

Adaptive and Maladaptive DNA Breaks in Neuronal Physiology and Alzheimer’s Disease


{kind=link}
Abstract
:1. Introduction
2. Adaptive Role of DNA Breaks in Learning and Memory
2.1. γH2AX as a Biomarker of DNA Damage
2.2. Evidence Establishing Neuronal Activity-Induced DNA Breaks
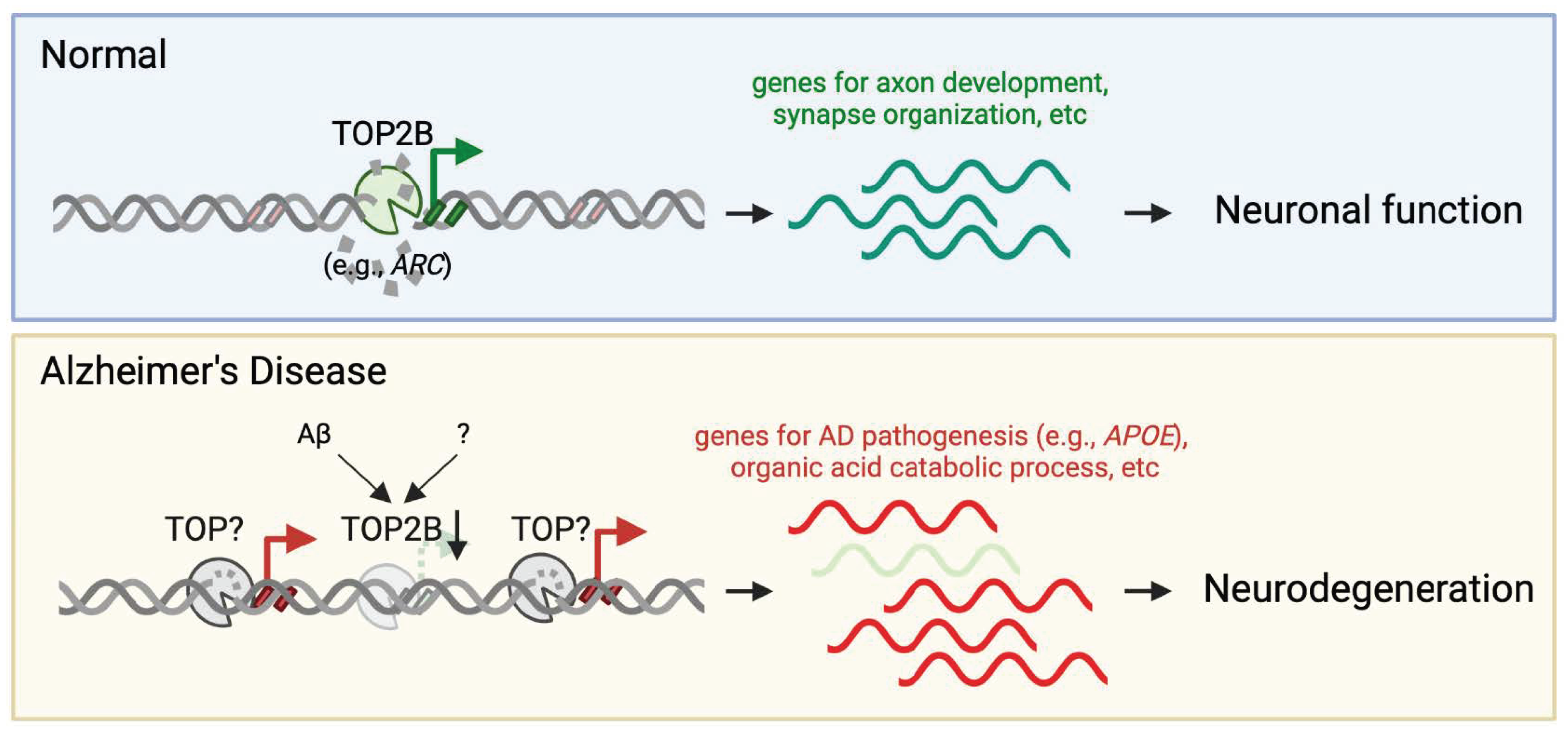
2.3. TOP2B—Catalyzing Activity-Dependent DNA Breaks for IEG Expression
2.4. DNA Breaks and Memory Formation through Inflammatory Response
2.5. Repair of Adaptive DNA Breaks
3. Accumulation of DNA Damage in AD
4. How Do the Positions of the DNA Damage Impact Neuronal Cell Function?
5. DNA Damage and Mitochondria Dysfunction
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, T.; Pan, Y.; Kao, S.Y.; Li, C.; Kohane, I.; Chan, J.; Yankner, B.A. Gene regulation and DNA damage in the ageing human brain. Nature 2004, 429, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Kawas, C.; Gray, S.; Brookmeyer, R.; Fozard, J.; Zonderman, A. Age-specific incidence rates of Alzheimer’s disease: The Baltimore Longitudinal Study of Aging. Neurology 2000, 54, 2072–2077. [Google Scholar] [CrossRef]
- Jellinger, K.A. Clinicopathological analysis of dementia disorders in the elderly—An update. J. Alzheimers Dis. 2006, 9 (Suppl. S3), 61–70. [Google Scholar] [CrossRef] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Steinerman, J.R.; Irizarry, M.; Scarmeas, N.; Raju, S.; Brandt, J.; Albert, M.; Blacker, D.; Hyman, B.; Stern, Y. Distinct pools of beta-amyloid in Alzheimer disease-affected brain: A clinicopathologic study. Arch. Neurol. 2008, 65, 906–912. [Google Scholar] [CrossRef]
- Adamec, E.; Vonsattel, J.P.; Nixon, R.A. DNA strand breaks in Alzheimer’s disease. Brain Res. 1999, 849, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Suberbielle, E.; Djukic, B.; Evans, M.; Kim, D.H.; Taneja, P.; Wang, X.; Finucane, M.; Knox, J.; Ho, K.; Devidze, N.; et al. DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat. Commun. 2015, 6, 8897. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, P.J. Genome integrity and disease prevention in the nervous system. Genes Dev. 2017, 31, 1180–1194. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, N.M.; Evans, M.D.; Mao, W.; Nana, A.L.; Seeley, W.W.; Adame, A.; Rissman, R.A.; Masliah, E.; Mucke, L. Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathol. Commun. 2019, 7, 77. [Google Scholar] [CrossRef] [PubMed]
- Thadathil, N.; Delotterie, D.F.; Xiao, J.; Hori, R.; McDonald, M.P.; Khan, M.M. DNA Double-Strand Break Accumulation in Alzheimer’s Disease: Evidence from Experimental Models and Postmortem Human Brains. Mol. Neurobiol. 2021, 58, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Asada-Utsugi, M.; Uemura, K.; Ayaki, T.; Uemura, M.T.; Minamiyama, S.; Hikiami, R.; Morimura, T.; Shodai, A.; Ueki, T.; Takahashi, R.; et al. Failure of DNA double-strand break repair by tau mediates Alzheimer’s disease pathology in vitro. Commun. Biol. 2022, 5, 358. [Google Scholar] [CrossRef] [PubMed]
- Welch, G.; Tsai, L.H. Mechanisms of DNA damage-mediated neurotoxicity in neurodegenerative disease. EMBO Rep. 2022, 23, e54217. [Google Scholar] [CrossRef] [PubMed]
- Dileep, V.; Boix, C.A.; Mathys, H.; Marco, A.; Welch, G.M.; Meharena, H.S.; Loon, A.; Jeloka, R.; Peng, Z.; Bennett, D.A.; et al. Neuronal DNA double-strand breaks lead to genome structural variations and 3D genome disruption in neurodegeneration. Cell 2023, 186, 4404–4421.e20. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Gao, F.; Pfenning, A.R.; Pan, L.; Yamakawa, S.; Seo, J.; Rueda, R.; Phan, T.X.; Yamakawa, H.; Pao, P.C.; et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 2015, 161, 1592–1605. [Google Scholar] [CrossRef] [PubMed]
- Delint-Ramirez, I.; Konada, L.; Heady, L.; Rueda, R.; Jacome, A.S.V.; Marlin, E.; Marchioni, C.; Segev, A.; Kritskiy, O.; Yamakawa, S.; et al. Calcineurin dephosphorylates topoisomerase IIbeta and regulates the formation of neuronal-activity-induced DNA breaks. Mol. Cell 2022, 82, 3794–3809.e8. [Google Scholar] [CrossRef] [PubMed]
- Weber Boutros, S.; Unni, V.K.; Raber, J. An Adaptive Role for DNA Double-Strand Breaks in Hippocampus-Dependent Learning and Memory. Int. J. Mol. Sci. 2022, 23, 8352. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Hill, S.E.; Nathan, W.J.; Paiano, J.; Callen, E.; Wang, D.; Shinoda, K.; van Wietmarschen, N.; Colon-Mercado, J.M.; Zong, D.; et al. Neuronal enhancers are hotspots for DNA single-strand break repair. Nature 2021, 593, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Reid, D.A.; Reed, P.J.; Schlachetzki, J.C.M.; Nitulescu, I.; Chou, G.; Tsui, E.C.; Jones, J.R.; Chandran, S.; Lu, A.T.; McClain, C.A.; et al. Incorporation of a nucleoside analog maps genome repair sites in postmitotic human neurons. Science 2021, 372, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Suberbielle, E.; Sanchez, P.E.; Kravitz, A.V.; Wang, X.; Ho, K.; Eilertson, K.; Devidze, N.; Kreitzer, A.C.; Mucke, L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-beta. Nat. Neurosci. 2013, 16, 613–621. [Google Scholar] [CrossRef]
- Neven, J.; Issayama, L.K.; Dewachter, I.; Wilson, D.M., 3rd. Genomic stress and impaired DNA repair in Alzheimer disease. DNA Repair. 2024, 139, 103678. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lautrup, S.; Caponio, D.; Zhang, J.; Fang, E.F. DNA Damage-Induced Neurodegeneration in Accelerated Ageing and Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 6748. [Google Scholar] [CrossRef] [PubMed]
- West, A.E.; Greenberg, M.E. Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold. Spring Harb. Perspect. Biol. 2011, 3, a005744. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.M.; Chen, J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [CrossRef] [PubMed]
- Podhorecka, M.; Skladanowski, A.; Bozko, P. H2AX Phosphorylation: Its Role in DNA Damage Response and Cancer Therapy. J. Nucleic Acids 2010, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Crowe, S.L.; Movsesyan, V.A.; Jorgensen, T.J.; Kondratyev, A. Rapid phosphorylation of histone H2A.X following ionotropic glutamate receptor activation. Eur. J. Neurosci. 2006, 23, 2351–2361. [Google Scholar] [CrossRef] [PubMed]
- Bellesi, M.; Bushey, D.; Chini, M.; Tononi, G.; Cirelli, C. Contribution of sleep to the repair of neuronal DNA double-strand breaks: Evidence from flies and mice. Sci. Rep. 2016, 6, 36804. [Google Scholar] [CrossRef] [PubMed]
- Minatohara, K.; Akiyoshi, M.; Okuno, H. Role of Immediate-Early Genes in Synaptic Plasticity and Neuronal Ensembles Underlying the Memory Trace. Front. Mol. Neurosci. 2015, 8, 78. [Google Scholar] [CrossRef] [PubMed]
- Guzowski, J.F.; Lyford, G.L.; Stevenson, G.D.; Houston, F.P.; McGaugh, J.L.; Worley, P.F.; Barnes, C.A. Inhibition of activity-dependent arc protein expression in the rat hippocampus impairs the maintenance of long-term potentiation and the consolidation of long-term memory. J. Neurosci. 2000, 20, 3993–4001. [Google Scholar] [CrossRef] [PubMed]
- Guzowski, J.F.; McNaughton, B.L.; Barnes, C.A.; Worley, P.F. Environment-specific expression of the immediate-early gene Arc in hippocampal neuronal ensembles. Nat. Neurosci. 1999, 2, 1120–1124. [Google Scholar] [CrossRef] [PubMed]
- Bramham, C.R.; Worley, P.F.; Moore, M.J.; Guzowski, J.F. The immediate early gene arc/arg3.1: Regulation, mechanisms, and function. J. Neurosci. 2008, 28, 11760–11767. [Google Scholar] [CrossRef] [PubMed]
- Pastuzyn, E.D.; Day, C.E.; Kearns, R.B.; Kyrke-Smith, M.; Taibi, A.V.; McCormick, J.; Yoder, N.; Belnap, D.M.; Erlendsson, S.; Morado, D.R.; et al. The Neuronal Gene Arc Encodes a Repurposed Retrotransposon Gag Protein that Mediates Intercellular RNA Transfer. Cell 2018, 172, 275–288.e18. [Google Scholar] [CrossRef] [PubMed]
- Stott, R.T.; Kritsky, O.; Tsai, L.H. Profiling DNA break sites and transcriptional changes in response to contextual fear learning. PLoS ONE 2021, 16, e0249691. [Google Scholar] [CrossRef] [PubMed]
- Bax, B.D.; Murshudov, G.; Maxwell, A.; Germe, T. DNA Topoisomerase Inhibitors: Trapping a DNA-Cleaving Machine in Motion. J. Mol. Biol. 2019, 431, 3427–3449. [Google Scholar] [CrossRef] [PubMed]
- Bunch, H.; Lawney, B.P.; Lin, Y.F.; Asaithamby, A.; Murshid, A.; Wang, Y.E.; Chen, B.P.; Calderwood, S.K. Transcriptional elongation requires DNA break-induced signalling. Nat. Commun. 2015, 6, 10191. [Google Scholar] [CrossRef] [PubMed]
- Jovasevic, V.; Wood, E.M.; Cicvaric, A.; Zhang, H.; Petrovic, Z.; Carboncino, A.; Parker, K.K.; Bassett, T.E.; Moltesen, M.; Yamawaki, N.; et al. Formation of memory assemblies through the DNA-sensing TLR9 pathway. Nature 2024, 628, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Bloodgood, B.L.; Hauser, J.L.; Lapan, A.D.; Koon, A.C.; Kim, T.K.; Hu, L.S.; Malik, A.N.; Greenberg, M.E. Activity-dependent regulation of inhibitory synapse development by Npas4. Nature 2008, 455, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Pollina, E.A.; Gilliam, D.T.; Landau, A.T.; Lin, C.; Pajarillo, N.; Davis, C.P.; Harmin, D.A.; Yap, E.L.; Vogel, I.R.; Griffith, E.C.; et al. A NPAS4-NuA4 complex couples synaptic activity to DNA repair. Nature 2023, 614, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Mullaart, E.; Boerrigter, M.E.; Ravid, R.; Swaab, D.F.; Vijg, J. Increased Levels of DNA Breaks in Cerebral Cortex of Alzheimer’s Disease Patients. Neurobiol. Aging 1990, 11, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Turunc Bayrakdar, E.; Uyanikgil, Y.; Kanit, L.; Koylu, E.; Yalcin, A. Nicotinamide treatment reduces the levels of oxidative stress, apoptosis, and PARP-1 activity in Abeta(1-42)-induced rat model of Alzheimer’s disease. Free Radic. Res. 2014, 48, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Gabbita, S.P.; Lovell, M.A.; Markesbery, W.R. Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J. Neurochem. 1998, 71, 2034–2040. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, J.P.; Swerdlow, R.H.; Miller, S.W.; Davis, R.E.; Parks, J.K.; Parker, W.D.; Tuttle, J.B. Calcium Homeostasis and Reactive Oxygen Species Production in Cells Transformed by Mitochondria from Individuals with Sporadic Alzheimer’s Disease. J. Neurosci. 1997, 17, 4612–4622. [Google Scholar] [CrossRef] [PubMed]
- Pao, P.C.; Patnaik, D.; Watson, L.A.; Gao, F.; Pan, L.; Wang, J.; Adaikkan, C.; Penney, J.; Cam, H.P.; Huang, W.C.; et al. HDAC1 modulates OGG1-initiated oxidative DNA damage repair in the aging brain and Alzheimer’s disease. Nat. Commun. 2020, 11, 2484. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Leon, J.; Sakumi, K.; Abolhassani, N.; Sheng, Z.; Tsuchimoto, D.; LaFerla, F.M.; Nakabeppu, Y. MTH1 and OGG1 maintain a low level of 8-oxoguanine in Alzheimer’s brain, and prevent the progression of Alzheimer’s pathogenesis. Sci. Rep. 2021, 11, 5819. [Google Scholar] [CrossRef] [PubMed]
- Dinkelmann, M.; Spehalski, E.; Stoneham, T.; Buis, J.; Wu, Y.; Sekiguchi, J.M.; Ferguson, D.O. Multiple functions of MRN in end-joining pathways during isotype class switching. Nat. Struct. Mol. Biol. 2009, 16, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Chiu, J.W.; Koller, B.H.; Jasin, M. Brca1 Controls Homology-Directed DNA Repair. Mol. Cell 1999, 4, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Lodato, M.A.; Woodworth, M.B.; Lee, S.; Evrony, G.D.; Mehta, B.K.; Karger, A.; Lee, S.; Chittenden, T.W.; D’Gama, A.M.; Cai, X.; et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 2015, 350, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.C.; Chang, A.N.; Kao, J.; Du, Z.; Meyers, R.M.; Alt, F.W.; Schwer, B. Long Neural Genes Harbor Recurrent DNA Break Clusters in Neural Stem/Progenitor Cells. Cell 2016, 164, 644–655. [Google Scholar] [CrossRef] [PubMed]
- McConnell, M.J.; Moran, J.V.; Abyzov, A.; Akbarian, S.; Bae, T.; Cortes-Ciriano, I.; Erwin, J.A.; Fasching, L.; Flasch, D.A.; Freed, D.; et al. Intersection of diverse neuronal genomes and neuropsychiatric disease: The Brain Somatic Mosaicism Network. Science 2017, 356, eaal1641. [Google Scholar] [CrossRef]
- Rodin, R.E.; Dou, Y.; Kwon, M.; Sherman, M.A.; D’Gama, A.M.; Doan, R.N.; Rento, L.M.; Girskis, K.M.; Bohrson, C.L.; Kim, S.N.; et al. The landscape of somatic mutation in cerebral cortex of autistic and neurotypical individuals revealed by ultra-deep whole-genome sequencing. Nat. Neurosci. 2021, 24, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, Y.; Huang, M.; Gunewardena, S.; Haeri, M.; Swerdlow, R.H.; Wang, N. Landscape of Double-Stranded DNA Breaks in Postmortem Brains from Alzheimer’s Disease and Non-Demented Individuals. J. Alzheimers Dis. 2023, 94, 519–535. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Haeri, M.; Swerdlow, R.H.; Wang, N. Loss of Adaptive DNA Breaks in Alzheimer’s Disease Brains. J. Alzheimers Dis. 2024, 97, 1861–1875. [Google Scholar] [CrossRef] [PubMed]
- Terzioglu-Usak, S.; Negis, Y.; Karabulut, D.S.; Zaim, M.; Isik, S. Cellular Model of Alzheimer’s Disease: Abeta1-42 Peptide Induces Amyloid Deposition and a Decrease in Topo Isomerase IIbeta and Nurr1 Expression. Curr. Alzheimer Res. 2017, 14, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Canela, A.; Sridharan, S.; Sciascia, N.; Tubbs, A.; Meltzer, P.; Sleckman, B.P.; Nussenzweig, A. DNA Breaks and End Resection Measured Genome-wide by End Sequencing. Mol. Cell 2016, 63, 898–911. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.X.; Mirzazadeh, R.; Garnerone, S.; Scott, D.; Schneider, M.W.; Kallas, T.; Custodio, J.; Wernersson, E.; Li, Y.; Gao, L.; et al. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat. Commun. 2017, 8, 15058. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, B.A.M.; Agostini, F.; Garnerone, S.; Petrosino, G.; Gothe, H.J.; Sayols, S.; Moor, A.E.; Itzkovitz, S.; Bienko, M.; Roukos, V.; et al. Genome-wide detection of DNA double-strand breaks by in-suspension BLISS. Nat. Protoc. 2020, 15, 3894–3941. [Google Scholar] [CrossRef] [PubMed]
- Norris, C.M. Calcineurin: Directing the damage in Alzheimer disease: An Editorial for ‘Neuronal calcineurin transcriptional targets parallel changes observed in Alzheimer disease brain’ on page 24. J. Neurochem. 2018, 147, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Segev, A.; Heady, L.; Crewe, M.; Madabhushi, R. Mapping catalytically engaged TOP2B in neurons reveals the principles of topoisomerase action within the genome. Cell Rep. 2024, 43, 113809. [Google Scholar] [CrossRef] [PubMed]
- Nikolac Perkovic, M.; Videtic Paska, A.; Konjevod, M.; Kouter, K.; Svob Strac, D.; Nedic Erjavec, G.; Pivac, N. Epigenetics of Alzheimer’s Disease. Biomolecules 2021, 11, 195. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.; Reddy, P.H. Defective Autophagy and Mitophagy in Aging and Alzheimer’s Disease. Front Neurosci 2020, 14, 612757. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. The Alzheimer’s Disease Mitochondrial Cascade Hypothesis: A Current Overview. J. Alzheimers Dis. 2023, 92, 751–768. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.M.; Kong, X.; Moncada, E.; Chen, Y.; Imamura, H.; Wang, P.; Berns, M.W.; Yokomori, K.; Digman, M.A. NAD+ consumption by PARP1 in response to DNA damage triggers metabolic shift critical for damaged cell survival. Mol. Biol. Cell 2019, 30, 2584–2597. [Google Scholar] [CrossRef] [PubMed]
- Ahel, D.; Horejsi, Z.; Wiechens, N.; Polo, S.E.; Garcia-Wilson, E.; Ahel, I.; Flynn, H.; Skehel, M.; West, S.C.; Jackson, S.P.; et al. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 2009, 325, 1240–1243. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Canto, C.; Oudart, H.; Brunyanszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Elesela, S.; Morris, S.B.; Narayanan, S.; Kumar, S.; Lombard, D.B.; Lukacs, N.W. Sirtuin 1 regulates mitochondrial function and immune homeostasis in respiratory syncytial virus infected dendritic cells. PLoS Pathog. 2020, 16, e1008319. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.Q.; Zhang, X.; Zhen, Y.; He, X.Y.; Zhao, S.; Li, X.F.; Yang, B.; Gao, F.; Guo, F.Y.; Fu, L.; et al. A novel small-molecule activator of Sirtuin-1 induces autophagic cell death/mitophagy as a potential therapeutic strategy in glioblastoma. Cell Death Dis. 2018, 9, 767. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roberts, A.; Swerdlow, R.H.; Wang, N. Adaptive and Maladaptive DNA Breaks in Neuronal Physiology and Alzheimer’s Disease. Int. J. Mol. Sci. 2024, 25, 7774. https://doi.org/10.3390/ijms25147774
Roberts A, Swerdlow RH, Wang N. Adaptive and Maladaptive DNA Breaks in Neuronal Physiology and Alzheimer’s Disease. International Journal of Molecular Sciences. 2024; 25(14):7774. https://doi.org/10.3390/ijms25147774
Chicago/Turabian StyleRoberts, Anysja, Russell H. Swerdlow, and Ning Wang. 2024. "Adaptive and Maladaptive DNA Breaks in Neuronal Physiology and Alzheimer’s Disease" International Journal of Molecular Sciences 25, no. 14: 7774. https://doi.org/10.3390/ijms25147774