Abstract
Acute leukemia is a group of aggressive hematological malignancies, with acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML) being the most common types. The biology of acute leukemia involves complex genetic and epigenetic alterations that lead to uncontrolled cell proliferation and resistance to apoptosis. Mitochondrial dysfunction is a feature of acute leukemia that results in altered energy production, unregulated cell death pathways, and increased cancer cell survival. Apoptosis, particularly via the mitochondrial pathway, is crucial for cellular homeostasis and cancer prevention. In acute leukemia, disruption of apoptosis is pivotal in disease development and progression, with elevated levels of anti-apoptotic proteins conferring a survival advantage to leukemia cells and promoting resistance to conventional therapies. Targeting mitochondrial apoptosis using BH3 mimetics and anti-apoptotic protein inhibitors is a viable therapeutic strategy. Alterations in the mitochondrial membrane potential, metabolism, and dynamics also contribute to the pathogenesis of acute leukemia. Continued research is vital for developing novel therapies and enhancing survival outcomes in patients with acute leukemia while minimizing the long-term adverse effects of treatment. In this narrative review, we provide a birds-eye view of the available scientific literature on the importance of mitochondria in acute leukemia, and discuss the role of BH3 mimetics in targeting the mitochondrial internal apoptotic machinery.
1. Introduction
Acute leukemia, a fast-growing cancer of immature blood cells, is of two main types: ALL and AML. Although acute leukemia can affect individuals of all ages, it exhibits specific prevalence patterns, with ALL being more common in children and AML being more common in adults [1]. Biological manifestations range from relatively well-differentiated forms to those with pronounced dysregulation and a lack of differentiation [2,3]. In ALL, malignant transformation occurs at various stages of lymphoid progenitor development, commonly affecting either the B- or the T-cell lineage. Conversely, AML originates from myeloid progenitor cells, leading to the accumulation of immature myeloblasts in the bone marrow and peripheral blood and rarely in soft tissues or skin. This unrestrained proliferation disrupts normal hematopoiesis, resulting in anemia, thrombocytopenia, and neutropenia.
The biology of acute leukemia involves a complex interplay between genetic and epigenetic alterations [4,5]. Mutations in genes that regulate the cell cycle, apoptosis, and differentiation, such as TP53, FLT3, and NPM1 in AML and NOTCH1 and TEL-AML1 in ALL, are frequently observed. These genetic abnormalities can lead to uncontrolled cell proliferation and resistance to apoptosis. Additionally, chromosomal translocations, such as the t(9;22) Philadelphia chromosome in ALL and t(15;17) in acute promyelocytic leukemia (a subtype of AML), are pivotal in leukemogenesis.
The prognosis of acute leukemia has significantly improved owing to sequential refinement and developments in chemotherapy, targeted therapies, and hematopoietic stem cell transplantation. However, outcomes vary considerably and are influenced by factors such as patient age, genetic mutations, and response to therapy. While pediatric ALL represents a success in cancer therapy with excellent outcomes, adult AML remains problematic with lower long-term survival rates. Continued research is vital for developing novel therapies and enhancing survival outcomes in patients with acute leukemia. Simultaneously, efforts must be made to minimize the long-term adverse effects of treatment, especially in pediatric patients.
2. Mitochondria and Acute Leukemia
Mitochondrial dysfunction is a feature of numerous malignancies, including acute leukemia. This malfunction results in altered energy production within cells, unregulated cell death, and increased survival of cancer cells [6,7]. Acute leukemia presents with several mitochondrial irregularities that facilitate cancer progression and resistance to treatment (Figure 1). Multiple mechanisms of mitochondrial dysregulation have been reported to cause the proliferation of leukemia clones, including higher mtDNA content, lower mitophagy, evasion of apoptosis, and metabolic shifts from the Krebs cycle (TCA) to fatty acid oxidation (FAO).
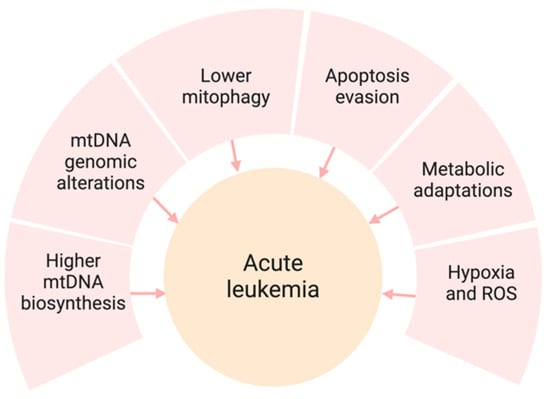
Figure 1.
Mitochondrial dysfunction and its role in acute leukemia. Several mitochondrial irregularities shown above contribute to leukemia progression and resistance to treatment.
Concepts of Mitochondrial Apoptosis
Apoptosis, also known as programmed cell death, is an essential biological process by which cells undergo systematic and controlled elimination. Unlike necrosis, which results from acute injury, apoptosis is energy-dependent and intricately regulated, and thereby maintains the cellular balance. This process involves cell shrinkage, nuclear fragmentation, chromatin condensation, and the formation of apoptotic bodies, which are subsequently engulfed by neighboring cells.
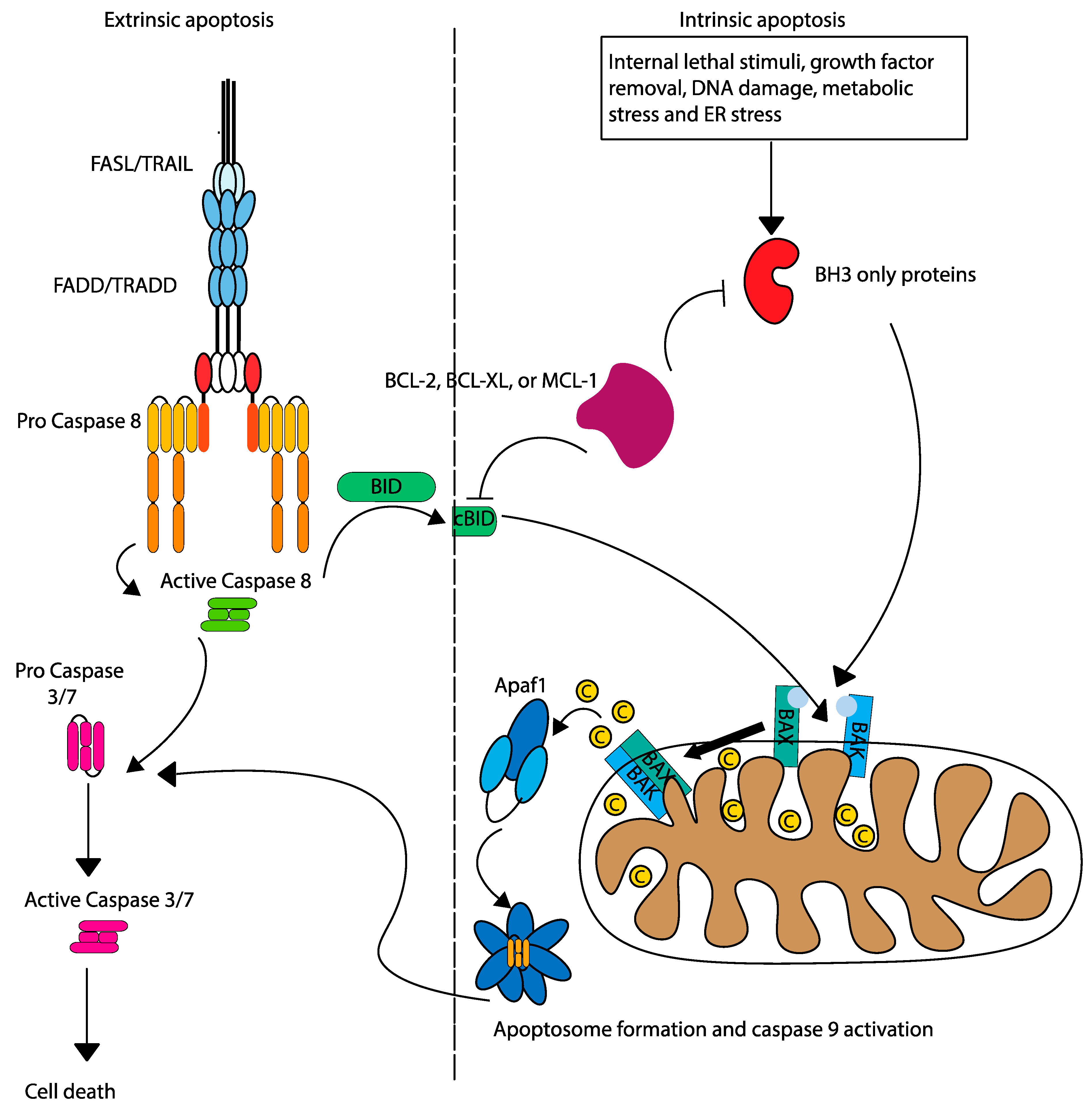
Two primary pathways govern apoptosis: extrinsic (death receptor), and intrinsic (mitochondrial). The extrinsic pathway is initiated by the binding of death ligands to surface receptors (FADD/TRADD), forming the death-inducing signalling complex (DISC) and activating caspase-8 (Figure 2). Caspase 8 cleaves caspase 3/7 to induce apoptosis directly, or can also cleave pro-apoptotic BID, leading to mitochondrial outer membrane permeabilization (MOMP) by oligomerization of BAK and BAX and linking the extrinsic pathway to the intrinsic apoptosis pathway [8]. The intrinsic pathway is mediated by a delicate balance between anti-apoptotic and pro-apoptotic BCL-2 family proteins. BCL-2 anti-apoptotic proteins (such as BCL-2, BCL-XL, and MCL-1) sequester pro-apoptotic activators (BIM, BID, and PUMA) and sensitizers (BAD, NOXA, and HRK) to inhibit oligomerization and activation of pro-apoptotic effectors (BAK and BAX). Triggered by internal stress (such as DNA damage or removal of growth factors), intrinsic apoptosis shifts the apoptosis pendulum towards pro-apoptotic sensitizers, which bind to anti-apoptotic BCL-2 proteins, thereby releasing activators. The pro-apoptotic activators bind to effectors, leading to their oligomerization, which leads to MOMP and release of cytochrome c into the cytoplasm, facilitating apoptosome formation and caspase-9 activation [9].
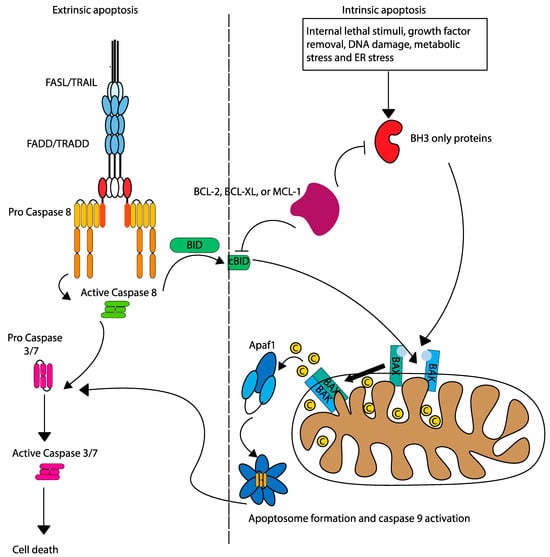
Figure 2.
Intrinsic and extrinsic apoptosis pathways. The intrinsic and extrinsic pathways of apoptosis are initiated by different mechanisms, but are linked by the activation of caspase 8, which cleaves pro-apoptotic BID.
3. Relevance of Mitochondrial Apoptosis in Acute Leukemia
In acute leukemia, the disruption of apoptosis is pivotal in disease development and progression. Mitochondrial dysfunction and the altered expression of Bcl-2 family proteins are central to this process. Acute leukemias often exhibit higher levels of anti-apoptotic proteins, such as Bcl-2, Bcl-XL, MCL-1 and others, which stabilize the mitochondrial membranes and prevent MOMP. This confers a survival advantage to leukemia cells and promotes resistance to conventional therapies [10]. Apoptotic priming, which is measured by the concentration of exogenous pro-apoptotic peptide required to release cytochrome c, is an important factor in determining tumor response to drugs. Myeloblasts exhibit greater susceptibility to apoptotic priming than normal hematopoietic stem cells when exposed to chemotherapeutic agents, including daunorubicin, etoposide, and mitoxantrone [11]. The correlation between the drug response and apoptotic priming was significantly stronger than that between cell proliferation and BAX expression. Subcellular heterogeneity in cytochrome c release in cancer cell lines dictates the heterogeneity in response to the chemotherapeutic drug etoposide and the reduction in apoptotic priming in myeloblasts after first-line chemotherapy, potentially leading to drug resistance [12].
Targeting mitochondrial apoptosis offers a potential therapeutic strategy for acute leukemia. BH3 mimetics and inhibitors of anti-apoptotic proteins, such as Venetoclax, aim to restore apoptotic balance and promote cell death in leukemic cells. These therapies provide hope for overcoming resistance mechanisms and improving patient outcomes [13].
In summary, apoptosis, particularly through the mitochondrial pathway, is crucial for cellular homeostasis and cancer prevention. In addition to mitochondrial apoptotic priming, dysregulation of other mitochondrial functions also plays an important role in acute leukemia.
3.1. Mitochondrial Membrane Potential
The mitochondrial membrane potential (Δψm) is a critical component of ATP generation through oxidative phosphorylation and the induction of apoptosis. In leukemia cells, alterations in Δψm can contribute to resistance to apoptosis. Specifically, elevated Δψm, as noted in acute promyelocytic leukemia, can result in an increased threshold for apoptotic signalling, making cells less likely to undergo programmed cell death. This alteration can be attributed to modifications in mitochondrial ion channels and transporters, which influence Δψm and, ultimately, cell survival [14].
3.2. Mitochondrial Metabolism and Bioenergetics
Leukemia cells undergo metabolic reprogramming to fulfil their energy and biosynthetic requirements. Reprogramming often involves a shift from oxidative phosphorylation to glycolysis even in the presence of oxygen, which is known as the Warburg effect. Consequently, leukemia cells reduce their reliance on mitochondrial ATP production and diminish the impact of mitochondrial dysfunction on energy production. However, leukemia cells retain functional mitochondria to produce critical metabolites and regulate reactive oxygen species (ROS) production [15]. The anti-apoptotic proteins not only regulate the apoptosis pathway but also have been shown to possess non-canonical functions mediating cell survival [16].
3.3. Mitochondrial Biogenesis and Dynamics
Mitochondrial biogenesis and dynamics, including fission, fusion, and mitophagy, are crucial for maintaining proper mitochondrial function and structure. In acute leukemia, there is evidence of disrupted mitochondrial dynamics, which can lead to an imbalance in cellular energy levels and impaired management of reactive oxygen species (ROS). Proteins involved in these processes, such as DRP1 (Dynamin-related protein 1) for fission and mitofusins for fusion, often show altered expression or activity in leukemia cells and can be potential targets for therapy [17]. Furthermore, impaired mitophagy can result in the accumulation of damaged mitochondria, exacerbating oxidative stress and promoting the development of leukemia [18].
3.4. Molecular Mechanisms and Genetic Mutations
3.4.1. Common Genetic Mutations Affecting Mitochondrial Function
Genetic alterations that affect mitochondrial function are widespread in patients with acute leukemia. Mutations in genes, including NOTCH1, TP53 and FLT3, not only promote oncogenesis but also affect mitochondrial pathways [19,20,21]. For instance, TP53 mutations, which are prevalent in various malignancies and acute leukemia, impair mitochondrial permeability and apoptosis regulation, resulting in increased cell survival [22]. FLT3 mutations, particularly FLT3-ITD mutations, are known to enhance cellular proliferation and metabolic reprogramming, partly through mitochondrial pathways [19].
3.4.2. Molecular Pathways Influenced by These Mutations
Genetic mutations in acute leukemia affect multiple molecular pathways, including those that regulate the mitochondrial function. The PI3K/AKT/mTOR pathway, which is often activated in leukemia, promotes cell growth and survival by modulating mitochondrial metabolism and biogenesis [23]. Additionally, mutations in epigenetic regulators, such as DNMT3A and TET2, can alter gene expression profiles, which may indirectly affect mitochondrial function and resilience [24,25].
In summary, mitochondrial dysfunction in acute leukemia includes alterations in the apoptotic pathways, metabolic reprogramming, and genetic mutations. Understanding these factors will offer valuable insights into potential therapeutic approaches targeting mitochondrial vulnerabilities in leukemia cells, ultimately improving the outcomes of patients with acute leukemia.
4. Role of Mitochondrial Apoptosis in Leukemia Initiation and Progression
The impact of mitochondrial apoptosis on leukemia initiation and progression is significant as it influences leukemia cell survival and therapeutic responses.
4.1. Early Apoptotic Changes
Mitochondrial apoptosis involves a sequence of molecular events that occurs in the outer mitochondrial membrane. Cellular stress or damage signals trigger disruption of the mitochondrial membrane, which is commonly mediated by pro-apoptotic proteins, such as Bax and Bak. This disruption causes the release of cytochrome c into the cytoplasm, triggering apoptosome formation and caspase activation, ultimately leading to cell death.
In the early stages of leukemia, mutations in certain oncogenes and tumor suppressor genes often support cell survival, despite damage that typically induces apoptosis. For example, overexpression of anti-apoptotic proteins such as Bcl-2 can inhibit mitochondrial membrane permeabilization, enabling leukemia cells to avoid apoptosis [26]. Resistance to early apoptotic signals is crucial for clonal expansion of leukemia cells, paving the way for disease progression.
4.2. Influence on Leukemic Stem Cells (LSCs)
LSCs are a subpopulation of leukemia cells capable of self-renewal and sustaining leukemia clones. These cells are known for their resistance to standard treatments, largely because of their quiescent nature and strong antiapoptotic defense. Mitochondrial pathways significantly contribute to the survival and maintenance of LSCs. LSCs often exhibit altered mitochondrial dynamics, including changes in fission and fusion and shifts in metabolic functions that favor survival [27]. Additionally, LSCs use mitochondrial protective mechanisms, such as the overexpression of Bcl-2 family proteins, to prevent the release of apoptotic factors [28]. This mitochondrial reinforcement aids the survival of LSCs and their resistance to oxidative stress, which is common in chemotherapeutic treatments, making mitochondrial apoptosis a critical therapeutic target for eliminating the root cause of leukemia proliferation.
4.3. Drug Resistance Mechanisms
Drug resistance poses a significant obstacle to effective treatment of leukemia, often leading to relapse and poor outcomes.
4.4. Evasion of Apoptosis in Resistant Leukemia
Leukemia cells often develop resistance to therapy by evading apoptosis, specifically via the intrinsic mitochondrial pathway. They achieve this by upregulating anti-apoptotic proteins, such as Bcl-2, Bcl-XL, and Mcl-1, which inhibit pro-apoptotic proteins and maintain mitochondrial integrity. These proteins prevent mitochondrial outer membrane permeabilization, blocking cytochrome c release and subsequent caspase activation and thereby averting cell death. Mutations in apoptotic regulators such as p53 can also contribute to resistance. Normally, p53 enhances apoptosis in response to DNA damage by promoting the transcription of pro-apoptotic proteins, such as PUMA and NOXA. When mutated, p53 fails to perform this function, leading to an impaired apoptotic response and allowing leukemia cells to survive despite chemotherapy [21].
4.5. Role of Mitochondria in Drug Resistance
The mitochondria are centrally involved in mediating drug resistance in leukemia. In addition to regulating apoptotic proteins, the mitochondria undergo functional and dynamic alterations that aid cell survival under therapeutic stress. For example, a metabolic shift towards oxidative phosphorylation (OXPHOS) over glycolysis can provide a survival advantage to leukemia cells. This metabolic reprogramming supports energy production and reduces the levels of reactive oxygen species (ROS), which are typically cytotoxic and induce apoptosis [29].
Mitochondrial dynamics, particularly fission and fusion processes, are also altered in drug-resistant leukemia cells. Proteins such as Drp1 drive mitochondrial fission, facilitating the removal of damaged mitochondria via mitophagy, which enhances cell survival [30].
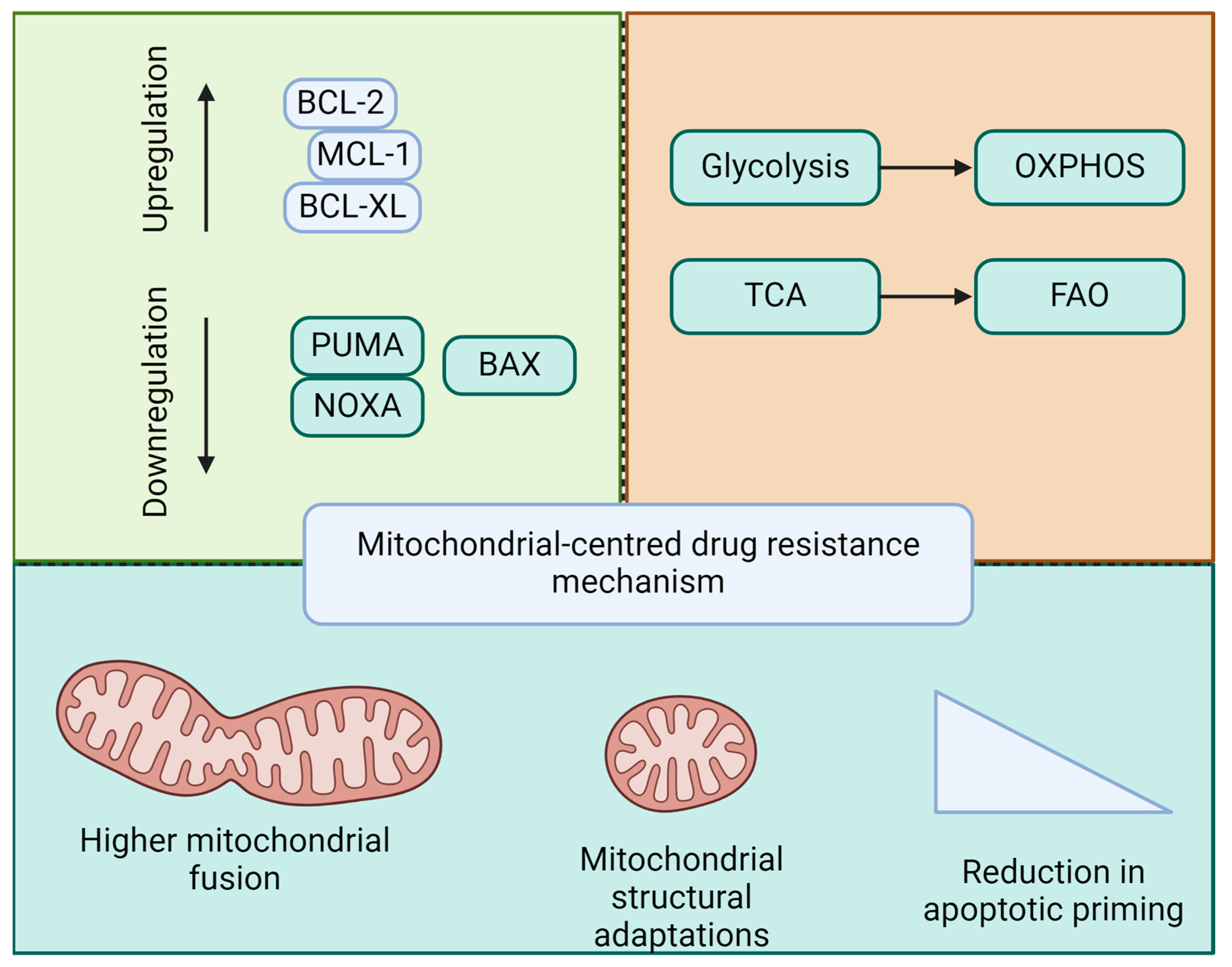
In summary, early apoptotic changes involving mitochondrial membrane permeabilization and anti-apoptotic defenses are key to leukemia transformation and clonal expansion. LSCs exploit mitochondrial mechanisms to ensure survival and resistance to therapies, posing a major challenge in achieving lasting remission. Drug resistance in leukemia is intricately linked to mitochondrial function and apoptosis (Figure 3). Understanding these mitochondrial pathways will provide the foundation for the development of novel therapeutic strategies. Targeting mitochondrial apoptotic pathways, either through the direct induction of apoptosis or the modulation of mitochondrial dynamics and bioenergetics, holds promise for overcoming resistance and improving outcomes in patients with leukemia.
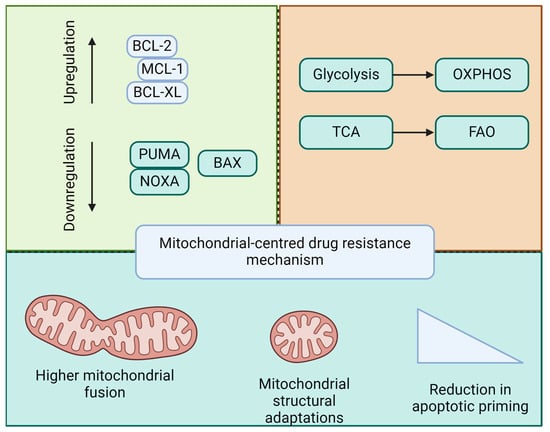
Figure 3.
Mitochondria-based mechanisms of drug resistance in leukemia. Drug resistance in acute leukemia and LSCs is linked to the regulation of pro- and anti-apoptotic balance, mitochondrial metabolic reprogramming, mitochondrial dynamics, and structural adaptations, thereby contributing to a reduction in apoptotic priming.
5. Mitochondria-Targeted Therapies in Acute Leukemia
Mitochondria-targeted therapies are gaining prominence in the treatment of both acute myeloid and acute lymphoblastic acute leukemia [31,32]. Their efficacy depends on targeting of apoptotic mechanisms and mitochondrial function, which are critical for cell survival. BH3 mimetics such as venetoclax, navitoclax, and other apoptotic modulators play crucial roles in these therapies. Combining these treatments with conventional chemotherapy or novel agents is a promising approach [33,34,35,36,37,38,39]. Here, we discuss the mechanisms, combination strategies, and clinical outcomes of these innovative therapies.
5.1. BH3 Mimetics
BH3 mimetics are a class of drugs that mimic the function of BH3-only proteins and are crucial for promoting apoptosis by antagonizing the anti-apoptotic Bcl-2 family proteins [40]. Venetoclax stands out for its role in inhibiting Bcl-2, a protein that typically prevents apoptosis and is overexpressed in many patients with leukemia. By binding to Bcl-2, venetoclax frees pro-apoptotic factors such as Bax and Bak, triggering cell death through the intrinsic mitochondrial pathway [41].
Venetoclax has shown significant efficacy in AML, especially in older patients or in those who are unable to endure intensive chemotherapy. When combined with hypomethylating agents, such as azacitidine or decitabine, venetoclax results in high response rates and extended survival by creating a pro-apoptotic environment that sensitizes leukemia cells [42]. Similarly, venetoclax in combination with chemotherapy has shown promising results in ALL [32].
The dual targeting of Bcl-2 and Bcl-XL appears to be promising for ALL treatment [37]. Mcl-1 inhibitors appear interesting and target Mcl-1, which is frequently upregulated in leukemia, thereby contributing to resistance to chemotherapy. Agents such as S63845 and AMG397 disrupt Mcl-1 interactions and promote mitochondria-mediated apoptosis [43].
5.2. Other Apoptotic Modulators
In addition to BH3 mimetics, other apoptotic modulators have been investigated. SMAC mimetics belong to another class of potential drugs [44]. These compounds neutralize the inhibitors of apoptosis proteins (IAPs), which normally prevent caspase activation and apoptosis. SMAC mimetics boost apoptotic signalling by deactivating IAPs, potentially enhancing the effectiveness of other apoptotic drugs.
5.3. Combination Therapies
5.3.1. Synergistic Effects with Chemotherapy
Combining mitochondria-targeted therapies with traditional chemotherapy has demonstrated synergistic effects that enhance the treatment efficacy [45,46]. Chemotherapy induces cellular stress and DNA damage by priming leukemia cells to induce apoptosis. BH3 mimetics and other apoptotic modulators increase this effect by inhibiting anti-apoptotic proteins, thereby promoting cancer cell death through the mitochondrial pathways.
One notable combination involves venetoclax with cytotoxic agents such as cytarabine or anthracycline compounds [47]. Chemotherapy-induced DNA damage increases the expression of pro-apoptotic proteins, thereby creating an optimal environment for BH3 mimetics to maximize their pro-apoptotic effects. Clinical trials have shown that such combinations yield higher complete remission and longer survival rates in patients with AML than chemotherapy alone. The VIALE-C trial is a prime example demonstrating the enhanced efficacy of venetoclax combined with low-dose cytarabine (LDAC) in older or unfit AML patients [48]. Similarly, the combination of venetoclax with fludarabine, cytarabine, and anthracycline-based chemotherapy in patients with newly diagnosed AML showed better rates of deeper remission and successful bridging to allogeneic bone marrow transplantation without an increase in therapy-related toxicity [46].
5.3.2. Potential in Combination with Novel Agents
The potential of mitochondria-targeted therapies when combined with novel targeted agents is another promising area. Such combinations can affect multiple survival mechanisms in leukemia cells, potentially overcoming resistance pathways and improving therapeutic outcomes. For instance, combining venetoclax with FLT3 inhibitors (such as gilteritinib or midostaurin) can be particularly effective in AML patients with FLT3 mutations [49,50]. FLT3 inhibitors disrupt critical signalling pathways that drive leukemia cell proliferation. Pairing these drugs with venetoclax, which induces apoptosis by targeting Bcl-2, provides an approach for concurrently inhibiting cell growth and promoting cell death.
Combining venetoclax with IDH inhibitors (such as ivosidenib or enasidenib) targets another subset of AML patients with IDH mutations [51]. These mutations hinder cellular differentiation and contribute to leukemogenesis. IDH inhibitors rectify these metabolic anomalies, whereas venetoclax enhances apoptosis, addressing multiple facets of leukemia cell survival and potentially yielding superior treatment results.
6. Clinical Trials and Outcomes
- Overview of Ongoing and Completed Clinical Trials
Numerous clinical trials have explored the efficacy and safety of mitochondria-targeted therapies for acute leukemia, with venetoclax being central to many pivotal studies. These trials typically investigated drugs in various combinations with chemotherapy and novel agents to expand their therapeutic reach.
6.1. VIALE-A Trial
The VIALE-A trial (NCT02993523) is a landmark study that evaluated the combination of venetoclax and azacitidine versus azacitidine alone in treatment-naïve patients with AML who were ineligible for intensive chemotherapy. The results showed significantly improved overall survival and higher complete remission rates in the combination therapy group, leading to FDA approval of venetoclax for treating newly diagnosed AML [52].
6.2. VIALE-C Trial
The VIALE-C trial (NCT03069352) studied the combination of venetoclax and low-dose cytarabine (LDAC) in a similar cohort. The study also reported better outcomes with the combination therapy, although the overall survival benefit was less pronounced than in VIALE-A. However, the response rates were notably higher, underscoring the potential of venetoclax to enhance the efficacy of low-dose chemotherapy regimens. The other trials that are currently recruiting patients are summarized in Table 1.

Table 1.
Ongoing recruitment of clinical trials employing BH3 mimetics in patients with AML and ALL.
6.3. Combinations with Intensive Chemotherapies and Novel Agents
Multiple trials have reported outcomes with the use of venetoclax in combination with more intensive chemotherapy regimens such as the ‘7 + 3’ regimen (cytarabine and daunorubicin), primarily targeting younger and better-fit patients with newly diagnosed AML [53,54,55,56]. These results indicate that integrating venetoclax into these protocols can be safely performed with promising response rates. Ongoing trials are also examining venetoclax with novel agents such as FLT3 and IDH inhibitors [57,58].
In summary, venetoclax has significantly altered the landscape of AML treatment, not only in older patients but also in those unsuitable for intensive chemotherapy. The VIALE-A and VIALE-C trials have highlighted the benefits of venetoclax in combination with hypomethylating agents and low-dose cytarabine. The improved survival and higher remission rates in these trials have established venetoclax as a critical component of AML therapy. Additionally, promising results from trials combining venetoclax with intensive chemotherapy regimens and novel targeted agents underscore the broad applicability and versatility of BH3 mimetics. These successes highlight the potential of mitochondria-targeted therapies to transform the existing treatment modalities and improve patient outcomes.
However, mitochondria-targeted therapies present significant challenges, particularly in terms of drug resistance and safety. Resistance to Bcl-2 inhibition, especially when used alone, can emerge through multiple mechanisms, including the upregulation of other anti-apoptotic proteins such as Mcl-1 and Bcl-XL, or mutations in the Bcl-2 gene [59]. Addressing these issues requires novel strategies to prevent the onset of resistance or overcome established resistance mechanisms.
The long-term safety implications associated with these medications will persist, given that they are novel, and our understanding of their interactions and potential toxicities is evolving with their increased usage. Additionally, other mitochondria-targeted agents, such as Mcl-1 inhibitors, face obstacles in clinical development, and some studies have been terminated owing to safety concerns. The narrow therapeutic window and potential off-target effects of these inhibitors underscore the challenge of precisely targeting Bcl-2 family proteins, while preserving normal cell function [60]. However, other agents are currently undergoing early phase trials [61,62].
7. Future Directions and Conclusions
To advance and enhance current therapeutic approaches, mitochondria-targeted therapies must be leveraged by building upon recent advancements and incorporating these new agents in an evolving medical landscape.
7.1. Development of Next-Generation Inhibitors
Dual Inhibitors: Ongoing research has focused on developing compounds that can simultaneously inhibit multiple anti-apoptotic proteins (e.g., dual Bcl-2/Bcl-XL inhibitors) to prevent resistance and promote sustained apoptosis in leukemia cells [63].
Improved Mcl-1 Inhibitors: Efforts are ongoing to optimize Mcl-1 inhibitors with improved efficacy and safety profiles, potentially through the development of selective inhibitors that minimize off-target effects [64].
7.2. Combination Therapies
Broadening Combinations: Expanding the range of combinations with other targeted therapies, immunotherapies, and novel agents can address multiple survival mechanisms in leukemia cells. For instance, combining BH3 mimetics with novel immune checkpoint inhibitors may enhance anti-tumor immune responses [65].
7.3. Overcoming Resistance Mechanisms
To combat acquired resistance, research strategies should focus on understanding and targeting the underlying mechanisms. One approach is to develop inhibitors that target Bcl-2, along with other pro-survival pathways, such as Mcl-1 [66].
7.4. Other Optimization Strategies
Incorporating diverse patient populations into clinical trials to collect real-world evidence on the efficacy and safety of mitochondria-targeted therapies will refine patient selection and enhance therapeutic strategies. Patient-derived xenografts (PDXs) and organoids, as advanced models, can more accurately represent leukemia heterogeneity and provide a more relevant context for novel therapy testing [67]. Using systems biology to map complex cellular signalling and apoptosis regulation networks in leukemia can inform the creation of multi-targeted strategies, incorporating functional and genetic methods for personalized treatments [68]. Identifying biomarkers is crucial for predicting and monitoring responses to mitochondria-targeted therapies, enabling personalized treatment regimens and early detection of resistance or relapse [69,70].
In summary, it is likely that in the future, we will utilize genomic, proteomic, and transcriptomic data to customize treatment combinations based on the molecular and functional profiles of individual patients with leukemia to ensure tailored and effective therapies under the umbrella of new trials that include diverse populations.
8. Conclusions
Mitochondria-targeted therapies have altered the paradigm for the treatment of acute leukemia. Recent successes with BH3 mimetics such as venetoclax, especially when used in combination, have led to better patient outcomes. However, challenges remain, such as treatment resistance and safety concerns. Overcoming these hurdles through innovative approaches, including combination therapies, personalized medicine, and new clinical trials, is crucial to the progress of these treatments. Utilizing the latest research and incorporating new therapeutic methods, we can look forward to developing more effective and tailored treatments that improve the quality of life and survival rates of patients with acute leukemia.
Author Contributions
P.I.: Conceptualization, Writing—original draft, Writing—review and editing. S.S.J.: Writing—original draft. K.B.: Writing—review and editing. S.B.: Writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Vakiti, A.; Reynolds, S.B.; Mewawalla, P. Acute Myeloid Leukemia. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK507875/ (accessed on 25 June 2024).
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Chen, S.-C.; Andersson, A.K.; Phillips, L.A.; Li, Y.; Sotzen, J.; Kundu, M.; Downing, J.R.; Melnick, A.; Mullighan, C.G. Integrated genetic and epigenetic analysis of childhood acute lymphoblastic leukemia. J. Clin. Investig. 2013, 123, 3099–3111. [Google Scholar] [CrossRef]
- Xu, H.; Wen, Y.; Jin, R.; Chen, H. Epigenetic modifications and targeted therapy in pediatric acute myeloid leukemia. Front. Pediatr. 2022, 10, 975819. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.L.; Chourasia, A.H.; Macleod, K.F. Mitochondrial Dysfunction in Cancer. Front. Oncol. 2013, 3, 292. [Google Scholar] [CrossRef]
- Luo, Y.; Ma, J.; Lu, W. The Significance of Mitochondrial Dysfunction in Cancer. Int. J. Mol. Sci. 2020, 21, 5598. [Google Scholar] [CrossRef] [PubMed]
- Jan, R.; Chaudhry, G.-S. Understanding Apoptosis and Apoptotic Pathways Targeted Cancer Therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Danial, N.N.; Korsmeyer, S.J. Cell Death: Critical Control Points. Cell 2004, 116, 205–219. [Google Scholar] [CrossRef]
- Tzifi, F.; Economopoulou, C.; Gourgiotis, D.; Ardavanis, A.; Papageorgiou, S.; Scorilas, A. The Role of BCL2 Family of Apoptosis Regulator Proteins in Acute and Chronic Leukemias. Adv. Hematol. 2012, 2012, 524308. [Google Scholar] [CrossRef]
- Vo, T.-T.; Ryan, J.; Carrasco, R.; Neuberg, D.; Rossi, D.J.; Stone, R.M.; DeAngelo, D.J.; Frattini, M.G.; Letai, A. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell 2012, 151, 344–355. [Google Scholar] [CrossRef]
- Bhola, P.D.; Mar, B.G.; Lindsley, R.C.; Ryan, J.A.; Hogdal, L.J.; Vo, T.T.; DeAngelo, D.J.; Galinsky, I.; Ebert, B.L.; Letai, A. Functionally identifiable apoptosis-insensitive subpopulations determine chemoresistance in acute myeloid leukemia. J. Clin. Investig. 2016, 126, 3827–3836. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Letai, A. BCL-2 inhibition in AML: An unexpected bonus? Blood 2018, 132, 1007–1012. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Romo-González, M.; Ijurko, C.; Hernández-Hernández, Á. Reactive Oxygen Species and Metabolism in Leukemia: A Dangerous Liaison. Front Immunol. 2022, 13, 889875. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.; Katz, S.G. Non-apoptotic functions of BCL-2 family proteins. Cell Death Differ. 2017, 24, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Larrue, C.; Mouche, S.; Lin, S.; Simonetta, F.; Scheidegger, N.K.; Poulain, L.; Birsen, R.; Sarry, J.-E.; Stegmaier, K.; Tamburini, J. Mitochondrial fusion is a therapeutic vulnerability of acute myeloid leukemia. Leukemia 2023, 37, 765–775. [Google Scholar] [CrossRef]
- Czegle, I.; Gray, A.L.; Wang, M.; Liu, Y.; Wang, J.; Wappler-Guzzetta, E.A. Mitochondria and Their Relationship with Common Genetic Abnormalities in Hematologic Malignancies. Life 2021, 11, 1351. [Google Scholar] [CrossRef]
- Ju, H.-Q.; Zhan, G.; Huang, A.; Sun, Y.; Wen, S.; Yang, J.; Lu, W.-H.; Xu, R.-H.; Li, J.; Li, Y.; et al. ITD mutation in FLT3 tyrosine kinase promotes Warburg effect and renders therapeutic sensitivity to glycolytic inhibition. Leukemia 2017, 31, 2143–2150. [Google Scholar] [CrossRef]
- Baran, N.; Lodi, A.; Dhungana, Y.; Herbrich, S.; Collins, M.; Sweeney, S.; Pandey, R.; Skwarska, A.; Patel, S.; Tremblay, M.; et al. Inhibition of mitochondrial complex I reverses NOTCH1-driven metabolic reprogramming in T-cell acute lymphoblastic leukemia. Nat. Commun. 2022, 13, 2801. [Google Scholar] [CrossRef]
- Nechiporuk, T.; Kurtz, S.E.; Nikolova, O.; Liu, T.; Jones, C.L.; D’Alessandro, A.; Culp-Hill, R.; d’Almeida, A.; Joshi, S.K.; Rosenberg, M.; et al. The TP53 Apoptotic Network is a Primary Mediator of Resistance to BCL2 inhibition in AML Cells. Cancer Discov. 2019, 9, 910–925. [Google Scholar] [CrossRef]
- Shin, D.-Y. TP53 Mutation in Acute Myeloid Leukemia: An Old Foe Revisited. Cancers 2023, 15, 4816. [Google Scholar] [CrossRef]
- Darici, S.; Alkhaldi, H.; Horne, G.; Jørgensen, H.G.; Marmiroli, S.; Huang, X. Targeting PI3K/Akt/mTOR in AML: Rationale and Clinical Evidence. J. Clin. Med. 2020, 9, 2934. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2011, 44, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C. Bcl-2–family proteins and hematologic malignancies: History and future prospects. Blood 2008, 111, 3322–3330. [Google Scholar] [CrossRef]
- Jones, C.L.; Inguva, A.; Jordan, C.T. Targeting Energy Metabolism in Cancer Stem Cells: Progress and Challenges in Leukemia and Solid Tumors. Cell Stem Cell 2021, 28, 378–393. [Google Scholar] [CrossRef]
- Khaldoyanidi, S.K.; Hindoyan, A.; Stein, A.; Subklewe, M. Leukemic stem cells as a target for eliminating acute myeloid leukemia: Gaps in translational research. Crit. Rev. Oncol. Hematol. 2022, 175, 103710. [Google Scholar] [CrossRef] [PubMed]
- de Beauchamp, L.; Himonas, E.; Helgason, G.V. Mitochondrial metabolism as a potential therapeutic target in myeloid leukaemia. Leukemia 2022, 36, 1–12. [Google Scholar] [CrossRef]
- Barbato, A.; Scandura, G.; Puglisi, F.; Cambria, D.; La Spina, E.; Palumbo, G.A.; Lazzarino, G.; Tibullo, D.; Di Raimondo, F.; Giallongo, C.; et al. Mitochondrial Bioenergetics at the Onset of Drug Resistance in Hematological Malignancies: An Overview. Front. Oncol. 2020, 10, 604143. [Google Scholar] [CrossRef]
- Griffioen, M.S.; de Leeuw, D.C.; Janssen, J.J.W.M.; Smit, L. Targeting Acute Myeloid Leukemia with Venetoclax; Biomarkers for Sensitivity and Rationale for Venetoclax-Based Combination Therapies. Cancers 2022, 14, 3456. [Google Scholar] [CrossRef]
- Aumann, S.; Shaulov, A.; Haran, A.; Gross Even-Zohar, N.; Vainstein, V.; Nachmias, B. The Emerging Role of Venetoclax-Based Treatments in Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2022, 23, 10957. [Google Scholar] [CrossRef]
- Jain, N.; Stevenson, K.E.; Winer, E.S.; Garcia, J.S.; Stone, R.M.; Jabbour, E.; Ravandi, F.; Stewart, J.M.; Legg, D.R.; Kantarjian, H.M.; et al. A Multicenter Phase I Study Combining Venetoclax with Mini-Hyper-CVD in Older Adults with Untreated and Relapsed/Refractory Acute Lymphoblastic Leukemia. Blood 2019, 134, 3867. [Google Scholar] [CrossRef]
- Richard-Carpentier, G.; Jabbour, E.; Short, N.J.; Rausch, C.R.; Savoy, J.M.; Bose, P.; Yilmaz, M.; Jain, N.; Borthakur, G.; Ohanian, M.; et al. Clinical Experience With Venetoclax Combined With Chemotherapy for Relapsed or Refractory T-Cell Acute Lymphoblastic Leukemia. Clin. Lymphoma Myeloma Leuk. 2020, 20, 212–218. [Google Scholar] [CrossRef]
- Gibson, B.E.; Webb, D.K.; Howman, A.J.; De Graaf, S.S.; Harrison, C.J.; Wheatley, K. Results of a randomized trial in children with Acute Myeloid Leukaemia: Medical research council AML12 trial. Br. J. Haematol. 2011, 155, 366–376. [Google Scholar] [CrossRef]
- Palmisiano, N.; Lee, J.-W.; Claxton, D.F.; Paietta, E.; Alkhateeb, H.B.; Park, J.H.; Podoltsev, N.; Atallah, E.L.; Schaar, D.G.; Dinner, S.; et al. Maximal Tolerated Dose Determined for Venetoclax in Combination with Liposomal Vincristine in Patients with Relapsed or Refractory Ph-Negative T-Cell or B-Cell Acute Lymphoblastic Leukemia: Results of Phase 1 Portion of ECOG-ACRIN EA9152. Blood 2021, 138, 3407. [Google Scholar] [CrossRef]
- Pullarkat, V.A.; Lacayo, N.J.; Jabbour, E.; Rubnitz, J.E.; Bajel, A.; Laetsch, T.W.; Leonard, J.; Colace, S.I.; Khaw, S.L.; Fleming, S.A.; et al. Venetoclax and Navitoclax in Combination with Chemotherapy in Patients with Relapsed or Refractory Acute Lymphoblastic Leukemia and Lymphoblastic Lymphoma. Cancer Discov. 2021, 11, 1440–1453. [Google Scholar] [CrossRef]
- Short, N.J.; Konopleva, M.; Kadia, T.; Kebriaei, P.; Daver, N.; Huang, X.; Masarova, L.; Cook, R.; Jain, N.; Jabbour, E.; et al. An Effective Chemotherapy-Free Regimen of Ponatinib plus Venetoclax for Relapsed/Refractory Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. Am. J. Hematol. 2021, 96, E229–E232. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, C.; Shi, T.; Zhang, Y.; Qian, J.; Wang, Y.; Hu, Y.; Mao, L.; Ye, X.; Liu, F.; et al. Venetoclax-ponatinib for T315I/compound-mutated Ph+ acute lymphoblastic leukemia. Blood Cancer J. 2022, 12, 20. [Google Scholar] [CrossRef]
- Chonghaile, T.N.; Letai, A. Mimicking the BH3 domain to kill cancer cells. Oncogene 2008, 27, S149–S157. [Google Scholar] [CrossRef]
- Montero, J.; Haq, R. Adapted to Survive: Targeting Cancer Cells with BH3 Mimetics. Cancer Discov. 2022, 12, 1217–1232. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Tiong, I.S.; Quaglieri, A.; MacRaild, S.; Loghavi, S.; Brown, F.C.; Thijssen, R.; Pomilio, G.; Ivey, A.; Salmon, J.M.; et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood 2020, 135, 791–803. [Google Scholar] [CrossRef]
- Bolomsky, A.; Vogler, M.; Köse, M.C.; Heckman, C.A.; Ehx, G.; Ludwig, H.; Caers, J. MCL-1 inhibitors, fast-lane development of a new class of anti-cancer agents. J. Hematol. Oncol. 2020, 13, 173. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.V.; Luger, S.; Mangan, J.; Zebrowski, A.; Loren, A.W.; Minderman, H.; Baird, J.; Porter, D.L.; Hexner, E.O.; Kumar, A.J.; et al. A Phase I Study Using Single Agent Birinapant in Patients with Relapsed Myelodysplastic Syndrome and Acute Myelogenous Leukemia. Blood 2014, 124, 3758. [Google Scholar] [CrossRef]
- Klener, P.; Sovilj, D.; Renesova, N.; Andera, L. BH3 Mimetics in Hematologic Malignancies. Int. J. Mol. Sci. 2021, 22, 10157. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Lachowiez, C.A.; Takahashi, K.; Loghavi, S.; Kadia, T.; Daver, N.; Xiao, L.; Adeoti, M.; Short, N.J.; Sasaki, K.; et al. Venetoclax combined with FLAG-IDA induction and consolidation in newly diagnosed acute myeloid leukemia. Am. J. Hematol. 2022, 97, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Karol, S.E.; Alexander, T.B.; Budhraja, A.; Pounds, S.B.; Canavera, K.; Wang, L.; Wolf, J.; Klco, J.M.; Mead, P.E.; Gupta, S.D.; et al. Venetoclax in combination with cytarabine with or without idarubicin in children with relapsed or refractory acute myeloid leukaemia: A phase 1, dose-escalation study. Lancet Oncol. 2020, 21, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Panayiotidis, P.; Montesinos, P.; Laribi, K.; Ivanov, V.; Kim, I.; Novak, J.; Champion, R.; Fiedler, W.; Pagoni, M.; et al. Long-term follow-up of VIALE-C in patients with untreated AML ineligible for intensive chemotherapy. Blood 2022, 140, 2754–2756. [Google Scholar] [CrossRef]
- Daver, N.; Perl, A.E.; Maly, J.; Levis, M.; Ritchie, E.; Litzow, M.; McCloskey, J.; Smith, C.C.; Schiller, G.; Bradley, T.; et al. Venetoclax Plus Gilteritinib for FLT3-Mutated Relapsed/Refractory Acute Myeloid Leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2022, 40, 4048–4059. [Google Scholar] [CrossRef]
- Li, Z.; Wang, J.; Ge, S.-S.; Qiu, Q.-C.; Du, J.-H.; Shan, S.-S.; Shen, X.-D.; Wan, C.-L.; Wang, B.-R.; Wu, D.-P.; et al. Combination of Venetoclax and Midostaurin Efficiently Suppressed Relapsed t(8;21)Acute Myeloid Leukemia With Mutant KIT After Failure of Venetoclax Plus Azacitidine Treatment. Front. Oncol. 2022, 12, 841276. [Google Scholar] [CrossRef]
- Lachowiez, C.A.; Loghavi, S.; Zeng, Z.; Tanaka, T.; Kim, Y.J.; Uryu, H.; Turkalj, S.; Jakobsen, N.A.; Luskin, M.R.; Duose, D.Y.; et al. A Phase Ib/II Study of Ivosidenib with Venetoclax ± Azacitidine in IDH1-Mutated Myeloid Malignancies. Blood Cancer Discov. 2023, 4, 276–293. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Suo, X.; Zheng, F.; Wang, D.; Zhao, L.; Liu, J.; Li, L.; Zhang, Z.; Zhang, C.; Li, Y.; Yang, S.; et al. Venetoclax combined with daunorubicin and cytarabine (2 + 6) as induction treatment in adults with newly diagnosed acute myeloid leukemia: A phase 2, multicenter, single-arm trial. Exp. Hematol. Oncol. 2023, 12, 45. [Google Scholar] [CrossRef]
- Wang, H.; Mao, L.; Yang, M.; Qian, P.; Lu, H.; Tong, H.; Xie, W.; Zhou, D.; Huang, X.; Wang, Y.; et al. Venetoclax plus 3 + 7 daunorubicin and cytarabine chemotherapy as first-line treatment for adults with acute myeloid leukaemia: A multicentre, single-arm, phase 2 trial. Lancet Haematol. 2022, 9, e415–e424. [Google Scholar] [CrossRef] [PubMed]
- Chua, C.C.; Roberts, A.W.; Reynolds, J.; Fong, C.Y.; Ting, S.B.; Salmon, J.M.; MacRaild, S.; Ivey, A.; Tiong, I.S.; Fleming, S.; et al. Chemotherapy and Venetoclax in Elderly Acute Myeloid Leukemia Trial (CAVEAT): A Phase Ib Dose-Escalation Study of Venetoclax Combined With Modified Intensive Chemotherapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 3506–3517. [Google Scholar] [CrossRef]
- Kadia, T.M.; Reville, P.K.; Borthakur, G.; Yilmaz, M.; Kornblau, S.; Alvarado, Y.; Dinardo, C.D.; Daver, N.; Jain, N.; Pemmaraju, N.; et al. Venetoclax plus intensive chemotherapy with cladribine, idarubicin, and cytarabine in patients with newly diagnosed acute myeloid leukaemia or high-risk myelodysplastic syndrome: A cohort from a single-centre, single-arm, phase 2 trial. Lancet Haematol. 2021, 8, e552–e561. [Google Scholar] [CrossRef] [PubMed]
- Astellas Pharma Global Development, Inc. A Phase 1/2, Multicenter, Open-Label, Randomized Dose Ranging and Expansion Study of the Combination of Gilteritinib, Venetoclax and Azacitidine in Patients With Newly Diagnosed FLT3 Mutated Acute Myeloid Leukemia (AML) Not Eligible for Intensive Induction Chemotherapy. clinicaltrials.gov; 2024 May. Report No.: NCT05520567. Available online: https://clinicaltrials.gov/study/NCT05520567 (accessed on 1 January 2024).
- M.D. Anderson Cancer Center. Phase 1b/2 Study of Oral Decitabine/Cedazuridine (ASTX727) and Venetoclax in Combination With the Targeted Mutant IDH1 Inhibitor Ivosidenib or the Targeted Mutant IDH2 Inhibitor Enasidenib. clinicaltrials.gov; 2024 May. Report No.: NCT04774393. Available online: https://clinicaltrials.gov/study/NCT04774393 (accessed on 1 January 2024).
- Garciaz, S.; Hospital, M.-A.; Collette, Y.; Vey, N. Venetoclax Resistance in Acute Myeloid Leukemia. Cancers 2024, 16, 1091. [Google Scholar] [CrossRef] [PubMed]
- Amgen. A Phase 1 First in Human Study Evaluating the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of AMG 176 in Subjects With Relapsed or Refractory Multiple Myeloma and Subjects With Relapsed or Refractory Acute Myeloid Leukemia. clinicaltrials.gov; 2024 Feb. Report No.: NCT02675452. Available online: https://clinicaltrials.gov/study/NCT02675452 (accessed on 1 January 2024).
- Institut de Recherches Internationales Servier. An International Phase Ib Multicentre Study to Characterize the Safety and Tolerability of Intravenously Administered S64315, a Selective Mcl-1 Inhibitor, in Combination with Orally Administered Venetoclax, a Selective Bcl-2 Inhibitor in Patients With Acute Myeloid Leukaemia (AML). clinicaltrials.gov; 2024 Feb. Report No.: NCT03672695. Available online: https://clinicaltrials.gov/study/NCT03672695 (accessed on 1 January 2024).
- Institut de Recherches Internationales Servier. Phase I, International, Multicentre, Open-Label, Non-Randomised, Non-Comparative Study of Intravenously Administered S64315, a Mcl-1 Inhibitor, in Patients with Acute Myeloid Leukaemia (AML) or Myelodysplastic Syndrome (MDS). clinicaltrials.gov; 2022 May. Report No.: NCT02979366. Available online: https://clinicaltrials.gov/study/NCT02979366 (accessed on 1 January 2024).
- Kargbo, R.B. Redefining Cancer Therapy: Toward BCL-XL/BCL-2 Dual Inhibitors with Diminished Platelet Toxicity. ACS Med. Chem. Lett. 2023, 14, 1156–1158. [Google Scholar] [CrossRef]
- Hird, A.W.; Tron, A.E. Recent advances in the development of Mcl-1 inhibitors for cancer therapy. Pharmacol. Ther. 2019, 198, 59–67. [Google Scholar] [CrossRef]
- Novartis Pharmaceuticals. A Phase II Multi-center, Single Arm, Safety and Efficacy Study of MBG453 in Combination with Azacitidine and Venetoclax for the Treatment of Acute Myeloid Leukemia (AML) in Adult Patients Unfit for Chemotherapy. clinicaltrials.gov; 2024 May. Report No.: NCT04150029. Available online: https://clinicaltrials.gov/study/NCT04150029 (accessed on 1 January 2024).
- Carter, B.Z.; Mak, P.Y.; Tao, W.; Ayoub, E.; Ostermann, L.B.; Huang, X.; Loghavi, S.; Boettcher, S.; Nishida, Y.; Ruvolo, V.; et al. Combined inhibition of BCL-2 and MCL-1 overcomes BAX deficiency-mediated resistance of TP53-mutant acute myeloid leukemia to individual BH3 mimetics. Blood Cancer J. 2023, 13, 57. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, W.; Cai, C.; Zhang, H.; Shen, H.; Han, Y. Patient-derived xenograft models in cancer therapy: Technologies and applications. Signal Transduct. Target. Ther. 2023, 8, 160. [Google Scholar] [CrossRef]
- Bhatia, K.; Sandhu, V.; Wong, M.H.; Iyer, P.; Bhatt, S. Therapeutic biomarkers in acute myeloid leukemia: Functional and genomic approaches. Front. Oncol. 2024, 14, 1275251. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Tierney, C.; Dunphy, K.; Miettinen, J.J.; Heckman, C.A.; Bazou, D.; O’Gorman, P. Identification of Protein Biomarker Signatures for Acute Myeloid Leukemia (AML) Using Both Nontargeted and Targeted Approaches. Proteomes 2021, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Zúñiga, C.D.; Garza-Veloz, I.; Martínez-Rendón, J.; Ureño-Segura, M.; Delgado-Enciso, I.; Martinez-Fierro, M.L. Circulating Biomarkers Associated with the Diagnosis and Prognosis of B-Cell Progenitor Acute Lymphoblastic Leukemia. Cancers 2023, 15, 4186. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).