancer showed high levels of circulating exomiR-1229, which correlates with tumor size, lymphatic metastasis, angiogenesis and poor survival [73]. In colon cancer tissues, HIPK2 mRNA expression was found to be significantly downregulated compared to the adjacent normal tissues and, mechanistically, exomiR-1229 was found to target the HIPK2 3’UTR. The reduction of the HIPK2 protein levels in of human umbilical vein endothelial cells (HUVECs) upregulated the downstream VEGFA, VEGFR1 and p-AKT, thereby stimulating angiogenesis [73] (Table 1). In agreement with earlier studies [37,45,62], HIPK2 reduction in colon cancer tissues, compared to the normal tissues, could be considered a potential novel prognostic marker of colon cancer progression, along with high production of exomiR-1229 [73]. Remarkably, exomiR-1229 was found to cause breast tumorigenesis by activating the Wnt/β-catenin pathway through targeting the key negative regulators of β-catenin, such as glycogen synthase kinase (GSK)-3 β, adenomatous polyposis coli (APC) and ICAT [124]. The β-catenin transcription factor is strongly involved in the early and stepwise events of the colon tumorigenesis by activating genes such as c-myc, cyclin D1 and VEGF [125]. Aberrant activation of Wnt/β-catenin signaling has been similarly linked to the progression of many other cancer types [125,126]. HIPK2 has been shown to phosphorylate and degrade β-catenin protein, therefore repressing the Wnt/β-catenin-mediated transactivation of target genes such as cyclin D1 [38]. The knockdown of endogenous HIPK2 in colon cancer cells augments the stability of β-catenin and the expression of β-catenin target genes, stimulating proliferation, increased wound healing and in vivo tumor growth [38]. These results are in agreement with earlier results showing that HIPK2 downregulates β-catenin levels through proteasomal degradation system, an effect dependent on HIPK2 catalytic activity and independent of p53 and GSK-3β activities [36,37]. Collectively, the above findings make it tempting to speculate that high levels of exomiR-1229 might induce tumor angiogenesis not only by blocking the HIPK2-mediated suppression of VEGF expression [73] but also by blocking the HIPK2-mediated inhibition of the β-catenin/VEGF pathway (Figure 3).

Figure 3.
Schematic representation of the HIPK2 role in the TME (tumor microenvironment). HIPK2 can be inhibited by exomiR-1299, HIF-1 and NRF2 (blocking black lines). HIF-1 and NRF2 can sustain their oncogenic ability (↑↓). HIPK2 inhibition leads to increased (red arrow) differentiation of CAF (cancer-associated fibroblasts); to induction of angiogenesis by targeting endothelial cells with secreted (red arrow) VEGF (vascular endothelial growth factor) by HIF-1, β-catenin or COX pathways; and to block (red arrow) dendritic cell (DC) maturation by means of PGE2 (prostaglandin E2) production.
In addition to β-catenin, another key transcription factor for VEGF expression is HIF-1 [118]. In vitro studies with colon cancer cells showed that HIPK2 binds, along with HDAC1, to the HIF-1α gene promoter repressing the HIF-1-mediated transcription of VEGF [85], in line with data showing that the HIPK2/HDAC1 complex leads to histone deacetylation and control of gene transcription [45,127]. As a proof-of-principle of the HIPK2/HIIF-1/VEGF regulation, conditioned media from HIPK2 knockdown colon cancer cells has been shown to enhance in vitro tube formation of HUVEC [85]. In agreement, tumor xenografts established from colon cancer cells depleted of HIPK2 function, compared to control ones, showed that HIPK2 knockdown increases tumor vascularity and blood density, as evaluated by immunohistochemical analysis and by HIF-1α and VEGF up-regulation at the mRNA levels [85]. These findings are in agreement with a study showing that HIPK2 induces HIF-1α proteasomal degradation and subsequent angiogenesis in liver cancer [128]. HIF-1α is mostly regulated at posttranscriptional levels by low oxygen conditions [129]; however, the regulation by HIPK2 at the transcriptional level [85] suggests that inhibition of HIPK2 can induce a pseudo-hypoxic phenotype with HIF-1 activation and an “angiogenic switch”, leading to tumor progression, invasion, angiogenesis and resistance to therapies, that can be applied to colon cancer and also to other solid cancers (Figure 3). Thus, HIF-1 activation has a special impact on cancer progression because of its effect on angiogenesis [82] and targeting hypoxia in tumors is a promising therapeutic strategy [130]. In this regard, our previous studies showed that zinc supplementation may downregulate HIF-1α and revert the hypoxia-induced changes of gene expression in colon cancer cells restoring HIPK2 and p53 activities [131,132,133]. Interestingly, hypoxia has been shown to downregulate HIPK2 through Siah-2-dependent proteasomal degradation, allowing the induction of a substantial fraction of hypoxia-induced genes [48], therefore, controlling tumor progression and response to therapies. To make the interplay even more complicated, an intriguing link between HIF-1 and NRF2 has been highlighted [134]. In xenograft models of tumors formed by colon cancer cells depleted of NRF2 function, tumors were smaller and developed fewer blood vessels, compared to control tumors, depending on the reduced expression of HIF-1α [135]. NRF2-inhibited cancer cells failed to accumulate HIF-1α protein in hypoxic conditions, likely due to reduced mitochondrial O2 consumption that enables PHD hydroxylation of HIF-1α and consequent protein degradation: this mechanism limits the expression of VEGF and other HIF-1 target genes [135]. These findings underscore an integrated interplay between HIF-1 and NRF2 in the cellular response to hypoxia and in tumor progression, highlighting NRF2 as a candidate molecular target to control angiogenesis by imposing a blockade of HIF-1 signaling (Figure 3). In addition, given the interplay between HIPK2 with HIF-1α [85,136], HIPK2 and NRF2 [42,44], and NRF2 and HIF-1, it can be speculated that HIPK2 apoptotic activity might be restrained by the HIF-1/NRF2 interplay; therefore, targeting either HIF-1- or NRF2-induced pathways or both is a possible strategy to control tumor progression, in part through reestablishing HIPK2 oncosuppressor activities.
In the TME, CAFs sustain cancer growth and support malignancy and tumor resistance to therapies in a crosstalk with cancer cells [137]. It was shown that HIPK2-modulated pathways derived from colon cancer cells depleted of HIPK2 function are involved in fibroblast transdifferentiation CAF-like [47]. Mechanistically, conditioned media of HIPK2-silenced colon cancer cells induced autophagy in fibroblasts that reduced caveolin-1 levels [47], a hallmark of the aggressive CAF phenotype in cancer patients [138]. Therefore, HIPK2 inhibition in cancer cells can also contribute to tumor progression and resistance to therapies through TME remodeling activities (Figure 3).
4. Conclusions
Although marked advances in colon cancer investigations have been made in the last years, colon cancer still remains the second leading cause of cancer-related mortality worldwide. Thus, additional diagnostic parameters and molecular determinants of clinical outcome and response to therapies are still required to help pave the way for the development of novel diagnostic and therapeutic strategies. This review provides information, mostly with pre-clinical evidence, about a potential candidate biomarker (HIPK2), that might aid in improving the diagnosis and help extend the treatment options of colon cancer cases, thus ameliorating the prognosis of colon cancer patients.
HIPK2 plays a key role in cancer biology thanks to the interaction with molecular pathways involved in cancer progression and response to therapies. Regarding colon cancer, several studies have demonstrated in vitro and in tumor xenografts that HIPK2 inhibition: (i) increases tumor progression and reduces tumor response to therapies through inactivation of the p53 apoptotic activity; (ii) induces VEGF production and angiogenesis by means of HIF-1, β-catenin or COX-2 activation; (iii) induces a pro-inflammatory phenotype that favors tumor progression and immune evasion; and (iv) induces CAF differentiation in the tumor–host interaction. Intriguingly, HIPK2 can be the target of miRNAs or hypoxia that inhibit its expression and increase tumor progression or can interact with NRF2 that impairs its apoptotic activity (Figure 3). The analyses of tissue samples or genomic data sets from colon cancer patients mostly agree with the molecular outcome, that is, that high HIPK2 expression correlates with low Dukes stages and with high patient survival, suggesting that the HIPK2 presence is important in restraining tumor progression. In addition, in few studies, high HIPK2 expression in patient samples has been found to correlate with better response of, for instance, stage II colon cancer to OXA and 5-FU, and also to a better prognosis for the patients, in agreement with the HIPK2 oncosuppressor role. In only one study, high HIPK2 expression in colon cancer tissues was found to correlate with tumor progression and TNM stages, although the molecular mechanisms were not completely unveiled. In summary, the collected findings indicate that HIPK2 expression could be a useful biomarker to evaluate colon cancer progression and response to therapies, also along with the molecule pathways that interact with it, as summarized above. The findings also suggest that it is worth further studying the mechanisms that control HIPK2 protein regulation or mRNA expression and dictate the pro-survival/apoptotic balance. In addition, more in vivo experiments and analyses of large patient groups, also according to tumor stage and response to therapies, will help to better decipher the prognostic role of HIPK2 and the translational potential in clinical practice.
Author Contributions
Conceptualization and writing, G.D. and A.V.; review and editing, A.G., V.D., A.V. and G.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors wish to thank people in the lab for helpful discussion.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Kasi, P.M.; Shahjehan, F.; Cochuyt, J.J.; Li, Z.; Colibaseanu, D.T.; Merchea, A. Rising proportion of young individuals with rectal and colon cancer. Clin. Colorectal Cancer 2019, 18, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Gastroenterol. Rev. 2019, 14, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.S.; Karuniawati, H.; Jairoun, A.A.; Urbi, Z.; Ooi, D.J.; John, A.; Lim, Y.C.; Kibria, K.M.K.; Mohiuddin, A.K.M.; Ming, L.C.; et al. Colorectal Cancer: A Review of Carcinogenesis, Global Epidemiology, Current Challenges, Risk Factors, Preventive and Treatment Strategies. Cancers 2022, 14, 1732. [Google Scholar] [CrossRef] [PubMed]
- Kanth, P.; Inadomi, J.M. Screening and prevention of colorectal cancer. BMJ 2021, 374, 1855. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Malki, A.; ElRuz, R.A.; Gupta, I.; Allouch, A.; Vranic, S.; Al Moustafa, A.E. Molecular mechanisms of colon cancer progression and metastasis: Recent insights and advancements. Int. J. Mol. Sci. 2021, 22, 130. [Google Scholar] [CrossRef] [PubMed]
- Ries, S.; Biederer, C.; Woods, D.; Shifman, O.; Shirasawa, S.; Sasazuki, T.; McMahon, M.; Oren, M.; McCormick, F. Opposing effects of Ras on p53: Transcriptional activation of mdm2 and induction of p19ARF. Cell 2000, 103, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, A.; Cecchinelli, B.; D’Angelo, M.; D’Orazi, G.; Crescenzi, M.; Sacchi, A.; Soddu, S. p53 can inhibit cell proliferation through caspase-mediated cleavage of ERK2/MAPK. Cell Death Differ. 2004, 11, 596–607. [Google Scholar] [CrossRef]
- Nakayama, M.; Oshima, M. Mutant p53 in colon cancer. J. Mol. Cell Biol. 2019, 11, 267–276. [Google Scholar] [CrossRef]
- Liebl, M.C.; Hofmann, T.G. The role of p53 signaling in colorectal cancer. Cancers 2021, 13, 2125. [Google Scholar] [CrossRef] [PubMed]
- Michel, M.; Kaps, L.; Maderer, A.; Galle, P.R.; Moehler, M. The Role of p53 Dysfunction in Colorectal Cancer and Its Implication for Therapy. Cancers 2021, 13, 2296. [Google Scholar] [CrossRef]
- Danac, J.M.C.; Uy, A.G.G.; Garcia, R.L. Exosomal microRNAs in colrectal cancer: Overcoming barriers of the metastatic cascade. Int. J. Mol. Med. 2021, 47, 112. [Google Scholar] [CrossRef]
- Li, J.; Chen, D.; Shen, M. Tumor microenvironment shapes colorectal cancer progression, metastasis, and treatment response. Front. Med. 2022, 9, 869010. [Google Scholar] [CrossRef]
- Van der Jeught, K.; Xu, H.C.; Li, Y.J.; Lu, X.B.; Ji, G. Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol. 2018, 24, 3834–3848. [Google Scholar] [CrossRef]
- Cornista, A.M.; Giolito, M.V.; Baker, K.; Hazime, H.; Dufait, I.; Datta, J.; Khumukcham, S.S.; De Ridder, M.; Roper, J.; Abreu, M.T.; et al. Colorectal cancer immunotherapy: State of art and future directions. Gastro Hep. Adv. 2023, 2, 1103–1119. [Google Scholar] [CrossRef]
- Di Nicolantonio, F.; Vitiello, P.P.; Marsoni, S.; Siena, S.; Tabernero, J.; Trusolino, L.; Bernards, R.; Bardelli, A. Precision oncology in metastatic colorectal cancer—from biology to medicine. Nat. Rev. Clin. Oncol. 2021, 18, 506–525. [Google Scholar] [CrossRef]
- Calzado, M.A.; Renner, F.; Roscic, A.; Schmitz, M.L. HIPK2: A versatile switchboard regulating the transcription machinery and cell death. Cell Cycle 2007, 6, 139–143. [Google Scholar] [CrossRef]
- Rinaldo, C.; Prodosmo, A.; Soddu, S. HIPK2: A multitalented partner for transcription factors in DNA damage response and development. Biochem. Cell Biol. 2007, 85, 411–418. [Google Scholar] [CrossRef]
- Feng, Y.; Zhou, L.; Sun, X.; Li, Q. Homeodomain-interacting protein kinase 2 (HIPK2): A promising target for anti-cancer therapies. Oncotarget 2017, 8, 20452–20461. [Google Scholar] [CrossRef]
- Garufi, A.; D’Orazi, V.; Pistritto, G.; Cirone, M.; D’Orazi, G. HIPK2 in angiogenesis: A promising biomarker in cancer progression and in angiogenic diseases. Cancers 2023, 15, 1566. [Google Scholar] [CrossRef]
- Garufi, A.; Pistritto, G.; D’Orazi, G. HIPK2 as a novel regulator of fibrosis. Cancers 2023, 15, 1059. [Google Scholar] [CrossRef]
- D’Orazi, G.; Rinaldo, C.; Soddu, S. Updates on HIPK2: A resourceful oncosuppressor for clearing cancer. J. Exp. Clin. Cancer Res. 2012, 31, 63. [Google Scholar] [CrossRef]
- Conte, A.; Pierantoni, G.M. Update on the regulation of HIPK1, HIPK2 and HIPK3 protein kinases by microRNAs. Microrna 2018, 7, 178–186. [Google Scholar] [CrossRef]
- Garufi, A.; D’Orazi, V.; Pistritto, G.; Cirone, M.; D’Orazi, G. The Sweet Side of HIPK2. Cancers 2023, 15, 2678. [Google Scholar] [CrossRef]
- D’Orazi, G.; Cecchinelli, B.; Bruno, T.; Manni, I.; Higashimoto, Y.; Saito, S.; Gostissa, M.; Coen, S.; Marchetti, A.; Del Sal, G.; et al. Homeodomain interacting protein kinase-2 phosphorylates p53 at Ser46 and mediates apoptosis. Nat. Cell Biol. 2002, 4, 11–19. [Google Scholar] [CrossRef]
- Hofmann, T.G.; Moller, A.; Sirma, H.; Zentgraf, H.; Droge, W.; Will, H.; Schmitz, M.L. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat. Cell Biol. 2002, 4, 1–10. [Google Scholar] [CrossRef]
- Hofmann, T.G.; Stollberg, N.; Schmitz, M.L.; Will, H. HIPK2 regulates transforming growth factor–beta-induced c-Jun NH(2)-terminal kinase activation and apoptosis in human hepatoma cells. Cancer Res. 2003, 63, 8271–8277. [Google Scholar]
- Zhang, J.; Pho, V.; Bonasera, S.J.; Holtzman, J.; Tang, A.T.; Hellmuth, J.; Tang, S.; Janak, P.H.; Tecott, L.H.; Huang, E.J. Essential function of HIPK2 in TGFbeta-dependent survival of midbrain dopamine neurons. Nat. Neurosci. 2007, 10, 77–86. [Google Scholar] [CrossRef]
- Wei, G.; Ku, S.; Ma, G.K.; Saito, S.; Tang, A.A.; Zhang, J.; Mao, J.H.; Appella, E.; Balmain, A.; Huang, E.J. HIPK2 represses β-catenin-mediated transcription, epidermal stem cell expansion, and skin tumorigenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 13040–13045. [Google Scholar] [CrossRef]
- Puca, R.; Nardinocchi, L.; D’Orazi, G. Regulation of vascular endothelial growth factor expression by homeodomain-interacting protein kinase-2. J. Exp. Clin. Cancer Res. 2008, 27, 22. [Google Scholar] [CrossRef]
- Kim, E.A.; Kim, J.E.; Sung, K.S.; Choi, D.W.; Lee, B.J.; Choi, C.Y. Homeodomain-interacting protein kinase 2 (HIPK2) targets beta-catenin for phosphorylation and proteasomal degradation. Biochem. Biophys. Res. Commun. 2010, 394, 966–971. [Google Scholar] [CrossRef]
- Hikasa, H.; Sokol, S.Y. Phosphorylation of TCF proteins by homeodomain-interacting protein kinase 2. J. Bio. Chem. 2011, 286, 12093–12100. [Google Scholar] [CrossRef]
- Shang, S.; Doan, C.N.; Arnold, T.D.; Lee, S.; Tang, A.A.; Reichardt, L.F.; Huang, E.J. Transcriptional corepressors HIPK1 and HIPK2 control angiogenesis via TGF-β-TAK-dependent mechanism. PLoS Biol. 2013, 11, e1001527. [Google Scholar] [CrossRef]
- Tomimaru, Y.; Koga, H.; Shin, T.H.; Xu, C.Q.; Wands, J.R.; Kim, M. The SxxSS motif of T-cell factor-4 isoforms modulates Wnt/b-catenin signal activation in hepatocellular carcinoma cells. Cancer Lett. 2013, 336, 359–369. [Google Scholar] [CrossRef]
- Torrente, L.; Sanchez, C.; Moreno, R.; Chowdhry, S.; Cabello, P.; Isono, K.; Koseki, H.; Honda, T.; Hayes, J.D.; Dinkova-Kostova, A.T.; et al. Crosstalk between NRF2 and HIPK2 shapes cytoprotective responses. Oncogene 2017, 36, 6204–6212. [Google Scholar] [CrossRef]
- D’Orazi, G.; Garufi, A.; Cirone, M. Nuclear factor erythroid 2 (NF-E 2) p45-related factor 2 interfere s with homeodomain-interacting protein kinase 2/p53 activity to impair solid tumor s chemosensitivity. IUBMB Life 2020, 72, 1634–1639. [Google Scholar] [CrossRef]
- Garufi, A.; Pistritto, G.; D’Orazi, V.; Cirone, M.; D’Orazi, G. The impact of NRF2 inhibition on drug-induced colon cancer cell death and p53 activity: A pilot study. Biomolecules 2022, 12, 461. [Google Scholar] [CrossRef]
- D’Orazi, G.; Sciulli, M.G.; Di Stefano, V.; Riccioni, S.; Frattini, M.; Falcioni, R.; Bertario, L.; Sacchi, A.; Patrignani, P. Homeodomain-interacting protein kinase-2 restrains cytosolic phospholipase A2-dependent prostaglandin E2 generation in human colorectal cancer cells. Clin. Cancer Res. 2006, 12, 735–741. [Google Scholar] [CrossRef]
- Zhang, F.; Qi, L.; Feng, Q.; Zhang, B.; Li, X.; Liu, C.; Li, W.; Liu, Q.; Yang, D.; Yin, Y.; et al. HIPK2 phosphorylates HDAC3 for NF-kB acetylation to ameliorate colitis-associated colorectal carcinoma and sepsis. Proc. Natl. Acad. Sci. USA 2021, 118, e2021798118. [Google Scholar] [CrossRef]
- Garufi, A.; Traversi, G.; Cirone, M.; D’Orazi, C. HIPK2 role in the tumor-host interaction: Impact on fibroblasts transdifferentiation CAF-like. IUBMB Life 2019, 71, 2055–2061. [Google Scholar] [CrossRef]
- Calzado, M.A.; de la Vega, L.; Moller, A.; Bowtell, D.D.; Schmitz, M.L. An inducible autoregulatory loop between HIPK2 and Siah2 at the apex of the hypoxic response. Nat. Cell Biol. 2009, 11, 85–91. [Google Scholar] [CrossRef]
- Baldari, S.; Garufi, A.; Granato, M.; Cuomo, L.; Pistritto, G.; Cirone, M.; D’Orazi, G. Hyperglycemia triggers HIPK2 protein degradation. Oncotarget 2017, 8, 1190–1203. [Google Scholar] [CrossRef]
- Muz, B.; de la Puerte, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapies. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef]
- Cheng, H.C.; Chang, T.K.; Su, W.C.; Tsai, H.L.; Wang, J.Y. Narrative review of the influence of diabetes mellitus and hyperglycemia on colorectal cancer risk and oncological outcomes. Transl. Oncol. 2021, 14, 101089. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef]
- Hassin, O.; Oren, M. Dragging p53 in cancer: One protein, many targets. Nat. Rev. Drug Discov. 2023, 22, 127–144. [Google Scholar] [CrossRef]
- Puca, R.; Nardinocchi, L.; Sacchi, A.; Rechavi, G.; Givol, D.; D’Orazi, G. HIPK2 modulates p53 activity towards pro-apoptotic transcription. Mol. Cancer 2009, 8, 85. [Google Scholar] [CrossRef]
- Puca, R.; Nardinocchi, L.; Gal, H.; Rechavi, G.; Amariglio, N.; Domany, E.; Notterman, D.A.; Scarsella, M.; Leonetti, C.; Sacchi, A.; et al. Reversible dysfunction of wild-type p53 following homeodomain-interacting protein kinase-2 knockdown. Cancer Res. 2008, 68, 3707–3714. [Google Scholar] [CrossRef]
- Puca, R.; Nardinocchi, L.; Bossi, G.; Sacchi, A.; Rechavi, G.; Givol, D.; D’Orazi, G. Restoring wtp53 activity in HIPK2 depleted MCF7 cells by modulating metallothionein and zinc. Exp. Cell Res. 2009, 315, 67–75. [Google Scholar] [CrossRef]
- Margalit, O.; Simon, A.J.; Yakubov, E.; Puca, R.; Yosepovich, A.; Avivi, C.; Jacob-Hirsch, J.; Gelernter, I.; Harmelin, A.; Barshack, I.; et al. Zinc supplementation augments in vivo antitumor effect of chemotherapy by restoring p53 function. Int. J. Cancer 2012, 131, E562–E568. [Google Scholar] [CrossRef]
- Li, X.L.; Arai, Y.; Harada, H.; Shima, Y.; Yoshida, H.; Rokudai, S.; Aikawa, Y.; Kimura, A.; Kitabayashi, I. Mutations of the HIPK2 gene in acute myeloid leukemia and myelodysplastic syndrome impair AML1- and p53-mediated transcription. Oncogene 2007, 26, 7231–7239. [Google Scholar] [CrossRef]
- Reed, S.M.; Quelle, D.E. P53 acetylation: Regulation and consequences. Cancers 2015, 7, 30–69. [Google Scholar] [CrossRef]
- Cirone, M.; Garufi, A.; Di Renzo, L.; Granato, M.; Faggioni, A.; D’Orazi, G. Zinc supplementation is required for the cytotoxic and immunogenic effects of chemotherapy in chemoresistant p53-functionally deficient cells. Oncoimmunol 2013, 2, e26198. [Google Scholar] [CrossRef]
- Soubeyran, I.; Mahouche, I.; Grigoletto, A.; Leste-Lasserre, T.; Drutel, G.; Rey, C.; Pedeboscq, S.; Blanchard, F.; Brouste, V.; Sabourin, J.C.; et al. Tissue Microarray Cytometry Reveals Positive Impact of Homeodomain Interacting Protein Kinase 2 in Colon Cancer Survival Irrespective of p53 Function. Am. J. Pathol. 2011, 178, 1986–1998. [Google Scholar] [CrossRef]
- Zhou, L.; Feng, Y.; Jin, Y.; Liu, X.; Sui, H.; Chai, N.; Chen, X.; Liu, N.; Ji, Q.; Wang, Y.; et al. Verbascoside promotes apoptosis by regulating HIPK2-p53 signaling in human colorectal cancer. BMC Cancer 2014, 14, 747. [Google Scholar] [CrossRef]
- Zhou, J.; Zheng, R.; Zhang, S.; Zeng, H.; Wang, S.; Chen, R.; Sun, K.; Li, J.; Zhuang, G.; Wei, W. Colorectal cancer burden and trends: Comparison between China and major burden countries in the world. Chin. J. Cancer Res. 2021, 33, 1–10. [Google Scholar] [CrossRef]
- Bon, G.; Di Carlo, S.E.; Folgiero, V.; Avetrani, P.; Lazzari, C.; D’Orazi, G.; Brizzi, M.F.; Sacchi, A.; Soddu, S.; Blandino, G.; et al. Negative regulation of beta (β) integrin transcription by homeodomain-interacting protein kinase e and p53 impairs tumor progression. Cancer Res. 2009, 69, 5978–5986. [Google Scholar] [CrossRef]
- Lavra, L.; Rinaldo, C.; Ulivieri, A.; Luciani, E.; Fidanza, P.; Giacomelli, L.; Bellotti, C.; Ricci, A.; Trovato, M.; Soddu, S.; et al. The loss of the p53 activator HIPK2 is responsible for Galectin-3 overexpression in well differentiated thyroid carcinomas. PLoS ONE 2011, 6, e20665. [Google Scholar] [CrossRef]
- Qin, Y.; Hu, Q.; Ji, S.; Xu, J.; Dai, W.; Liu, W.; Xu, W.; Sun, Q.; Zhang, Z.; Ni, Q.; et al. Homeodomain-interacting protein kinase 2 suppresses proliferation and aerobic glycolysis via ERK/cMyc axis in pancreatic cancer. Cell Prolif. 2019, 52, e12603. [Google Scholar] [CrossRef]
- Attia, Y.M.; El-Kersh, D.M.; Wagdy, H.A.; Elmazar, M.M. Verbascoside: Identification, quantification and potential sensitization of colorectal cancer cells to 5-FU targeting PI3K/AKT pathway. Sci. Rep. 2018, 8, 16939. [Google Scholar] [CrossRef]
- Khalaf, H.A.A.; Jasim, R.A.; Ibrahim, I.T. Verbascoside—A review of its antitumor activities. Pharmacol. Pharm. 2021, 12, 109–126. [Google Scholar] [CrossRef]
- Garufi, A.; Pistritto, G.; Ceci, C.; Di Renzo, L.; Santararelli, R.; Faggioni, A.; Cirone, M.; D’Orazi, G. Targeting COX-2/PGE(2) pathway in HIPK2 knockdown cancer cells: Impact on dendritic cell maturation. PLoS ONE 2012, 7, e48342. [Google Scholar] [CrossRef][Green Version]
- Di Segni, M.; Virdia, I.; Verdina, A.; Amoreo, C.A.; Baldari, S.; Toietta, G.; Diodoro, M.G.; Mottolese, M.; Sperduti, I.; Moretti, F.; et al. HIPK2 cooperates with KRAS signaling and associates with colorectal cancer progression. Mol. Cancer Res. 2022, 20, 686–689. [Google Scholar] [CrossRef]
- Verdina, A.; Di Segni, M.; Amoreo, C.A.; Sperduti, I.; Buglioni, S.; Mottolese, M.; Di Rocco, G.; Soddu, S. HIPK2 is a potential predictive marker of a favorable response for adjuvant chemotherapy in stage II colorectal cancer. Oncol. Rep. 2021, 45, 899–910. [Google Scholar] [CrossRef]
- Li, S.N.; Yang, S.; Wang, H.Q.; Hui, T.L.; Cheng, M.; Zhang, X.; Li, B.K.; Wang, G.Y. Upregulated lncRNA PRNT promotes progression and oxaliplatin resistance of colorectal cancer cells by regulating HIPK2 transcription. World J. Gastrointest. Oncol. 2024, 16, 1564–1577. [Google Scholar] [CrossRef]
- Hu, H.Y.; Yu, C.H.; Zhang, H.H.; Zhang, S.Z.; Yu, W.Y.; Yang, Y.; Chen, Q. Exosomal miR-1229 derived from colorectal cancer cells promotes angiogenesis by targeting HIPK2. Int. J. Biol. Macromol. 2019, 132, 470–477. [Google Scholar] [CrossRef]
- Finetti, F.; Travelli, C.; Ercoli, J.; Colombo, G.; Buoso, E.; Trabalzini, L. Prostaglandin E2 and cancer: Insight into tumor progression and imunity. Biology 2020, 9, 434. [Google Scholar] [CrossRef]
- Santiso, A.; Heinemann, A.; Kargl, J. Prostaglandin E2 in the tumor microenvironment, a convoluted affair mediated by EP receptors 2 and 4. Pharmacol. Rev. 2024, 76, 388–413. [Google Scholar] [CrossRef]
- Chan, T.A. Prostaglandin and the colon cancer connection. Trends Mol. Med. 2006, 12, 240–244. [Google Scholar] [CrossRef]
- Wang, D.; DuBois, R.N. Cyclooxygenase 2-derived prostaglandin E2 regulates the angiogenic switch. Proc. Natl. Acad. Sci. USA 2004, 101, 415–416. [Google Scholar] [CrossRef]
- Nunez, F.; Bravo, S.; Cruzat, F.; Montecino, M.; De Ferrari, G.V. Wnt/b-catenin signaling enhances cyclooxygenase-2 (COX2) transcriptional activity in gastric cancer cells. PLoS ONE 2011, 6, e18562. [Google Scholar] [CrossRef]
- Kaidi, A.; Qualtrough, D.; Williams, A.C.; Paraskeva, C. Direct transcriptional up-regulation of cyclooxygenase-2 by hypoxia-inducible factor (HIF)-1 promotes colorectal tumor cell survival and enhances HIF-1 transcriptional activity during hypoxia. Cancer Res. 2006, 66, 6683–6691. [Google Scholar] [CrossRef]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef]
- Tsujii, M.; Kawano, S.; Tsuji, S.; Sawaoka, H.; Hori, M.; DuBois, R.N. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell 1998, 93, 705–716. [Google Scholar] [CrossRef]
- Semenza, G.L. Involvement of hypoxia-inducible factor 1 in human cancer. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Hussain, S.P.; Harris, C.C. Inflammation and cancer: An ancient link with novel potentials. Int. J. Cancer 2007, 121, 2373–2380. [Google Scholar] [CrossRef]
- Sheng, K.C.; Wright, M.D.; Apostolopoulos, V. Inflammatory mediators hold the key to dendritic cell suppression and tumor progression. Curr. Med. Chem. 2011, 18, 5507–5518. [Google Scholar] [CrossRef]
- Nardinocchi, L.; Puca, R.; Guidolin, D.; Belloni, A.S.; Bossi, G.; Michiels, C.; Sacchi, A.; Onisto, M.; D’Orazi, G. Transcriptional regulation of hypoxia-inducible factor 1alpha by HIPK2 suggests a novel mechanism to restrain tumor growth. Biochim. Biophys. Acta-Mol. Cell Res. 2009, 1793, 368–377. [Google Scholar] [CrossRef]
- Muthusami, S.; Ramachandran, I.K.; Babu, K.N.; Krishnamoorthy, S.; Guruswamy, A.; Queimado, L.; Chaudhuri, G.; Ramachandran, I. Role of inflammation in the development of colorectal cancer. Endocr. Metab. Immune Disord. Drug Targets 2021, 21, 77–90. [Google Scholar] [CrossRef]
- Rychter, A.M.; Lykowska-Szuber, L.; Zawada, A.; Szymczak-Tomczak, A.; Ratajczak, A.E.; Skoracka, K.; Kolan, M.; Dobrowolska, A.; Krela-Kazmierczak, I. Why does obesity as an inflamamtory condition predispose to colorectal cancer? J. Clin. Med. 2023, 12, 2451. [Google Scholar] [CrossRef]
- Long, A.G.; Lundsmith, E.T.; Hamilton, K.E. Inflammation and colon cancer. Curr. Colorectal Cancer Rep. 2017, 13, 341–351. [Google Scholar] [CrossRef]
- Mao, J.H.; Wu, D.; Kim, I.J.; Kang, H.C.; Wei, G.; Climent, J.; Kumar, A.; Pelorosso, F.G.; SelRosario, R.; Huang, E.J.; et al. Hipk2 cooperates with p53 to suppress g-ray radiation-induced mouse thymic lymphoma. Oncogene 2012, 31, 1176–1189. [Google Scholar] [CrossRef]
- Li, R.; Shang, J.; Zhou, W.; Jiang, L.; Xie, D.; Tu, G. Overexpression of HIPK2 attenuates spinal cord injury in rats by modulating apoptosis, oxidative stress, and inflammation. Biomed. Pharmacother. 2018, 103, 127–134. [Google Scholar] [CrossRef]
- Jiang, Z.; Bo, L.; Meng, Y.; Wang, C.; Chen, T.; Wang, C.; Yu, X.; Deng, X. Overexpression of homeodomain-interacting protein kinase 2 (HIPK2) attenuates sepsis-mediated liver injury by restoring autophagy. Cell Death Dis. 2018, 9, 847. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, Z.; Wang, Y.; Wen, X.; Amador, E.H.; Yuan, L.; Ran, X.; Xiong, L.; Ran, Y.; Wen, Y. Colorectal liver metastasis: Molecular mechanisms and interventional therapy. Sig. Transduc. Target. Ther. 2022, 7, 70. [Google Scholar] [CrossRef]
- Yi, H.; Liao, Z.W.; Chen, J.J.; Shi, X.Y.; Chen, G.L.; Wu, G.T.; Zhou, D.Y.; Zhou, G.Q.; Huang, J.Y.; Lian, L.; et al. Genome variation in colorectal cancer patient with liver metastasis measured by whole-exome sequencing. J. Gastrointest. Oncol. 2021, 12, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Wang, S.; Moghaddam, S.J.; Ooi, A.; Chapman, E.; Wong, P.K.; Zhang, D.D. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 2014, 74, 7430–7441. [Google Scholar] [CrossRef] [PubMed]
- Saul, V.V.; Schmitz, M.L. Posttranslational modifications regulate HIPK2, a driver of proliferative diseases. J. Mol. Med. 2013, 91, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Siepi, F.; Gatti, V.; Camerini, S.; Crescenzi, M.; Soddu, S. HIPK2 catalytic activity and subcellular localization are regulated by activation-loop Y354 autophosphorylation. Biochim. Biophys. Acta 2013, 1833, 1443–1453. [Google Scholar] [CrossRef] [PubMed]
- De la Vega, L.; Grishina, I.; Moreno, R.; Krüger, M.; Braun, T.; Schmitz, M.L. A redox-regulated SUMO/acetylation switch of HIPK2 controls the survival threshold to oxidative stress. Mol. Cell 2012, 46, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W.; Choi, C.Y. HIPK2 modification code for cell death and survival. Mol. Cell Oncol. 2014, 1, e955999. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Chu, E. Adjuvant chemotherapy for stage II colon cancer: The debate goes on. J. Oncol. Pract. 2017, 13, 245–246. [Google Scholar] [CrossRef]
- Kumar, A.; Gautam, V.; Sandhu, A.; Rawat, K.; Sharma, A.; Saha, L. Current and emerging therapeutic approaches for colorectal cancer: A comprehensive review. World J. Gastrontest. Surg. 2023, 15, 495–519. [Google Scholar] [CrossRef]
- Sadeghi, M.R.; Jeddi, F.; Soozangar, N.; Somi, M.H.; Samadi, N. The role of Nrf2-Keap1 axis in colorectal cancer, progression, and chemoresistance. Tumour Biol. 2017, 39, 6. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Donquiles, C.; Alonso-Molero, J.; Fernandez-Villa, T.; Vilorio-Marques, L.; Molina, A.J.; Martin, V. The NRF2 transcription factor plays a dual role in colorectal caner: A systematic review. PLoS ONE 2017, 12, e0177549. [Google Scholar] [CrossRef] [PubMed]
- McMahonm, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Chen, J.; Liu, X.M.; Zhe, H. Nrf2-mediated metabolic reprogramming in cancer. Oxid. Med. Cell Longev. 2018, 2018, 9304091. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef] [PubMed]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the hallmarks of cancer. Cancer Cell 2018, 34, 20–43. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Kim, M.Y.; Godoy, L.C.; Thiantanawat, A.; Trudel, L.J.; Wogan, G.N. Nitric oxide activation of Keap1/Nrf2 signaling in human colon carcinoma cells. Proc. Natl. Acad. Sci. USA 2009, 106, 14547–14551. [Google Scholar] [CrossRef]
- Hu, T.; Yao, Y.; Yu, S.; Guo, H.; Han, L.; Wang, W.; Tian, T.; Hao, Y.; Liu, Z.; Nan, K.; et al. Clinicopathologic significance of CXCR4 and Nrf2 in colorectal cancer. J. Biomed. Res. 2013, 27, 283–290. [Google Scholar]
- Lee, Y.J.; Kim, W.I.; Bae, J.H.; Cho, M.K.; Lee, S.H.; Nam, H.S.; Choi, I.H.; Cho, S.W. Overexpression of Nrf2 promotes colon cancer progression via ERK and AKT signaling pathways. Ann. Surg. Treat. Res. 2020, 98, 159–167. [Google Scholar] [CrossRef]
- Torrente, L.; Maan, G.; Rezig, A.O.; Quinn, J.; Jackson, A.; Grilli, A.; Casares, L.; Zhang, Y.; Kulesskiy, E.; Saarela, J.; et al. High NRF2 levels correlate with poor prognosis in colorectal cancer patients and with sensitivity to the kinase inhibitor AT9283 in vitro. Biomolecules 2020, 10, 1365. [Google Scholar] [CrossRef]
- No, J.H.; Kim, Y.B.; Song, Y.S. Targeting Nrf2 signaling to combat chemoresistance. J. Cancer Prev. 2014, 19, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.L.; Wang, C.M.; Yao, C.L.; Chen, A.C.; Nien, C.Y.; Sun, Y.H.; Tseng, T.Y.; Luo, Y.H. Blockage of Nrf2 and autophagy by L-selenocystine induces selective death in Nrf2-addicted colorectal cancer cells through p62-Keap-1-Nrf2 axis. Cell Death Dis. 2022, 13, 1060. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Yang, W.; Yang, W.; Yang, L.; Wang, T.; Li, C.L.; Zhang, P.; Ruidong, Y.Y.; Tao, K. Nrf2 inhibition increases sensitivity to chemotherapy of colorectal cancer by promoting ferroptosis and pyroptosis. Sci. Rep. 2023, 13, 14359. [Google Scholar] [CrossRef] [PubMed]
- La Mendola, D.; Trincavelli, M.L.; Martini, C. Angiogenesis in disease. Int. J. Mol. Sci. 2022, 23, 10962. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Han, F.; Du, Y.; Shi, H.; Zhou, W. Hypoxic microenvironment in cancer: Molecular mechanisms and therapeutic interventions. Sig. Transduct. Target. Ther. 2023, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in signaling and disease: Beyond discovery and development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.H.; Semenza, G.L.; Bauer, H.; Marti, H. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am. J. Physiol. 1996, 271, C1172–C1180. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Dor, Y.; Herbert, J.M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.; et al. Role of HIF-1α in hypoxia-mediated apoptosis, cell proliferation and tumor angiogenesis. Nature 1998, 394, 485–490. [Google Scholar] [CrossRef]
- Dai, J.; Su, Y.; Zhong, S.; Cong, L.; Liu, B.; Yang, J.; Tao, Y.; He, Z.; Chen, C.; Jiang, Y. Exosomes: Key players in cancer and potential therapeutic strategy. Sig. Transduct. Target. Ther. 2020, 5, 145. [Google Scholar] [CrossRef]
- Maia, J.; Caja, S.; Strano Moraes, M.C.; Couto, N.; Costa-Silva, B. Exosome-based cell-cell communication in the tumor microenvironment. Front. Cell Dev. Biol. 2018, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.L.; Chen, K.C.; Hsieh, J.T.; Shen, T.L. Exosomes in cancer development and clinical applications. Cancer Sci. 2018, 109, 2364–2374. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Weng, X.; Liu, C.; Li, X.; Chen, C. Hypoxia enhances activity and malignant behaviors of colorectal cancer cells through the STAT3/MicroRNA-19a/PTEN/PI3K/AKT axis. Anal. Cell Pathol. 2021, 2021, 4132488. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Zheng, H.; Liu, X.; Zhang, W.; Zhu, J.; Wu, G.; Cao, L.; Song, J.; Wu, S.; Song, L.; et al. MicroRNA-1229 overexpression promotes cell proliferation and tumorigenicity and activates Wnt/β-catenin signaling in breast cancer. Oncotarget 2016, 17, 24076–24087. [Google Scholar] [CrossRef] [PubMed]
- Lecarpentier, Y.; Schussler, O.; Henert, J.L.; Vallée, A. Multiple targets of the canonical WNT/β-catenin signaling in cancers. Front. Oncol. 2019, 9, 1248. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Yu, C.; Li, F.; Zuo, Y.; Wang, Y.; Yao, L.; Wu, C.; Wang, C.; Ye, L. Wnt/β-catenin signaling in cancers and targeted therapies. Signal Transduct. Target. Ther. 2021, 6, 307. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.Y.; Kim, Y.H.; Kwon, H.J.; Kim, Y. The homeodomain protein NK-3 recruits Groucho and a histone deacetylase complex to repress transcription. J. Biol. Chem. 1999, 274, 33194–33197. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Duan, X.; Li, X.; Li, J.; Ba, Q.; Wang, H. HIPK2 suppresses tumor growth and progression of hepatocellular carcinoma through promoting the degradation of HIF-1α. Oncogene 2020, 39, 2863–2876. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix–loop–helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef]
- Bosco, M.C.; D’Orazi, G.; Del Bufalo, D. Targeting hypoxia in tumor: A new promising therapeutic strategy. J. Exp. Clin. Cancer Res. 2020, 39, 8. [Google Scholar] [CrossRef]
- Nardinocchi, L.; Puca, R.; Sacchi, A.; Rechavi, G.; Givol, D.; D’Orazi, G. Targeting hypoxia in cancer cells by restoring homeodomain-interacting protein kinase 2 and p53 activity and suppressing HIF-1alpha. PLoS ONE 2009, 4, e6819. [Google Scholar] [CrossRef] [PubMed]
- Nardinocchi, L.; Pantisano, V.; Puca, R.; Porru, M.; Aiello, A.; Grasselli, A.; Leonetti, C.; Safran, M.; Rechavi, G.; Givol, D.; et al. Zinc downregulates HIF-1α and inhibits its activity in tumor cells in vitro and in vivo. PLoS ONE 2010, 5, e15048. [Google Scholar] [CrossRef] [PubMed]
- Sheffer, S.; Simon, A.J.; Jacob-Hirsch, J.; Rechavi, G.; Domany, E.; Givol, D.; D’Orazi, G. Genome-wide analysis discloses reversal of the hypoxia-induced changes of gene expression in colon cancer cells by zinc supplementation. Oncotarget 2011, 2, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Toth, R.K.; Warfel, N.A. Strange bedfellows: Nuclear factor, Erythroid 2-like 2 (Nrf2) and hypoxia-inducible factor 1 (HIF-1) in tumor hypoxia. Antioxidants 2017, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Hur, E.G.; Kang, S.J.; Kim, J.A.; Thapa, D.; Mie Lee, Y.; Ku, S.K.; Kwak, M.K. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Cancer Res. 2011, 71, 2260–2275. [Google Scholar] [CrossRef] [PubMed]
- Calzado, M.A.; de La Vega, L.; Munoz, E.; Schmitz, M.L. From top to bottom: The two faces of HIPK2 for regulation of the hypoxic responses. Cell Cycle 2009, 8, 1659–1664. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, M.; Nguyen, T.; Gundre, E.; Ogunlusi, O.; El-Sobky, M.; Giri, B.; Sarkar, T.R. Cancer-associated fibroblasts: The chief architect in the tumor microenvironment. Front. Cell Dev. Biol. 2023, 11, 1089068. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Whitaker-Menezes, D.; Daumer, K.M.; Milliman, J.N.; Chiavarina, B.; Migneco, G.; Witkiewicz, A.K.; Martinez-Cantarin, M.P.; Flomenberg, N.; et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation. Cell Cycle 2010, 12, 2423–2433. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).