
Trans-[Pt(amine)Cl2(PPh3)] Complexes Target Mitochondria and Endoplasmic Reticulum in Gastric Cancer Cells

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
2.1. Phospine-Based Agents P1 and P2 Decrease Cell Proliferation
2.2. P1 and P2 Generate ROS and Produce DNA Damage
2.3. P1 and P2 Target Mitochondria and Induce Apoptosis
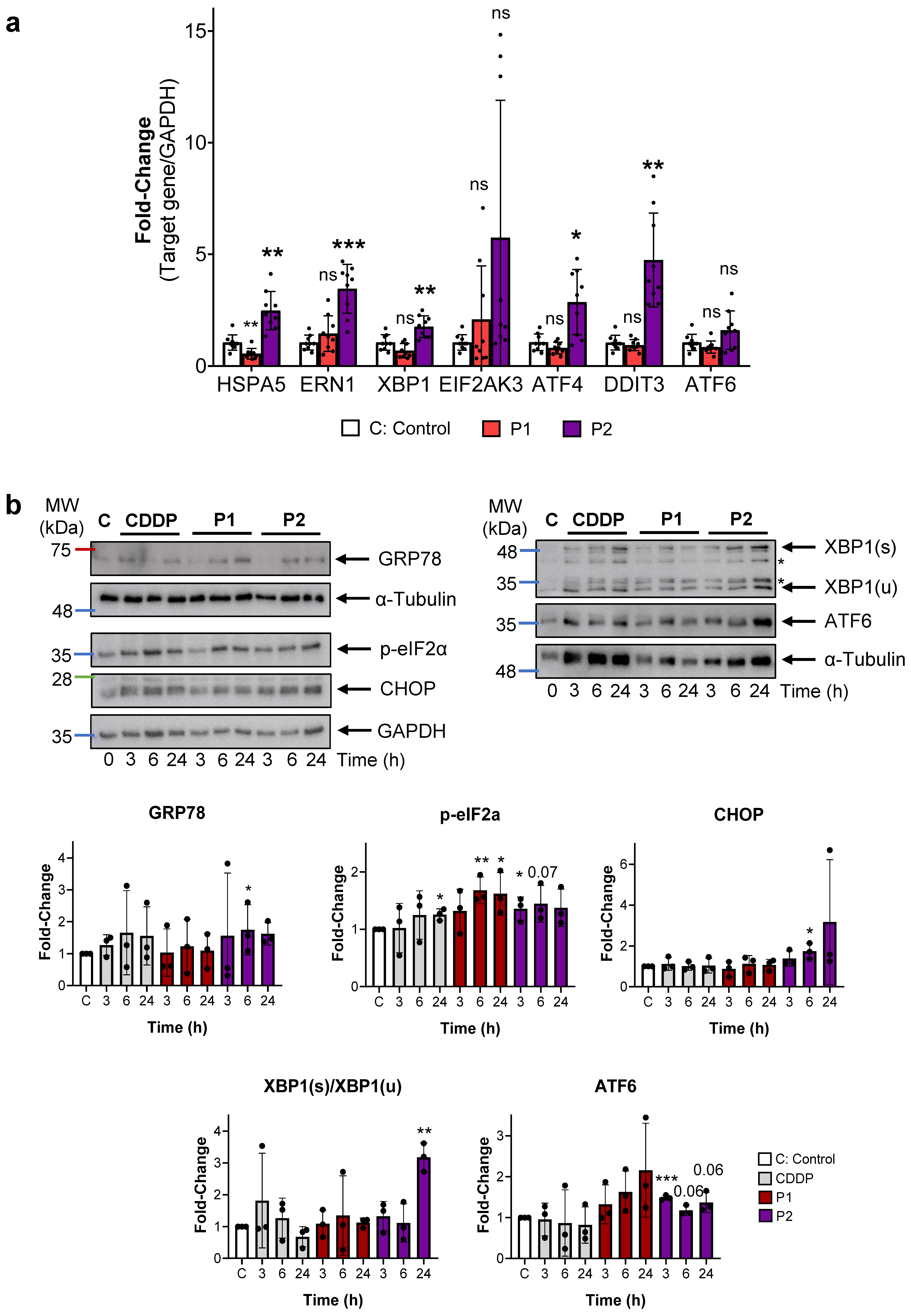
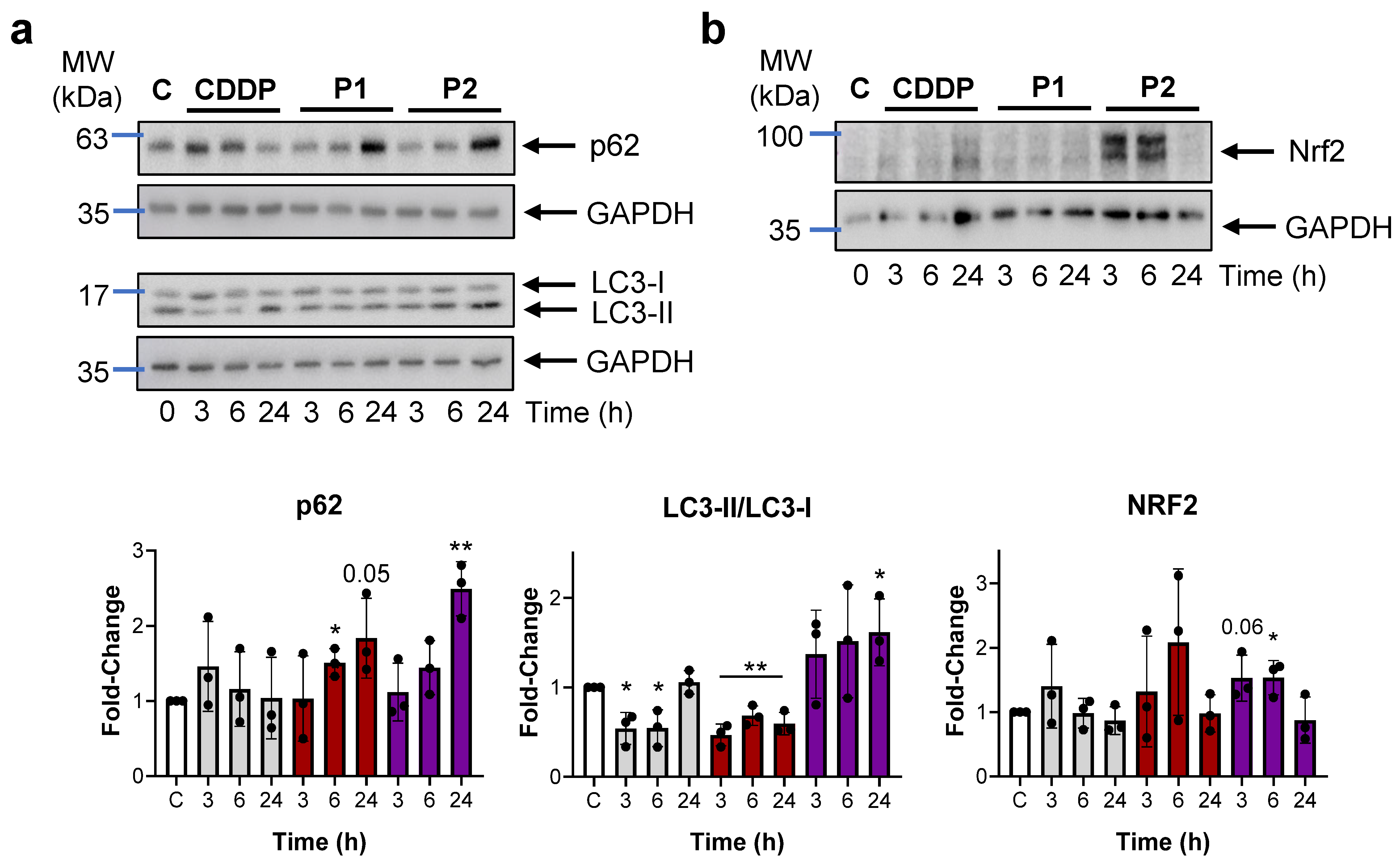
2.4. P1 and P2 Produce ER Stress and Block Autophagy
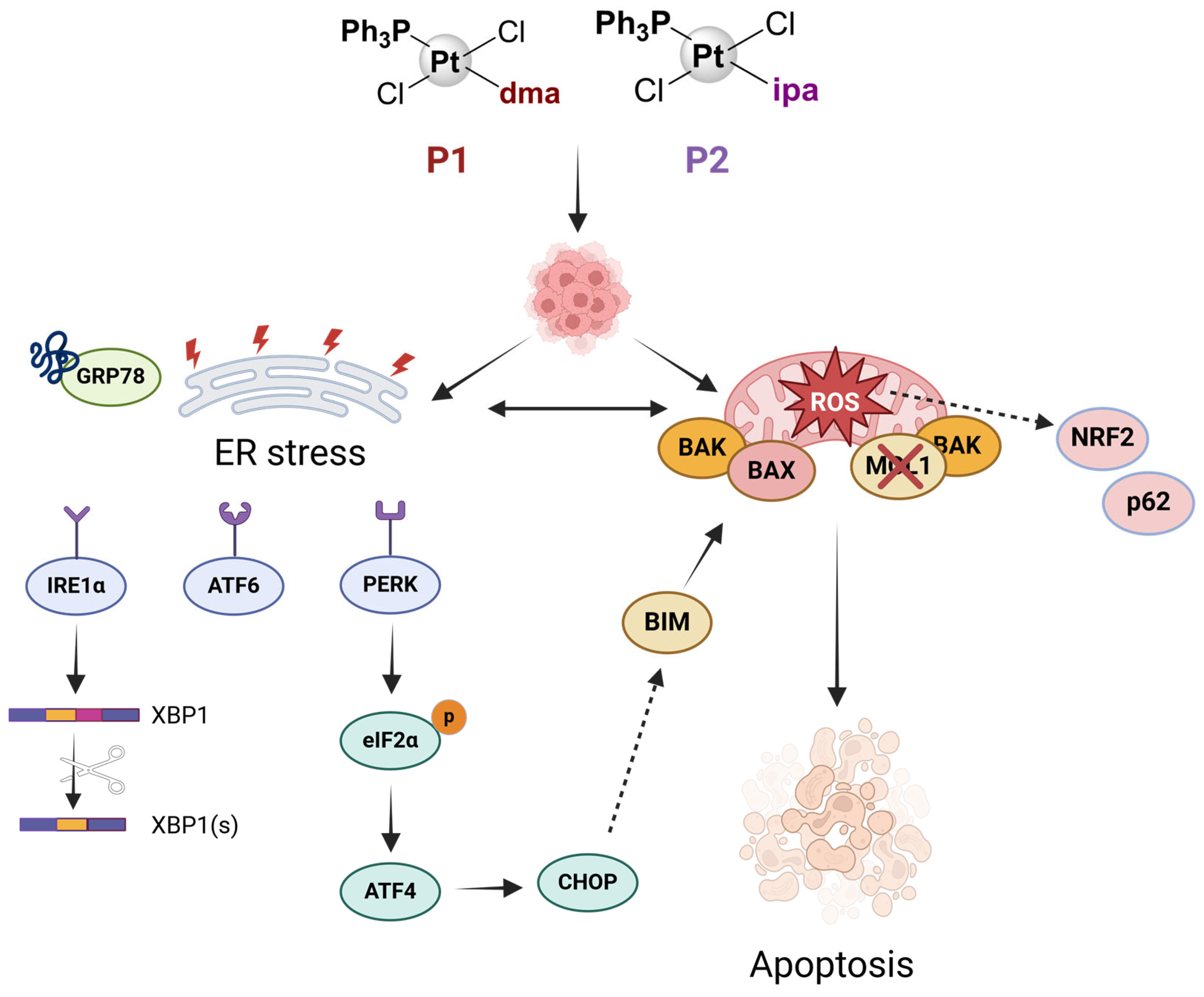
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Chemicals
4.3. Cell Viability
4.4. Clonogenic Assay
4.5. Wound Healing (Cell Migration) Assay
4.6. Cellular Uptake and Distribution by ICP-MS
4.7. Cell Cycle Profile Analysis
4.8. ROS Detection by Confocal Microscopy
4.9. Ratio GSH/GSSG
4.10. Immunofluorescence Assay
4.11. Mitochondrial Membrane Potential
4.12. Western Blotting
4.13. RT-qPCR
4.14. Statistics and Reproducibility
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Bargiela-Iparraguirre, J.; Herrero, J.M.; Pajuelo-Lozano, N.; Perez, M.; Perona, R.; Quiroga, A.; Calés, C.; Sanchez-Perez, I. Regulatory effects of miR-19a on MAD2 expression and tumorigenesis in gastric cancer. Genes Dis. 2023, 10, 1180–1182. [Google Scholar] [CrossRef]
- Cunningham, D.; Allum, W.H.; Stenning, S.P.; Thompson, J.N.; Van de Velde, C.J.; Nicolson, M.; Scarffe, J.H.; Lofts, F.J.; Falk, S.J.; Iveson, T.J.; et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N. Engl. J. Med. 2006, 355, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Al-Batran, S.E.; Homann, N.; Pauligk, C.; Goetze, T.O.; Meiler, J.; Kasper, S.; Kopp, H.G.; Mayer, F.; Haag, G.M.; Luley, K.; et al. Perioperative chemotherapy with fluorouracil plus leucovorin, oxaliplatin, and docetaxel versus fluorouracil or capecitabine plus cisplatin and epirubicin for locally advanced, resectable gastric or gastro-oesophageal junction adenocarcinoma (FLOT4): A randomised, phase 2/3 trial. Lancet 2019, 393, 1948–1957. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.L.; He, Y.; Xu, R.H. Gastric cancer treatment: Recent progress and future perspectives. J. Hematol. Oncol. 2023, 16, 57. [Google Scholar] [CrossRef] [PubMed]
- Anfossi, S.; Babayan, A.; Pantel, K.; Calin, G.A. Clinical utility of circulating non-coding RNAs—An update. Nat. Rev. Clin. Oncol. 2018, 15, 541–563. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Hartinger, C.; Gabbiani, C.; Mini, E.; Dyson, P.J.; Keppler, B.K.; Messori, L. Gold(III) compounds as anticancer agents: Relevance of gold-protein interactions for their mechanism of action. J. Inorg. Biochem. 2008, 102, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Ma, X.; Chang, X.; Liang, Z.; Lv, L.; Shan, M.; Lu, Q.; Wen, Z.; Gust, R.; Liu, W. Recent development of gold(I) and gold(III) complexes as therapeutic agents for cancer diseases. Chem. Soc. Rev. 2022, 51, 5518–5556. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, T.J.; Martins, A.S.; Marques, M.P.M.; Gil, A.M. Metabolic Aspects of Palladium(II) Potential Anti-Cancer Drugs. Front. Oncol. 2020, 10, 590970. [Google Scholar] [CrossRef]
- Herrero, J.M.; Fabra, D.; Matesanz, A.I.; Hernandez, C.; Sanchez-Perez, I.; Quiroga, A.G. Dithiobiureas Palladium(II) complexes’ studies: From their synthesis to their biological action. J. Inorg. Biochem. 2023, 246, 112261. [Google Scholar] [CrossRef]
- Kaur, P.; Johnson, A.; Northcote-Smith, J.; Lu, C.; Suntharalingam, K. Immunogenic Cell Death of Breast Cancer Stem Cells Induced by an Endoplasmic Reticulum-Targeting Copper(II) Complex. Chembiochem 2020, 21, 3618–3624. [Google Scholar] [CrossRef] [PubMed]
- Zehra, S.; Tabassum, S.; Arjmand, F. Biochemical pathways of copper complexes: Progress over the past 5 years. Drug Discov. Today 2021, 26, 1086–1096. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Ramirez, P.; Berger, S.A.; Josa, D.; Aguila, D.; Caballero, A.B.; Fontova, P.; Soto-Cerrato, V.; Martinez, M.; Gamez, P. Steric hindrance, ligand ejection and associated photocytotoxic properties of ruthenium(II) polypyridyl complexes. J. Biol. Inorg. Chem. 2023, 28, 403–420. [Google Scholar] [CrossRef]
- Monro, S.; Colon, K.L.; Yin, H.; Roque, J., 3rd; Konda, P.; Gujar, S.; Thummel, R.P.; Lilge, L.; Cameron, C.G.; McFarland, S.A. Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433. Chem. Rev. 2019, 119, 797–828. [Google Scholar] [CrossRef] [PubMed]
- Alcala, S.; Villarino, L.; Ruiz-Canas, L.; Couceiro, J.R.; Martinez-Calvo, M.; Palencia-Campos, A.; Navarro, D.; Cabezas-Sainz, P.; Rodriguez-Arabaolaza, I.; Cordero-Barreal, A.; et al. Targeting cancer stem cell OXPHOS with tailored ruthenium complexes as a new anti-cancer strategy. J. Exp. Clin. Cancer Res. 2024, 43, 33. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Zhu, M.; Jiang, M.; Yang, F.; Zhang, Z. Current status of iridium-based complexes against lung cancer. Front. Pharmacol. 2022, 13, 1025544. [Google Scholar] [CrossRef] [PubMed]
- Vigueras, G.; Markova, L.; Novohradsky, V.; Marco, A.; Cutillas, N.; Kostrhunova, H.; Kasparkova, J.; Ruiz, J.; Brabec, V. A photoactivated Ir (iii) complex targets cancer stem cells and induces secretion of damage-associated molecular patterns in melanoma cells characteristic of immunogenic cell death. Inorg. Chem. Front. 2021, 8, 4696–4711. [Google Scholar] [CrossRef]
- Wu, Q.N.; Qi, J.; Liu, Z.K.; Luo, X.J.; Yu, K.; Lu, Y.X.; Wang, Y.; Jin, Y.; Liu, J.; Huang, L.Y.; et al. HIPK3 maintains sensitivity to platinum drugs and prevents disease progression in gastric cancer. Cancer Lett. 2024, 584, 216643. [Google Scholar] [CrossRef]
- Huang, J.; Zhao, Y.; Xu, Y.; Zhu, Y.; Huang, J.; Liu, Y.; Zhao, L.; Li, Z.; Liu, H.; Wang, Q.L.; et al. Comparative effectiveness and safety between oxaliplatin-based and cisplatin-based therapy in advanced gastric cancer: A meta-analysis of randomized controlled trials. Oncotarget 2016, 7, 34824–34831. [Google Scholar] [CrossRef]
- Tham, M.J.R.; Babak, M.V.; Ang, W.H. PlatinER: A Highly Potent Anticancer Platinum(II) Complex that Induces Endoplasmic Reticulum Stress Driven Immunogenic Cell Death. Angew. Chem. Int. Ed. Engl. 2020, 59, 19070–19078. [Google Scholar] [CrossRef]
- Matesanz, A.I.; Herrero, J.M.; Faraco, E.J.; Cubo, L.; Quiroga, A.G. New Platinum(II) Triazole Thiosemicarbazone Complexes: Analysis of Their Reactivity and Potential Antitumoral Action. Chembiochem 2020, 21, 1226–1232. [Google Scholar] [CrossRef] [PubMed]
- McGhie, B.S.; Sakoff, J.; Gilbert, J.; Gordon, C.P.; Aldrich-Wright, J.R. Synthesis and Characterisation of Platinum(II) Diaminocyclohexane Complexes with Pyridine Derivatives as Anticancer Agents. Int. J. Mol. Sci. 2023, 24, 17150. [Google Scholar] [CrossRef] [PubMed]
- Melones-Herrero, J.; Alcala, S.; Ruiz-Canas, L.; Benitez-Buelga, C.; Batres-Ramos, S.; Cales, C.; Lorenzo, O.; Perona, R.; Quiroga, A.G.; Sainz, B., Jr.; et al. Platinum iodido drugs show potential anti-tumor activity, affecting cancer cell metabolism and inducing ROS and senescence in gastrointestinal cancer cells. Commun. Biol. 2024, 7, 353. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Lima, F.J.; Quiroga, A.G.; Garcia-Serrelde, B.; Blanco, F.; Carnero, A.; Navarro-Ranninger, C. New trans-platinum drugs with phosphines and amines as carrier ligands induce apoptosis in tumor cells resistant to cisplatin. J. Med. Chem. 2007, 50, 2194–2199. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhu, C.; He, Y.; Zhang, Z.; Zhou, W.; Muhammad, N.; Guo, Y.; Wang, X.; Guo, Z. Restraining Cancer Cells by Dual Metabolic Inhibition with a Mitochondrion-Targeted Platinum(II) Complex. Angew Chem. Int. Ed. Engl. 2019, 58, 4638–4643. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef] [PubMed]
- Komarnicka, U.K.; Pucelik, B.; Wojtala, D.; Lesiow, M.K.; Stochel, G.; Kyziol, A. Evaluation of anticancer activity in vitro of a stable copper(I) complex with phosphine-peptide conjugate. Sci. Rep. 2021, 11, 23943. [Google Scholar] [CrossRef] [PubMed]
- King, A.P.; Wilson, J.J. Endoplasmic reticulum stress: An arising target for metal-based anticancer agents. Chem. Soc. Rev. 2020, 49, 8113–8136. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518. [Google Scholar] [CrossRef]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef] [PubMed]
- Cutillas, N.; Martinez, A.; Yellol, G.S.; Rodriguez, V.; Zamora, A.; Pedreno, M.; Donaire, A.; Janiak, C.; Ruiz, J. Anticancer C,N-cycloplatinated(II) complexes containing fluorinated phosphine ligands: Synthesis, structural characterization, and biological activity. Inorg. Chem. 2013, 52, 13529–13535. [Google Scholar] [CrossRef] [PubMed]
- Frezza, M.; Dou, Q.P.; Xiao, Y.; Samouei, H.; Rashidi, M.; Samari, F.; Hemmateenejad, B. In vitro and in vivo antitumor activities and DNA binding mode of five coordinated cyclometalated organoplatinum(II) complexes containing biphosphine ligands. J. Med. Chem. 2011, 54, 6166–6176. [Google Scholar] [CrossRef] [PubMed]
- Icsel, C.; Yilmaz, V.T.; Cevatemre, B.; Aygun, M.; Ulukaya, E. Cytotoxic platinum(II) complexes derived from saccharinate and phosphine ligands: Synthesis, structures, DNA cleavage, and oxidative stress-induced apoptosis. J. Biol. Inorg. Chem. 2020, 25, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Icsel, C.; Yilmaz, V.T.; Aygun, M.; Ulukaya, E. Trans-Pd/Pt(II) saccharinate complexes with a phosphine ligand: Synthesis, cytotoxicity and structure-activity relationship. Bioorg. Med. Chem. Lett. 2020, 30, 127077. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, V.T.; Icsel, C.; Turgut, O.R.; Aygun, M.; Erkisa, M.; Turkdemir, M.H.; Ulukaya, E. Synthesis, structures and anticancer potentials of platinum(II) saccharinate complexes of tertiary phosphines with phenyl and cyclohexyl groups targeting mitochondria and DNA. Eur. J. Med. Chem. 2018, 155, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Icsel, C.; Yilmaz, V.T.; Aygun, M.; Cevatemre, B.; Alper, P.; Ulukaya, E. Palladium(ii) and platinum(ii) saccharinate complexes with bis(diphenylphosphino)methane/ethane: Synthesis, S-phase arrest and ROS-mediated apoptosis in human colon cancer cells. Dalton Trans. 2018, 47, 11397–11410. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Reeder, E.; Parkin, S.; Awuah, S.G. Gold(I/III)-Phosphine Complexes as Potent Antiproliferative Agents. Sci. Rep. 2019, 9, 12335. [Google Scholar] [CrossRef] [PubMed]
- Compagno, N.; Piccolo, R.; Bortolamiol, E.; Demitri, N.; Rizzolio, F.; Visentin, F.; Scattolin, T. Platinum (0)-η2-1, 2-(E) ditosylethene Complexes Bearing Phosphine, Isocyanide and N-Heterocyclic Carbene Ligands: Synthesis and Cytotoxicity towards Ovarian and Breast Cancer Cells. Molecules 2024, 29, 1119. [Google Scholar] [CrossRef]
- Henkenius, K.; Greene, B.H.; Barckhausen, C.; Hartmann, R.; Marken, M.; Kaiser, T.; Rehberger, M.; Metzelder, S.K.; Parak, W.J.; Neubauer, A.; et al. Maintenance of cellular respiration indicates drug resistance in acute myeloid leukemia. Leuk. Res. 2017, 62, 56–63. [Google Scholar] [CrossRef]
- Medrano, M.A.; Alvarez-Valdes, A.; Perles, J.; Lloret-Fillol, J.; Munoz-Galvan, S.; Carnero, A.; Navarro-Ranninger, C.; Quiroga, A.G. Oxidation of anticancer Pt(II) complexes with monodentate phosphane ligands: Towards stable but active Pt(IV) prodrugs. Chem. Commun. 2013, 49, 4806–4808. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, T.; Su, W.; Dou, Z.; Zhao, D.; Jin, X.; Lei, H.; Wang, J.; Xie, X.; Cheng, B.; et al. Mutant p53 in cancer: From molecular mechanism to therapeutic modulation. Cell Death Dis. 2022, 13, 974. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Bratton, S.B. Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxid. Redox Signal. 2013, 19, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Gillissen, B.; Richter, A.; Sinnberg, T.; Schlaak, M.S.; Eberle, J. Effective Targeting of Melanoma Cells by Combination of Mcl-1 and Bcl-2/Bcl-x(L)/Bcl-w Inhibitors. Int. J. Mol. Sci. 2024, 25, 3453. [Google Scholar] [CrossRef] [PubMed]
- Pajuelo-Lozano, N.; Bargiela-Iparraguirre, J.; Dominguez, G.; Quiroga, A.G.; Perona, R.; Sanchez-Perez, I. XPA, XPC, and XPD Modulate Sensitivity in Gastric Cisplatin Resistance Cancer Cells. Front. Pharmacol. 2018, 9, 1197. [Google Scholar] [CrossRef] [PubMed]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Wali, J.A.; Rondas, D.; McKenzie, M.D.; Zhao, Y.; Elkerbout, L.; Fynch, S.; Gurzov, E.N.; Akira, S.; Mathieu, C.; Kay, T.W.; et al. The proapoptotic BH3-only proteins Bim and Puma are downstream of endoplasmic reticulum and mitochondrial oxidative stress in pancreatic islets in response to glucotoxicity. Cell Death Dis. 2014, 5, e1124. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Li, G.; Zhu, B.; Yang, J.; Cui, S.; Jiang, R.; Wang, B. p20BAP31 Induces Autophagy in Colorectal Cancer Cells by Promoting PERK-Mediated ER Stress. Int. J. Mol. Sci. 2024, 25, 5101. [Google Scholar] [CrossRef] [PubMed]
- Kapuy, O. Mechanism of Decision Making between Autophagy and Apoptosis Induction upon Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2024, 25, 4368. [Google Scholar] [CrossRef]
- Moscat, J.; Karin, M.; Diaz-Meco, M.T. p62 in Cancer: Signaling Adaptor Beyond Autophagy. Cell 2016, 167, 606–609. [Google Scholar] [CrossRef]
- Komatsu, M.; Ichimura, Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Lett. 2010, 584, 1374–1378. [Google Scholar] [CrossRef] [PubMed]
- Rusten, T.E.; Stenmark, H. p62, an autophagy hero or culprit? Nat. Cell Biol. 2010, 12, 207–209. [Google Scholar] [CrossRef]
- ES11/11013, ISO 9001:2015; Quality Certificate for the Design, Development and Testing of Centers of Assistance to Research (CAI, among Them, the Unit of Geological Techniques) of the University Complutense of Madrid. SGS International Certification Services Iberica, S.A.U.: Madrid, Spain, 2023.
- Sanchez-Perez, I.; Murguia, J.R.; Perona, R. Cisplatin induces a persistent activation of JNK that is related to cell death. Oncogene 1998, 16, 533–540. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AGS | MKN45 | PANC1 | AC16 | HPDE | |
|---|---|---|---|---|---|
| CDDP | 24.95 ± 2.78 (0.1) | 7.22 ± 0.72 (0.2) | 9.71 ± 1.21 (0.2) | 6.51 ± 1.87 | 1.58 ± 0.89 |
| P1 | 9.12 ± 0.59 (>2.7) | 20.91 ± 1.73 (>1.2) | 10.57 ± 2.43 (>2.4) | 19.91 ± 2.89 | >25 |
| P2 | 5.66 ± 0.41 (3.9) | 10.15 ± 0.13 (2.2) | 9.74 ± 0.50 (2.3) | 7.57 ± 1.64 | 22.09 ± 0.53 |
| Pt (ng·mL−1) | ||
|---|---|---|
| Nucleus | Cytoplasm | |
| Control | <0.05 | <0.05 |
| P1 | 0.54 ± 0.24 | 0.87 ± 0.18 |
| P2 | 1.35 ± 0.07 | 2.25 ± 0.64 |
| Primer | Sequence |
|---|---|
| HSPA5 | F: TGCCCAACGCCAAGCAACC R: AATAGCAGCTGCCGTAGGCTCG |
| ERN1 | F: ATCTTGGGCGAACAGAATACACC R: CACCGGAGCTCTCGGGTTTTG |
| XBP1 | F: GCTTCTGTCGGGGCAGC R: ACTCTGTTTTTCAGTTTCCTCCTCA |
| EIF2AK3 | F: GGGAGCAGGGAAGAAAAGGTCA R: ACACCAAGGAACCGGATCCCAC |
| ATF4 | F: GGGCTCCTCCGAATGGCTGG R: CGGAGAAGGCATCCTCCTTGC |
| DDIT3 | F: ACCTCCTGGAAATGAAGAGGAAGAA R: GGGCTCTGGGAGGTGCTTGT |
| ATF6 | F: AACCTGCACCCACTAAAGGCCA R: TCCCCCAGCAACAGCAAGGAC |
| GAPDH | F: GAGAGACCCTCACTGCTG R: GATGGTACATGACAAGGTGG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melones-Herrero, J.; Delgado-Aliseda, P.; Figueiras, S.; Velázquez-Gutiérrez, J.; Quiroga, A.G.; Calés, C.; Sánchez-Pérez, I. Trans-[Pt(amine)Cl2(PPh3)] Complexes Target Mitochondria and Endoplasmic Reticulum in Gastric Cancer Cells. Int. J. Mol. Sci. 2024, 25, 7739. https://doi.org/10.3390/ijms25147739
Melones-Herrero J, Delgado-Aliseda P, Figueiras S, Velázquez-Gutiérrez J, Quiroga AG, Calés C, Sánchez-Pérez I. Trans-[Pt(amine)Cl2(PPh3)] Complexes Target Mitochondria and Endoplasmic Reticulum in Gastric Cancer Cells. International Journal of Molecular Sciences. 2024; 25(14):7739. https://doi.org/10.3390/ijms25147739
Chicago/Turabian StyleMelones-Herrero, Jorge, Patricia Delgado-Aliseda, Sofía Figueiras, Javier Velázquez-Gutiérrez, Adoración Gomez Quiroga, Carmela Calés, and Isabel Sánchez-Pérez. 2024. "Trans-[Pt(amine)Cl2(PPh3)] Complexes Target Mitochondria and Endoplasmic Reticulum in Gastric Cancer Cells" International Journal of Molecular Sciences 25, no. 14: 7739. https://doi.org/10.3390/ijms25147739