
Metabolic Derangement of Essential Transition Metals and Potential Antioxidant Therapies
 , , and
, , and
Abstract
:1. Introduction
2. Essential Transition Metals in Metabolism and Disease
2.1. Iron (Fe)
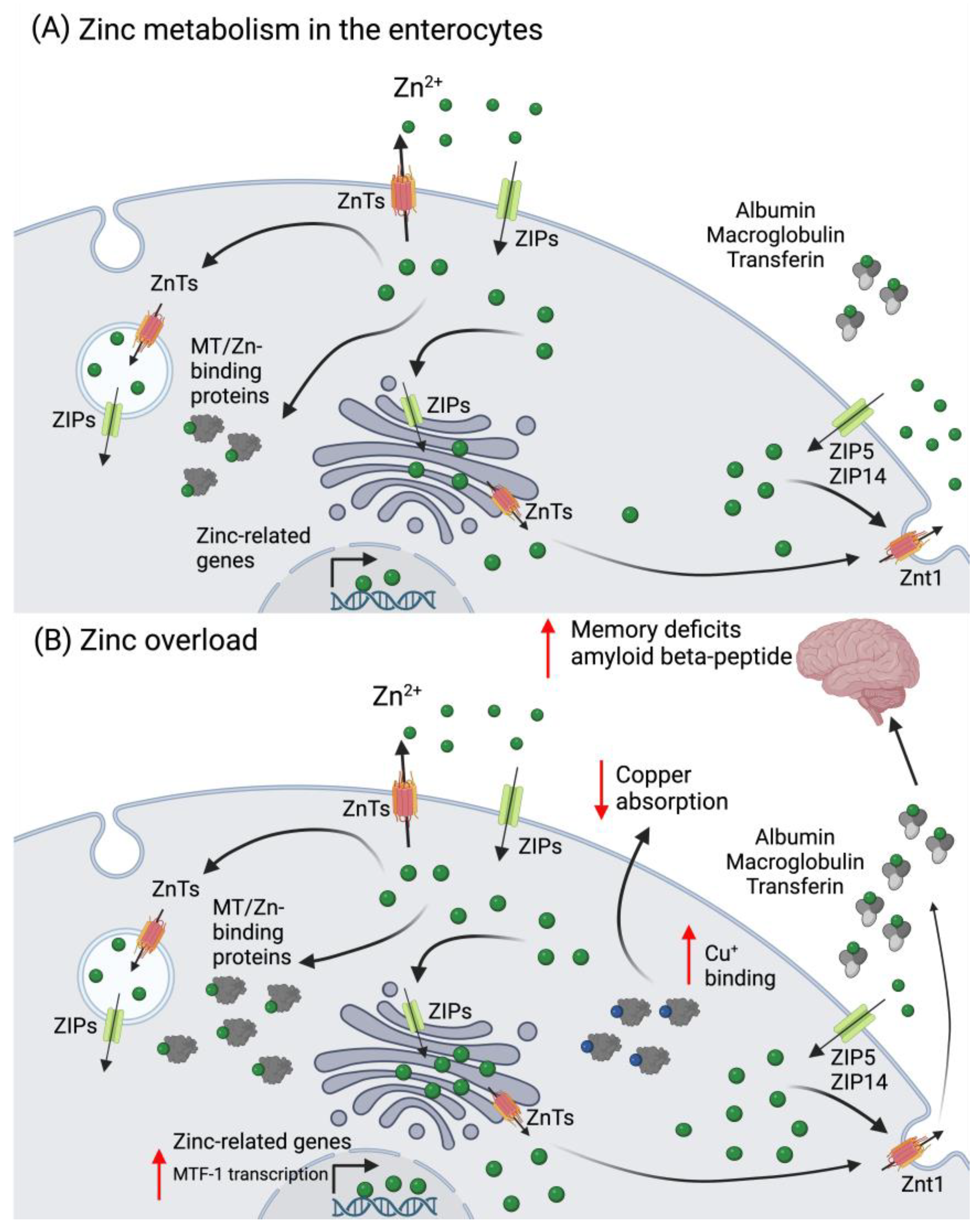
2.2. Zinc (Zn)
2.3. Copper (Cu)
2.4. Manganese (Mn)
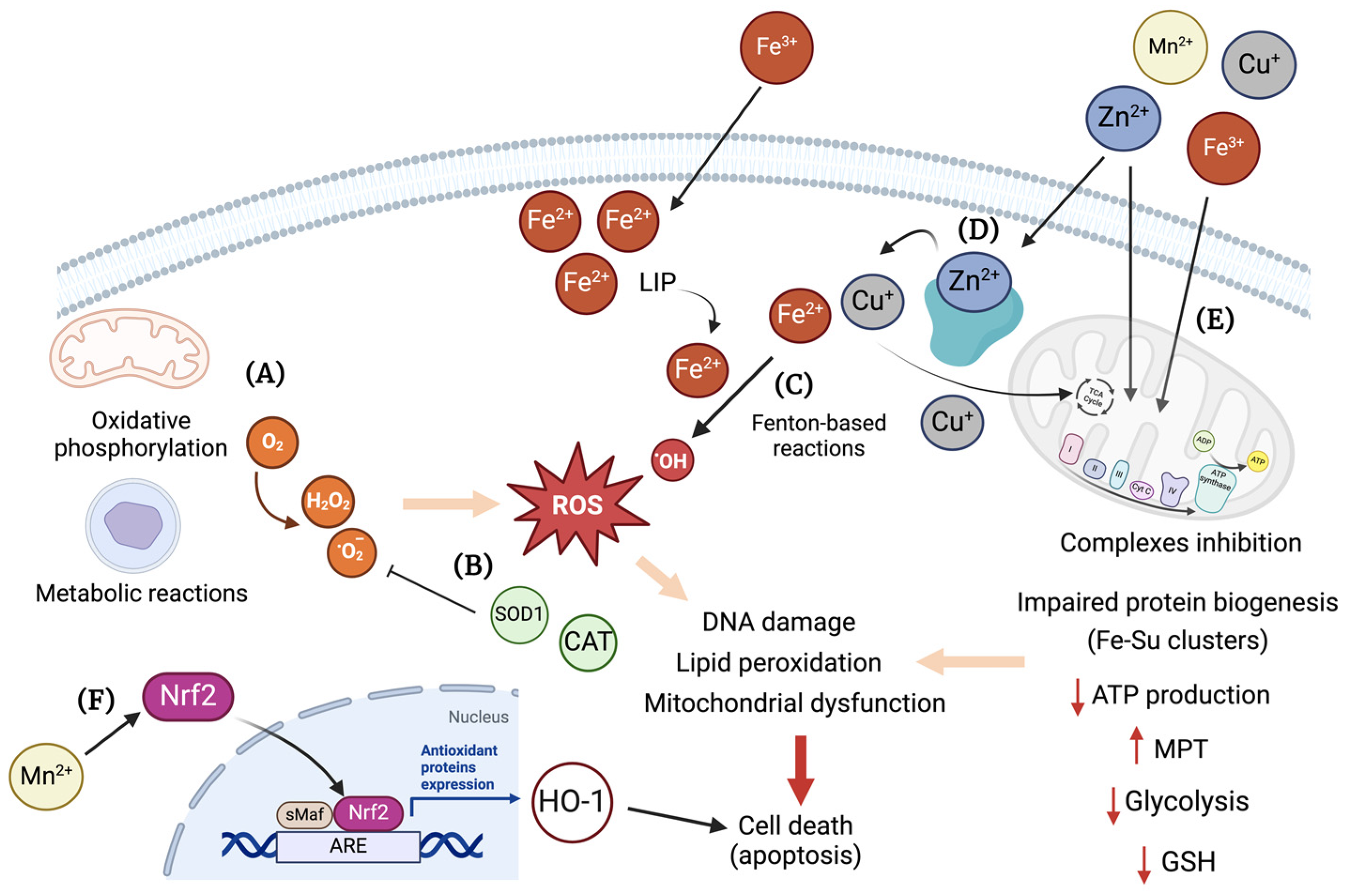
3. Essential Transition Metals and Oxidative Stress
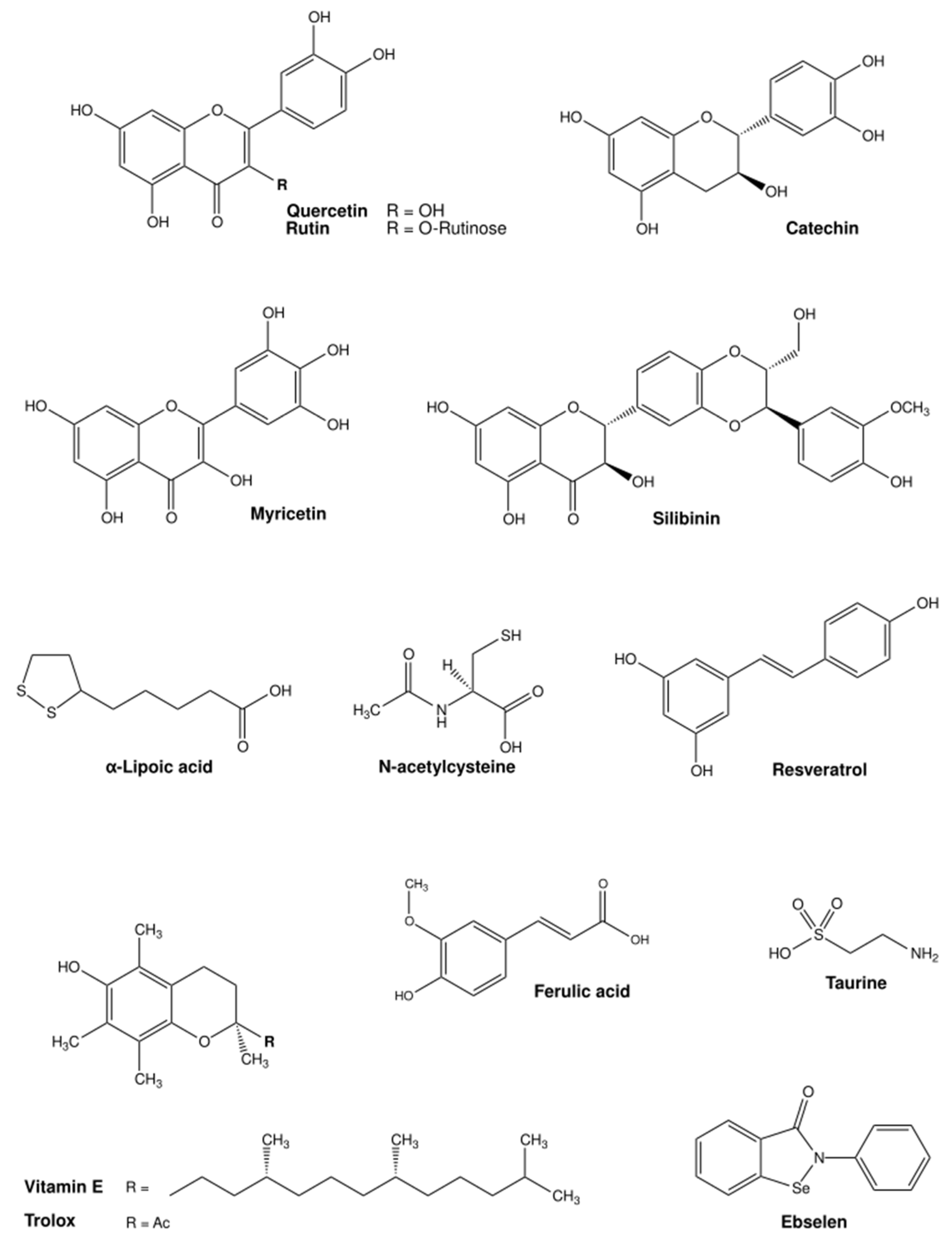
4. Antioxidant Therapies for Essential Transition Metals Toxicity
4.1. Iron (Fe)
4.2. Zinc (Zn)
4.3. Copper (Cu)
4.4. Manganese (Mn)
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADD | Average daily dose |
| AChE | Acetylcholinesterase |
| Atox1 | Antioxidant protein 1 |
| ALT | Alanine transaminase |
| AST | Aspartate transaminase |
| ATP7A | ATPase copper-transporting alpha |
| ATP7B | ATPase copper-transporting beta |
| BDNF | Brain-derived neurotrophic factor |
| BMI | Body mass index |
| bw | Body weight |
| CaNa2EDTA | Calcium disodium edetate |
| CAT | Catalase |
| CCO | Cytochrome C oxidase |
| CCS | Copper chaperone for Cu/Zn superoxide dismutase |
| Cp | Ceruloplasmin |
| CTR1 | High-affinity copper transporter 1 |
| CTR2 | Low-affinity copper transporter 2 |
| Cu | Copper |
| DCYTB | Ferrireductase duodenal cytochrome b |
| DFO | Deferoxamine |
| DMT1 | Divalent metal transporter 1 |
| DTPA | Diethylentriamene pentaacetate |
| EDTA | Ethylenediaminetetraacetic acid |
| ERK1/2 | Extracellular signal-regulated kinases 1 and 2 |
| Fe | Iron |
| FDA | Food and Drug Administration |
| FPN1 | Ferroportin-1 |
| GIT | Gastrointestinal tract |
| GPx | Glutathione peroxidase |
| GR | Glutathione reductase |
| GSH | Glutathione |
| GSSG | Glutathione disulfide |
| GST | Glutathione S-transferase |
| HCP1 | Heme carrier protein 1 |
| HFD | High-fat diet |
| HFE | Homeostatic iron regulator |
| His | Histidine |
| HJV | Hemojuvelin |
| HO-1 | Heme oxygenase 1 |
| IL | Interleukin |
| iNOS | Inducible nitric oxide synthase |
| iPSC-CMS | Induced pluripotent stem cell-derived cardiomyocytes |
| MAPKs | Mitogen-activated protein kinases |
| MDA | Malondialdehyde |
| Mn | Manganese |
| MnSOD | Manganese superoxide dismutase |
| MPT | Mitochondrial permeability transition pore |
| MT | Metallothionein |
| MTF-1 | Metal regulatory transcription factor 1 |
| NAC | N-acetylcysteine |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NF-kB | Nuclear factor kappa B |
| Nrf2 | Nuclear factor erythroid 2 |
| NO | Nitric oxide |
| PAS | Para-amino salicylic acid |
| PARP-1 | Poly(ADP-ribose)-polymerase 1 |
| PD | Parkinson’s disease |
| PKC | Protein kinase C |
| PPIs | Proton pump inhibitors |
| ROS | Reactive oxygen species |
| RNS | Reactive nitrogen species |
| STEAP | Six-transmembrane epithelial antigen of the prostate |
| SOD1 | Cu/Zn superoxide dismutase |
| TDI | Tolerable daily intake |
| TGN | Trans-Golgi network |
| Tf | Transferrin |
| TfR1 | Transferrin receptor 1 |
| TfR2 | Transferrin receptor 2 |
| TNF-α | Tumor necrosis factor alpha |
| WD | Wilson disease |
| ZIP | Zrt-, Irt- like protein family |
| Zn | Zinc |
| ZnT | Zinc transporter |
| ZnONP | Zinc oxide nanoparticles |
References
- Jomova, K.; Makova, M.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Rhodes, C.J.; Valko, M. Essential metals in health and disease. Chem. Biol. Interact. 2022, 367, 110173. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yang, X. The Essential Element Manganese, Oxidative Stress, and Metabolic Diseases: Links and Interactions. Oxidative Med. Cell. Longev. 2018, 2018, 7580707. [Google Scholar] [CrossRef] [PubMed]
- Tchounwou, P.B.; Yedjou, C.G.; Patlolla, A.K.; Sutton, D.J. Heavy Metal Toxicity and the Environment. Mol. Clin. Environ. Toxicol. 2012, 101, 133–164. [Google Scholar]
- Ackerman, C.M.; Chang, C.J. Copper Signaling in the Brain and Beyond. J. Biol. Chem. 2018, 293, 4628–4635. [Google Scholar] [CrossRef] [PubMed]
- Bashir, K.; Rasheed, S.; Kobayashi, T.; Seki, M.; Nishizawa, N.K. Regulating Subcellular Metal Homeostasis: The Key to Crop Improvement. Front. Plant Sci. 2016, 7, 1192. [Google Scholar] [CrossRef] [PubMed]
- Briffa, J.; Sinagra, E.; Blundell, R. Heavy metal pollution in the environment and their toxicological effects on humans. Heliyon 2020, 6, e04691. [Google Scholar] [CrossRef] [PubMed]
- Ijomone, O.M.; Ifenatuoha, C.W.; Aluko, O.M.; Ijomone, O.K.; Aschner, M. The aging brain: Impact of heavy metal neurotoxicity. Crit. Rev. Toxicol. 2020, 50, 801–814. [Google Scholar] [CrossRef]
- Morel, J.-D.; Sauzéat, L.; Goeminne, L.J.E.; Jha, P.; Williams, E.; Houtkooper, R.H.; Aebersold, R.; Auwerx, J.; Balter, V. The mouse metallomic landscape of aging and metabolism. Nat. Commun. 2022, 13, 607. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Tian, Y.; Yuan, Z.; Zeng, Y.; Wang, S.; Fan, X.; Yang, D.; Yang, M. Iron Metabolism in Aging and Age-Related Diseases. Int. J. Mol. Sci. 2022, 23, 3612. [Google Scholar] [CrossRef] [PubMed]
- Huat, T.J.; Camats-Perna, J.; Newcombe, E.A.; Valmas, N.; Kitazawa, M.; Medeiros, R. Metal Toxicity Links to Alzheimer’s Disease and Neuroinflammation. J. Mol. Biol. 2019, 431, 1843–1868. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J. Risks of Copper and Iron Toxicity During Aging in Humans. Chem. Res. Toxicol. 2009, 23, 319–326. [Google Scholar] [CrossRef]
- Parmalee, N.L.; Aschner, M. Manganese and aging. Neurotoxicology 2016, 56, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Wang, J.; Feng, J.; Cao, B. Zinc in Cognitive Impairment and Aging. Biomolecules 2022, 12, 1000. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Mao, X.; Li, X.; Ouyang, H. Association Between Iron Status and Incident Coronary Artery Disease: A Population Based-Cohort Study. Sci. Rep. 2022, 12, 17490. [Google Scholar] [CrossRef] [PubMed]
- Silvestre, O.M.; Gonçalves, A.; Nadruz, W., Jr.; Claggett, B.; Couper, D.; Eckfeldt, J.H.; Pankow, J.S.; Anker, S.D.; Solomon, S.D. Ferritin Levels and Risk of Heart Failure—The Atherosclerosis Risk in Communities Study. Eur. J. Heart Fail. 2017, 19, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Miao, J. An Emerging Role of Defective Copper Metabolism in Heart Disease. Nutrients 2022, 14, 700. [Google Scholar] [CrossRef] [PubMed]
- Martineta, M.; Siregar, Y.; Ahmad, H. Association Between Blood Copper Levels and the Incidence of Ischemic Heart Disease. Open Access Maced. J. Med. Sci. 2022, 10, 1212–1217. [Google Scholar] [CrossRef]
- Yassine, A.A.; MacDougall, K.; Sasso, R.; Shammaa, Y.; Alsheikh, M.; Abureesh, M.; Dahabra, L.; Alshami, M.; Mulrooney, S. The Evolution of Iron-Related Comorbidities and Hospitalization in Patients with Hemochromatosis: An Analysis of the Nationwide Inpatient Sample. Blood Sci. 2023, 5, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.W.; Lee, Y.-C.; Kuo, C.-S.; Chiang, C.-H.; Chang, H.-H.; Huang, K.-C. Association of Serum Levels of Zinc, Copper, and Iron with Risk of Metabolic Syndrome. Nutrients 2021, 13, 548. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Shao, Y.; Yan, K.; Yao, T.; Liu, L.; Sun, F.; Wu, J.; Huang, Y. The Link between Trace Metal Elements and Glucose Metabolism: Evidence from Zinc, Copper, Iron, and Manganese-Mediated Metabolic Regulation. Metabolites 2023, 13, 1048. [Google Scholar] [CrossRef] [PubMed]
- Porcu, C.; Antonucci, L.; Barbaro, B.; Illi, B.; Nasi, S.; Martini, M.; Licata, A.; Miele, L.; Grieco, A.; Balsano, C. Copper/Myc/Ctr1 Interplay: A Dangerous Relationship in Hepatocellular Carcinoma. Oncotarget 2018, 9, 9325–9343. [Google Scholar] [CrossRef] [PubMed]
- Michalczyk, K.; Cymbaluk-Płoska, A. The Role of Zinc and Copper in Gynecological Malignancies. Nutrients 2020, 12, 3732. [Google Scholar] [CrossRef] [PubMed]
- Saleh, S.A.K.; Adly, H.M.; Abdelkhaliq, A.A.; Nassir, A.M. Serum Levels of Selenium, Zinc, Copper, Manganese, and Iron in Prostate Cancer Patients. Curr. Urol. 2020, 14, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Bagherzadeh, F.; Horestani, M.K.; Sadeghi, M.; Ahmadi, A.; Bahreini, R.; Fadaei, A.; Forouzandeh, S.; Hemati, S.; Mohammadi-Moghadam, F. Influence of metal ions concentration in drinking water in the development of ulcerative colitis. Int. J. Environ. Sci. Technol. 2022, 19, 3539–3546. [Google Scholar] [CrossRef]
- Bagherzadeh, F.; Mohammadi-Moghadam, F. New insights into the role of metal(loid)s in the development of ulcerative colitis: A systematic review. Environ. Sci. Pollut. Res. Int. 2023, 30, 66486–66493. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Zhang, B. The Impact of Zinc and Zinc Homeostasis on the Intestinal Mucosal Barrier and Intestinal Diseases. Biomolecules 2022, 12, 900. [Google Scholar] [CrossRef] [PubMed]
- Młyniec, K. Zinc in the Glutamatergic Theory of Depression. Curr. Neuropharmacol. 2015, 13, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.R.; Ahmed, M.U.; Mitu, S.A.; Islam, M.S.; Rahman, G.K.M.M.; Qusar, M.M.A.S.; Hasnat, A. Comparative Analysis of Serum Zinc, Copper, Manganese, Iron, Calcium, and Magnesium Level and Complexity of Interelement Relations in Generalized Anxiety Disorder Patients. Biol. Trace Elem. Res. 2013, 154, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; You, Y.; Chen, J.; Zhang, L. Copper in depressive disorder: A systematic review and meta-analysis of observational studies. Psychiatry Res. 2018, 267, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.; Khoshkhat, P.; Chamani, M.; Shahsavari, S.; Dorkoosh, F.A.; Rajabi, A.; Maniruzzaman, M.; Nokhodchi, A. In-depth multidisciplinary review of the usage, manufacturing, regulations & market of dietary supplements. J. Drug Deliv. Sci. Technol. 2022, 67, 102985. [Google Scholar]
- Augustsson, A.; Qvarforth, A.; Engström, E.; Paulukat, C.; Rodushkin, I. Trace and major elements in food supplements of different origin: Implications for daily intake levels and health risks. Toxicol. Rep. 2021, 8, 1067–1080. [Google Scholar] [CrossRef] [PubMed]
- Hurrell, R.; Egli, I. Iron bioavailability and dietary reference values1234. Am. J. Clin. Nutr. 2010, 91, 1461S–1467S. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, R.S.; Han, S.M.; Leeuwenburgh, C.; Xiao, R. Iron homeostasis and organismal aging. Ageing Res. Rev. 2021, 72, 101510. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.; Richardson, D.R. The active role of vitamin C in mammalian iron metabolism: Much more than just enhanced iron absorption! Free Radic. Biol. Med. 2014, 75, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Yiannikourides, A.; Latunde-Dada, G.O. A Short Review of Iron Metabolism and Pathophysiology of Iron Disorders. Medicines 2019, 6, 85. [Google Scholar] [CrossRef] [PubMed]
- Alshehri, A.A.; Albahli, O.M.; Alturki, A.M.; Alwasaidi, T.A.; Alfaris, N.F. Correlation of Anemia Due to Poor Iron Status with Obesity at King Fahad Medical City, Riyadh, Saudi Arabia. Cureus 2024, 16, e52424. [Google Scholar] [CrossRef] [PubMed]
- Laudisio, D.; de Alteriis, G.; Vetrani, C.; Aprano, S.; Pugliese, G.; Zumbolo, F.; Colao, A.; Savastano, S. Iron Levels and Markers of Inflammation in a Population of Adults with Severe Obesity, a Cross-Sectional Study. Nutrients 2023, 15, 4702. [Google Scholar] [CrossRef] [PubMed]
- Alshwaiyat, N.M.; Ahmad, A.; Hassan, W.M.R.W.; Al-Jamal, H.A.N. Association between obesity and iron deficiency (Review). Exp. Ther. Med. 2021, 22, 1268. [Google Scholar] [CrossRef]
- Cepeda-Lopez, A.C.; Melse-Boonstra, A.; Zimmermann, M.B.; Herter-Aeberli, I. In overweight and obese women, dietary iron absorption is reduced and the enhancement of iron absorption by ascorbic acid is one-half that in normal-weight women. Am. J. Clin. Nutr. 2015, 102, 1389–1397. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, N.U.; El-Mallah, C.; Herter-Aeberli, I.; Bissani, N.; Wehbe, N.; Obeid, O.; Zimmermann, M.B. The effect of central obesity on inflammation, hepcidin, and iron metabolism in young women. Int. J. Obes. 2020, 44, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Uña, M.; López-Mancheño, Y.; Diéguez, C.; Fernández-Rojo, M.A.; Novelle, M.G. Unraveling the Role of Leptin in Liver Function and Its Relationship with Liver Diseases. Int. J. Mol. Sci. 2020, 21, 9368. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Kuragano, T.; Kimura, T.; Nanami, M.; Hasuike, Y.; Nakanishi, T. Interplay of adipocyte and hepatocyte: Leptin upregulates hepcidin. Biochem. Biophys. Res. Commun. 2018, 495, 1548–1554. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Chen, Y.; Li, C. Serum iron concentration and leptin inversely relate, partially mediated by body mass index in American adults. Nutr. Res. 2024, 124, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dev, S.; Babitt, J.L. Overview of Iron Metabolism in Health and Disease. Hemodial. Int. 2017, 21, S6–S20. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. Iron and the Liver. Liver Int. 2016, 36, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Shahinfar, H.; Jayedi, A.; Shab-Bidar, S. Dietary iron intake and the risk of type 2 diabetes: A systematic review and dose-response meta-analysis of prospective cohort studies. Eur. J. Nutr. 2022, 61, 2279–2296. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Shen, H.-C.; Lian, T.-H.; Mao, L.; Tang, S.-X.; Sun, L.; Huang, X.-Y.; Guo, P.; Cao, C.-J.; Yu, S.-Y.; et al. Iron deposition in substantia nigra: Abnormal iron metabolism, neuroinflammatory mechanism and clinical relevance. Sci. Rep. 2017, 7, 14973. [Google Scholar] [CrossRef] [PubMed]
- Ghassaban, K.; He, N.; Sethi, S.K.; Huang, P.; Chen, S.; Yan, F.; Haacke, E.M. Regional High Iron in the Substantia Nigra Differentiates Parkinson’s Disease Patients from Healthy Controls. Front. Aging Neurosci. 2019, 11, 106. [Google Scholar] [CrossRef] [PubMed]
- Spotorno, N.; Acosta-Cabronero, J.; Stomrud, E.; Lampinen, B.; Strandberg, O.T.; van Westen, D.; Hansson, O. Relationship between cortical iron and tau aggregation in Alzheimer’s disease. Brain 2020, 143, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- van Duijn, S.; Bulk, M.; van Duinen, S.G.; Nabuurs, R.J.; van Buchem, M.A.; van der Weerd, L.; Natté, R. Cortical Iron Reflects Severity of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 60, 1533–1545. [Google Scholar]
- Bacon, B.R.; Adams, P.C.; Kowdley, K.V.; Powell, L.W.; Tavill, A.S.; American Association for the Study of Liver Diseases. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011, 54, 328–343. [Google Scholar] [CrossRef] [PubMed]
- Kowdley, K.V.; Brown, K.E.; Ahn, J.; Sundaram, V. ACG Clinical Guideline: Hereditary Hemochromatosis. Am. J. Gastroenterol. 2019, 114, 1202–1218. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. Hereditary hemochromatosis: Pathogenesis, diagnosis, and treatment. Gastroenterology 2010, 139, 393–408.e2. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Senussi, N.H.; Fertrin, K.Y.; Kowdley, K.V. Iron overload disorders. Hepatol. Commun. 2022, 6, 1842–1854. [Google Scholar] [CrossRef] [PubMed]
- Kowdley, K.V.; Modi, N.B.; Peltekian, K.; Vierling, J.M.; Ferris, C.; Valone, F.H.; Gupta, S. Rusfertide for the treatment of iron overload in HFE-related haemochromatosis: An open-label, multicentre, proof-of-concept phase 2 trial. Lancet Gastroenterol. Hepatol. 2023, 8, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Mercadante, C.J.; Prajapati, M.; Parmar, J.H.; Conboy, H.L.; Dash, M.E.; Pettiglio, M.A.; Herrera, C.; Bu, J.T.; Stopa, E.G.; Mendes, P.; et al. Gastrointestinal Iron Excretion and Reversal of Iron Excess in a Mouse Model of Inherited Iron Excess. Haematologica 2018, 104, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Kalleda, N.; Flace, A.; Altermatt, P.; Ingoglia, G.; Doucerain, C.; Nyffenegger, N.; Dürrenberger, F.; Manolova, V. Ferroportin inhibitor vamifeport ameliorates ineffective erythropoiesis in a mouse model of β-thalassemia with blood transfusions. Haematologica 2023, 108, 2703–2714. [Google Scholar] [CrossRef] [PubMed]
- Nyffenegger, N.; Flace, A.; Doucerain, C.; Dürrenberger, F.; Manolova, V. The Oral Ferroportin Inhibitor VIT-2763 Improves Erythropoiesis without Interfering with Iron Chelation Therapy in a Mouse Model of β-Thalassemia. Int. J. Mol. Sci. 2021, 22, 873. [Google Scholar] [CrossRef] [PubMed]
- Richard, F.; van Lier, J.J.; Roubert, B.; Haboubi, T.; Göhring, U.M.; Dürrenberger, F. Oral ferroportin inhibitor VIT-2763: First-in-human, phase 1 study in healthy volunteers. Am. J. Hematol. 2020, 95, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Maares, M.; Haase, H. A Guide to Human Zinc Absorption: General Overview and Recent Advances of In Vitro Intestinal Models. Nutrients 2020, 12, 762. [Google Scholar] [CrossRef]
- Roohani, N.; Hurrell, R.; Kelishadi, R.; Schulin, R. Zinc and its importance for human health: An integrative review. J. Res. Med. Sci. 2013, 18, 144–157. [Google Scholar] [PubMed]
- Chen, B.; Yu, P.; Chan, W.N.; Xie, F.; Zhang, Y.; Liang, L.; Leung, K.T.; Lo, K.W.; Yu, J.; Tse, G.M.K.; et al. Cellular zinc metabolism and zinc signaling: From biological functions to diseases and therapeutic targets. Signal Transduct. Target. Ther. 2024, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc in Cellular Regulation: The Nature and Significance of “Zinc Signals”. Int. J. Mol. Sci. 2017, 18, 2285. [Google Scholar] [CrossRef] [PubMed]
- Ruz, M.; Carrasco, F.; Rojas, P.; Basfi-Fer, K.; Hernández, M.C.; Pérez, A. Nutritional Effects of Zinc on Metabolic Syndrome and Type 2 Diabetes: Mechanisms and Main Findings in Human Studies. Biol. Trace Elem. Res. 2019, 188, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.L.; Failla, M.L.; Smith, J.C. Influence of Genetic Obesity on Tissue Concentrations of Zinc, Copper, Manganese and Iron in Mice. J. Nutr. 1986, 116, 1432–1441. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.L.; Failla, M.L. Zinc Metabolism in Genetically Obese (ob/ob) Mice2. J. Nutr. 1987, 117, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-D.; Lin, P.-Y.; Cheng, V.; Lin, W.-H. Zinc supplementation aggravates body fat accumulation in genetically obese mice and dietary-obese mice. Biol. Trace Elem. Res. 1996, 52, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Övermöhle, C.; Rimbach, G.; Waniek, S.; Strathmann, E.A.; Liedtke, T.; Stürmer, P.; Both, M.; Weber, K.S.; Lieb, W. Association of Plasma Zinc and Copper with Body Composition, Lipids and Inflammation in a Cross-Sectional General Population Sample from Germany. Nutrients 2023, 15, 4460. [Google Scholar] [CrossRef]
- Bolatimi, O.E.; Head, K.Z.; Luo, J.; Gripshover, T.C.; Lin, Q.; Adiele, N.V.; Wason, W.H.; Wilkerson, C.; Cave, M.C.; Young, J.L. Can Zinc Supplementation Attenuate High Fat Diet-Induced Non-Alcoholic Fatty Liver Disease? Int. J. Mol. Sci. 2023, 24, 1763. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Zhang, Z.; Liu, S.; Aluo, Z.; Zhang, L.; Yu, L.; Li, Y.; Song, Z.; Zhou, L. Zinc Supplementation Alleviates Lipid and Glucose Metabolic Disorders Induced by a High-Fat Diet. J. Agric. Food Chem. 2020, 68, 5189–5200. [Google Scholar] [CrossRef] [PubMed]
- Khorsandi, H.; Nikpayam, O.; Yousefi, R.; Parandoosh, M.; Hosseinzadeh, N.; Saidpour, A.; Ghorbani, A. Zinc supplementation improves body weight management, inflammatory biomarkers and insulin resistance in individuals with obesity: A randomized, placebo-controlled, double-blind trial. Diabetol. Metab. Syndr. 2019, 11, 101. [Google Scholar] [CrossRef] [PubMed]
- Bashandy, S.A.E.; El-Seidy, A.M.A.; Ibrahim, F.A.A.; Abdelrahman, S.S.; Moussa, S.A.A.; ElBaset, M.A. Zinc nanoparticles ameliorated obesity-induced cardiovascular disease: Role of metabolic syndrome and iron overload. Sci. Rep. 2023, 13, 16010. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cao, D.; Huang, Y.; Chen, B.; Chen, Z.; Wang, R.; Dong, Q.; Wei, Q.; Liu, L. Zinc Intakes and Health Outcomes: An Umbrella Review. Front. Nutr. 2022, 9, 798078. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jing, X.-P.; Zhang, S.-P.; Gu, R.-X.; Tang, F.-X.; Wang, X.-L.; Xiong, Y.; Qiu, M.; Sun, X.-Y.; Ke, D.; et al. High Dose Zinc Supplementation Induces Hippocampal Zinc Deficiency and Memory Impairment with Inhibition of BDNF Signaling. PLoS ONE 2013, 8, e55384. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.B.; Kim, J.A.; Park, S.; Lee, J.Y. Associative Interactions among Zinc, Apolipoprotein E, and Amyloid-β in the Amyloid Pathology. Int. J. Mol. Sci. 2020, 21, 802. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, B.H.; Orhan, F.; Bruno, S.; Oliveira, A.O.; Sparding, T.; Landen, M.; Sellgren, C.M. Serum concentration of zinc is elevated in clinically stable bipolar disorder patients. Brain Behav. 2022, 12, e2472. [Google Scholar] [CrossRef] [PubMed]
- Doğan, E.; Güzel, A.; Çiftçi, T.; Aycan, İ.; Çelik, F.; Çetin, B.; Kavak, G.Ö. Zinc Phosphide Poisoning. Case Rep. Crit. Care 2014, 2014, 589712. [Google Scholar] [CrossRef] [PubMed]
- Sagah, G.; Oreby, M.M.; El-Gharbawy, R.M.; Fathy, A.S.A. Evaluation of Potential Oxidative Stress in Egyptian Patients with Acute Zinc Phosphide Poisoning and the Role of Vitamin C. Int. J. Health Sci. 2015, 9, 373–382. [Google Scholar] [CrossRef]
- Nriagu, J. Zinc Toxicity in Humans; School of Public Health, University of Michigan: Ann Arbor, MI, USA, 2007; pp. 1–7. [Google Scholar]
- Plum, L.M.; Rink, L.; Haase, H. The essential toxin: Impact of zinc on human health. Int. J. Environ. Res. Public Health 2010, 7, 1342–1365. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Carmichael, M.F. Zinc-Induced Copper Deficiency as a Rare Cause of Neurological Deficit and Anemia. Cureus 2023, 15, e43856. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.; Yacoubian, C.; Watson, N.; Morrison, I. The risk of copper deficiency in patients prescribed zinc supplements. J. Clin. Pathol. 2015, 68, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Agnew, U.M.; Slesinger, T.L. Zinc Toxicity. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2024. [Google Scholar]
- Linder, M.C. Copper Homeostasis in Mammals, with Emphasis on Secretion and Excretion. A Review. Int. J. Mol. Sci. 2020, 21, 4932. [Google Scholar] [CrossRef] [PubMed]
- Bost, M.; Houdart, S.; Oberli, M.; Kalonji, E.; Huneau, J.-F.; Margaritis, I. Dietary copper and human health: Current evidence and unresolved issues. J. Trace Elem. Med. Biol. 2016, 35, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Blockhuys, S.; Celauro, E.; Hildesjö, C.; Feizi, A.; Stål, O.; Fierro-González, J.C.; Wittung-Stafshede, P. Defining the human copper proteome and analysis of its expression variation in cancers. Metallomics 2017, 9, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Tsang, T.; Davis, C.I.; Brady, D.C. Copper biology. Curr. Biol. 2021, 31, R421–R427. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.H.; Maryon, E.B. How Mammalian Cells Acquire Copper: An Essential but Potentially Toxic Metal. Biophys. J. 2016, 110, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jiang, Y.; Shi, H.; Peng, Y.; Fan, X.; Li, C. The molecular mechanisms of copper metabolism and its roles in human diseases. Pflüg. Arch. Eur. J. Physiol. 2020, 472, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Ciofi-Baffoni, S.; Kozyreva, T.; Zovo, K.; Palumaa, P. Affinity gradients drive copper to cellular destinations. Nature 2010, 465, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Członkowska, A.; Litwin, T.; Dusek, P.; Ferenci, P.; Lutsenko, S.; Medici, V.; Rybakowski, J.K.; Weiss, K.H.; Schilsky, M.L. Wilson disease. Nat. Rev. Dis. Primers 2018, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.A.; Schilsky, M.L.; American Association for the Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: An update. Hepatology 2008, 47, 2089–2111. [Google Scholar] [CrossRef] [PubMed]
- Kirsipuu, T.; Zadorožnaja, A.; Smirnova, J.; Friedemann, M.; Plitz, T.; Tõugu, V.; Palumaa, P. Copper(II)-binding equilibria in human blood. Sci. Rep. 2020, 10, 5686. [Google Scholar] [CrossRef] [PubMed]
- Lutsenko, S. Human copper homeostasis: A network of interconnected pathways. Curr. Opin. Chem. Biol. 2010, 14, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.C.; Hazegh-Azam, M. Copper biochemistry and molecular biology. Am. J. Clin. Nutr. 1996, 63, 797S–811S. [Google Scholar] [CrossRef] [PubMed]
- Przybyłkowski, A.; Gromadzka, G.; Wawer, A.; Grygorowicz, T.; Cybulska, A.; Członkowska, A. Intestinal Expression of Metal Transporters in Wilson’s Disease. Biometals 2013, 26, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Scheinberg, I.H.; Sternlieb, I. Wilson Disease and Idiopathic Copper Toxicosis. Am. J. Clin. Nutr. 1996, 63, 842S–845S. [Google Scholar] [CrossRef] [PubMed]
- Hedera, P. Clinical Management of Wilson Disease. Ann. Transl. Med. 2019, 7, S66. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Wilson’s disease. J. Hepatol. 2012, 56, 671–685. [Google Scholar] [CrossRef] [PubMed]
- Kasztelan-Szczerbinska, B.; Cichoz-Lach, H. Wilson’s Disease: An Update on the Diagnostic Workup and Management. J. Clin. Med. 2021, 10, 5097. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Müller, W.; Feichtinger, H. Idiopathic Copper Toxicosis. Am. J. Clin. Nutr. 1998, 67, 1082S–1086S. [Google Scholar] [CrossRef]
- Taylor, A.A.; Tsuji, J.S.; Garry, M.R.; McArdle, M.E.; Goodfellow, W.L.; Adams, W.J.; Menzie, C.A. Critical Review of Exposure and Effects: Implications for Setting Regulatory Health Criteria for Ingested Copper. Environ. Manag. 2020, 65, 131–159. [Google Scholar] [CrossRef]
- Liu, J.; Chen, C.; Liu, Y.; Sun, X.; Ding, X.; Qiu, L.; Han, P.; Kang, Y.J. Trientine Selectively Delivers Copper to the Heart and Suppresses Pressure Overload-Induced Cardiac Hypertrophy in Rats. Exp. Biol. Med. 2018, 243, 1141–1152. [Google Scholar] [CrossRef]
- Rahimzadeh, M.R.; Kazemi, S.; Moghadamnia, A.A. Copper Poisoning with Emphasis on Its Clinical Manifestations and Treatment of Intoxication. Adv. Public Health 2024, 2024, 6001014. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, H.; Amarsingh, G.V.; Cheung, C.C.H.; Hogl, S.; Narayanan, U.; Zhang, L.; McHarg, S.; Xu, J.; Gong, D.; et al. Diabetic Cardiomyopathy Is Associated with Defective Myocellular Copper Regulation and Both Defects Are Rectified by Divalent Copper Chelation. Cardiovasc. Diabetol. 2014, 13, 100. [Google Scholar] [CrossRef]
- Sailer, J.; Nagel, J.; Akdogan, B.; Jauch, A.T.; Engler, J.; Knolle, P.A.; Zischka, H. Deadly excess copper. Redox Biol. 2024, 75, 103256. [Google Scholar] [CrossRef] [PubMed]
- Perestrelo, A.P.; Miranda, G.F.L.; Gonçalves, M.I.; Belino, C.; Ballesteros, R. Chronic Copper Sulfate Poisoning. Eur. J. Case Rep. Intern. Med. 2021, 8, 002309. [Google Scholar] [PubMed]
- Perumal, V.; Narayanan, N.; Mohanan, J.; Rangarajan, J.; Kalifa, M.; Perincheri, U.M.; Palanisamy, E.; Selvaraj, P. S Study on Clinical Profile, Complications and Outcome of Copper Sulphate Poisoning at a Tertiary Care Centre in South India. J. Evolut. Med. Dent. Sci. 2016, 5, 3440–3444. [Google Scholar] [CrossRef]
- Tharmaraj, D.; Kerr, P.G. Haemolysis in haemodialysis. Nephrology 2017, 22, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Alengebawy, A.; Abdelkhalek, S.T.; Qureshi, S.R.; Wang, M.-Q. Heavy Metals and Pesticides Toxicity in Agricultural Soil and Plants: Ecological Risks and Human Health Implications. Toxics 2021, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, C. Review of Copper Provision in the Parenteral Nutrition of Adults. Nutr. Clin. Pract. 2017, 32, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Singer, P.; Berger, M.M.; Berghe, G.V.D.; Biolo, G.; Calder, P.; Forbes, A.; Griffiths, R.; Kreyman, G.; Leverve, X.; Pichard, C. ESPEN Guidelines on Parenteral Nutrition: Intensive care. Clin. Nutr. 2009, 28, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, L.; Shan, R.; Wang, C. Associations between dietary copper intake, general obesity and abdominal obesity risk: A nationwide cohort study in China. Front. Nutr. 2022, 9, 1009721. [Google Scholar] [CrossRef] [PubMed]
- Bulka, C.M.; Persky, V.W.; Daviglus, M.L.; Durazo-Arvizu, R.A.; Argos, M. Multiple metal exposures and metabolic syndrome: A cross-sectional analysis of the National Health and Nutrition Examination Survey 2011–2014. Environ. Res. 2019, 168, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Zhang, C.; Bu, J. Relationship between Selected Serum Metallic Elements and Obesity in Children and Adolescent in the U.S. Nutrients 2017, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.-L.; Wang, C.-W.; Wu, D.-W.; Chen, S.-C.; Hung, C.-H.; Kuo, C.-H. Associations of Heavy Metals with Metabolic Syndrome and Anthropometric Indices. Nutrients 2020, 12, 2666. [Google Scholar] [CrossRef] [PubMed]
- Baj, J.; Flieger, W.; Barbachowska, A.; Kowalska, B.; Flieger, M.; Forma, A.; Teresiński, G.; Portincasa, P.; Buszewicz, G.; Radzikowska-Büchner, E.; et al. Consequences of Disturbing Manganese Homeostasis. Int. J. Mol. Sci. 2023, 24, 14959. [Google Scholar] [CrossRef] [PubMed]
- Winslow, J.W.; Limesand, K.H.; Zhao, N. The Functions of ZIP8, ZIP14, and ZnT10 in the Regulation of Systemic Manganese Homeostasis. Int. J. Mol. Sci. 2020, 21, 3304. [Google Scholar] [CrossRef] [PubMed]
- O’Neal, S.L.; Zheng, W. Manganese Toxicity Upon Overexposure: A Decade in Review. Curr. Environ. Health Rep. 2015, 2, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Fitsanakis, V.A.; Zhang, N.; Garcia, S.; Aschner, M. Manganese (Mn) and iron (Fe): Interdependency of transport and regulation. Neurotoxicol. Res. 2010, 18, 124–131. [Google Scholar] [CrossRef]
- Gurol, K.C.; Aschner, M.; Smith, D.R.; Mukhopadhyay, S. Role of excretion in manganese homeostasis and neurotoxicity: A historical perspective. Am. J. Physiol. Gastrointest. Liver Physiol. 2022, 322, G79–G92. [Google Scholar] [CrossRef] [PubMed]
- Oulhote, Y.; Mergler, D.; Bouchard, M.F. Sex- and age-differences in blood manganese levels in the U.S. general population: National health and nutrition examination survey 2011–2012. Environ. Health 2014, 13, 87. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Bornhorst, J.; Aschner, M. Manganese Metabolism in Humans. Front. Biosci. Elite 2018, 23, 1655–1679. [Google Scholar]
- Chesi, A.; Kilaru, A.; Fang, X.; Cooper, A.A.; Gitler, A.D. The Role of the Parkinson’s Disease Gene PARK9 in Essential Cellular Pathways and the Manganese Homeostasis Network in Yeast. PLoS ONE 2012, 7, e34178. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Xia, N.; Wong, A.; Bakshi, R.; Cahill, C.M. Targeting the Iron-Response Elements of the mRNAs for the Alzheimer’s Amyloid Precursor Protein and Ferritin to Treat Acute Lead and Manganese Neurotoxicity. Int. J. Mol. Sci. 2019, 20, 994. [Google Scholar] [CrossRef] [PubMed]
- Sidoryk-Węgrzynowicz, M.; Aschner, M. Manganese Toxicity in the Central Nervous System: The Glutamine/Glutamate-γ-aminobutyric Acid Cycle. J. Int. Med. 2013, 273, 466–477. [Google Scholar] [CrossRef]
- Smith, M.R.; Fernandes, J.; Go, Y.-M.; Jones, D.P. Redox dynamics of manganese as a mitochondrial life-death switch. Biochem. Biophys. Res. Commun. 2017, 482, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Yang, H.; Tian, X.; Wang, H.; Zhou, T.; Zhang, S.; Yu, J.; Zhang, T.; Fan, D.; Guo, X.; et al. High Manganese, a Risk for Alzheimer’s Disease: High Manganese Induces Amyloid-Β Related Cognitive Impairment. J. Alzheimer’s Dis. 2014, 42, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Studer, J.M.; Schweer, W.P.; Gabler, N.K.; Ross, J.W. Functions of manganese in reproduction. Anim. Reprod. Sci. 2022, 238, 106924. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, D.; Rudrashetti, A.P.; Rajasekaran, S. The impact of environmental and occupational exposures of manganese on pulmonary, hepatic, and renal functions. J. Appl. Toxicol. 2022, 42, 103–129. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Su, X.; Su, D.; Zeng, F.; Wang, M.H.; Huang, L.; Huang, E.; Zhu, Y.; Zhao, D.; He, D.; et al. Dietary intake of manganese and the risk of the metabolic syndrome in a Chinese population. Br. J. Nutr. 2016, 116, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Tao, C.; Huang, Y.; Huang, X.; Li, Z.; Fan, Y.; Zhang, Y.; Wan, T.; Lu, L.; Xu, Q.; Wu, W.; et al. Association between Blood Manganese Levels and Visceral Adipose Tissue in the United States: A Population-Based Study. Nutrients 2022, 14, 4770. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Karvonen-Gutierrez, C.A.; Herman, W.H.; Mukherjee, B.; Park, S.K. Metals and risk of incident metabolic syndrome in a prospective cohort of midlife women in the United States. Environ. Res. 2022, 210, 112976. [Google Scholar] [CrossRef] [PubMed]
- Quadri, M.; Federico, A.; Zhao, T.; Breedveld, G.J.; Battisti, C.; Delnooz, C.; Severijnen, L.-A.; Mammarella, L.D.T.; Mignarri, A.; Monti, L.; et al. Mutations in SLC30A10 Cause Parkinsonism and Dystonia with Hypermanganesemia, Polycythemia, and Chronic Liver Disease. Am. J. Hum. Genet. 2012, 90, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Das, A.P.; Ghosh, S.; Mohanty, S.; Sukla, L. Consequences of manganese compounds: A review. Toxicol. Environ. Chem. 2014, 96, 981–997. [Google Scholar] [CrossRef]
- Sobańska, Z.; Roszak, J.; Kowalczyk, K.; Stępnik, M. Applications and Biological Activity of Nanoparticles of Manganese and Manganese Oxides in in Vitro and in Vivo Models. Nanomaterials 2021, 11, 1084. [Google Scholar] [CrossRef] [PubMed]
- Stamelou, M.; Tuschl, K.; Chong, W.K.; Burroughs, A.K.; Mills, P.B.; Bhatia, K.P.; Clayton, P.T. Dystonia with brain manganese accumulation resulting from SLC30A10 mutations: A new treatable disorder. Mov. Disord. 2012, 27, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Tuschl, K.; Mills, P.B.; Clayton, P.T. Chapter Twelve—Manganese and the Brain. In International Review of Neurobiology; Bhatia, K.P., Schneider, S.A., Eds.; Academic Press: Cambridge, MA, USA, 2013; pp. 277–312. [Google Scholar]
- Jiang, Y.M.; Mo, X.-A.M.; Du, F.-Q.; Fu, X.; Zhu, X.-Y.; Gao, H.-Y.; Xie, J.-L.; Liao, F.-L.; Pira, E.; Zheng, W. Effective treatment of manganese-induced occupational Parkinsonism with p-aminosalicylic acid: A case of 17-year follow-up study. J. Occup. Environ. Med. 2006, 48, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Baros, S.; Valko, M. Redox active metal-induced oxidative stress in biological systems. Trans. Met. Chem. 2012, 37, 127–134. [Google Scholar] [CrossRef]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Checa, J.; Aran, J.M. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J. Inflamm. Res. 2020, 13, 1057–1073. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Shang, P. The significance, trafficking and determination of labile iron in cytosol, mitochondria and lysosomes. Metallomics 2018, 10, 899–916. [Google Scholar] [CrossRef] [PubMed]
- Robello, E.; Galatro, A.; Puntarulo, S. Iron and Its Catalytic Properties on Radical Generation. In Reactive Oxygen, Nitrogen and Sulfur Species in Plants; Wiley: Hoboken, NJ, USA, 2019; pp. 39–52. [Google Scholar]
- Ikeda, Y.; Satoh, A.; Horinouchi, Y.; Hamano, H.; Watanabe, H.; Imao, M.; Imanishi, M.; Zamami, Y.; Takechi, K.; Izawa-Ishizawa, Y.; et al. Iron Accumulation Causes Impaired Myogenesis Correlated with MAPK Signaling Pathway Inhibition by Oxidative Stress. FASEB J. 2019, 33, 9551–9564. [Google Scholar] [CrossRef] [PubMed]
- Messner, D.J.; Rhieu, B.H.; Kowdley, K.V. Iron Overload Causes Oxidative Stress and Impaired Insulin Signaling in AML-12 Hepatocytes. Dig. Dis. Sci. 2013, 58, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Molinari, C.; Morsanuto, V.; Ghirlanda, S.; Ruga, S.; Notte, F.; Gaetano, L.; Uberti, F. Role of Combined Lipoic Acid and Vitamin D3 on Astrocytes as a Way to Prevent Brain Ageing by Induced Oxidative Stress and Iron Accumulation. Oxidative Med. Cell. Longev. 2019, 2019, 2843121. [Google Scholar] [CrossRef] [PubMed]
- Guan, P.; Sun, Z.-M.; Luo, L.-F.; Zhao, Y.-S.; Yang, S.-C.; Yu, F.-Y.; Wang, N.; Ji, E.-S. Hydrogen Gas Alleviates Chronic Intermittent Hypoxia-Induced Renal Injury Through Reducing Iron Overload. Molecules 2019, 24, 1184. [Google Scholar] [CrossRef] [PubMed]
- Rathore, K.I.; Kerr, B.J.; Redensek, A.; López-Vales, R.; Jeong, S.Y.; Ponka, P.; David, S. Ceruloplasmin Protects Injured Spinal Cord from Iron-Mediated Oxidative Damage. J. Neurosci. 2008, 28, 12736–12747. [Google Scholar] [CrossRef] [PubMed]
- Hernansanz-Agustín, P.; Enríquez, J.A. Generation of Reactive Oxygen Species by Mitochondria. Antioxidants 2021, 10, 415. [Google Scholar] [CrossRef]
- Rydz, L.; Wróbel, M.; Jurkowska, H. Sulfur Administration in Fe–S Cluster Homeostasis. Antioxidants 2021, 10, 1738. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Chan, G.C.; Ye, J.; Lian, Q.; Chen, J.; Yang, M. Thrombopoietin Protects Cardiomyocytes from Iron-Overload Induced Oxidative Stress and Mitochondrial Injury. Cell. Physiol. Biochem. 2015, 36, 2063–2071. [Google Scholar] [CrossRef] [PubMed]
- Gordan, R.; Fefelova, N.; Gwathmey, J.K.; Xie, L.-H. Iron Overload, Oxidative Stress and Calcium Mishandling in Cardiomyocytes: Role of the Mitochondrial Permeability Transition Pore. Antioxidants 2020, 9, 758. [Google Scholar] [CrossRef] [PubMed]
- Volani, C.; Doerrier, C.; Demetz, E.; Haschka, D.; Paglia, G.; Lavdas, A.A.; Gnaiger, E.; Weiss, G. Dietary iron loading negatively affects liver mitochondrial function†. Metallomics 2017, 9, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, A.; Chakraborty, K.; Shukla, A. Cellular copper homeostasis: Current concepts on its interplay with glutathione homeostasis and its implication in physiology and human diseases. Metallomics 2017, 9, 1376–1388. [Google Scholar] [CrossRef] [PubMed]
- Krężel, A.; Maret, W. The Functions of Metamorphic Metallothioneins in Zinc and Copper Metabolism. Int. J. Mol. Sci. 2017, 18, 1237. [Google Scholar] [CrossRef] [PubMed]
- Zischka, H.; Lichtmannegger, J. Chapter 6—Cellular Copper Toxicity: A Critical Appraisal of Fenton-Chemistry—Based Oxidative Stress in Wilson Disease. In Wilson Disease; Weiss, K.H., Schilsky, M., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 65–81. [Google Scholar]
- Barber, R.G.; Grenier, Z.A.; Burkhead, J.L. Copper Toxicity Is Not Just Oxidative Damage: Zinc Systems and Insight from Wilson Disease. Biomedicines 2021, 9, 316. [Google Scholar] [CrossRef] [PubMed]
- Rae, T.D.; Schmidt, P.J.; Pufahl, R.A.; Culotta, V.C.; O’Halloran, T.V. Undetectable intracellular free copper: The requirement of a copper chaperone for superoxide dismutase. Science 1999, 284, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Borchard, S.; Bork, F.; Rieder, T.; Eberhagen, C.; Popper, B.; Lichtmannegger, J.; Schmitt, S.; Adamski, J.; Klingenspor, M.; Weiss, K.-H.; et al. The exceptional sensitivity of brain mitochondria to copper. Toxicology 2018, 51, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Zischka, H.; Lichtmannegger, J.; Schmitt, S.; Jägemann, N.; Schulz, S.; Wartini, D.; Jennen, L.; Rust, C.; Larochette, N.; Galluzzi, L.; et al. Liver mitochondrial membrane crosslinking and destruction in a rat model of Wilson disease. J. Clin. Investig. 2011, 121, 1508–1518. [Google Scholar] [CrossRef] [PubMed]
- Falcone, E.; Stellato, F.; Vileno, B.; Bouraguba, M.; Lebrun, V.; Ilbert, M.; Morante, S.; Faller, P. Revisiting the pro-oxidant activity of copper: Interplay of ascorbate, cysteine, and glutathione. Metallomics 2023, 15, mfad040. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Coy, S. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Zhu, H.; Sheng, F.; Tian, Y.; Zhou, J.; Chen, Y.; Song, L.; Lin, J. Activation of the MAPK11/12/13/14 (P38 MAPK) Pathway Regulates the Transcription of Autophagy Genes in Response to Oxidative Stress Induced by a Novel Copper Complex in HeLa Cells. Autophagy 2014, 10, 1285–1300. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Guo, H.; Jian, Z.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Copper Induces Oxidative Stress and Apoptosis in the Mouse Liver. Oxidative Med. Cell. Longev. 2020, 2020, 1359164. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Pei, R.; Zhang, Z.; Liao, J.; Yu, W.; Qiao, N.; Han, Q.; Li, Y.; Hu, L.; Guo, J.; et al. Copper induces oxidative stress and apoptosis through mitochondria-mediated pathway in chicken hepatocytes. Toxicology 2019, 54, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R. Critical Role of Zinc as Either an Antioxidant or a Prooxidant in Cellular Systems. Oxidative Med. Cell. Longev. 2018, 2018, 9156285. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Lai, C.; Chen, F.; Ding, Y.; Zhou, Y.; Su, S.; Ni, R.; Tang, Z. Emodin Protects SH-SY5Y Cells Against Zinc-Induced Synaptic Impairment and Oxidative Stress Through the ERK1/2 Pathway. Front. Pharmacol. 2022, 13, 821521. [Google Scholar] [CrossRef] [PubMed]
- Slepchenko, K.G.; Lu, Q.; Li, Y.V. Zinc wave during the treatment of hypoxia is required for initial reactive oxygen species activation in mitochondria. Int. J. Physiol. Pathophysiol. Pharmacol. 2016, 8, 44–51. [Google Scholar] [PubMed]
- Noh, K.M.; Koh, J.Y. Induction and Activation by Zinc of NADPH Oxidase in Cultured Cortical Neurons and Astrocytes. J. Neurosci. 2000, 20, RC111. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Liu, K.J.; Qi, Z. Zinc causes the death of hypoxic astrocytes by inducing ROS production through mitochondria dysfunction. Biophys. Rep. 2019, 5, 209–217. [Google Scholar] [CrossRef]
- Neely, M.D.; Davison, C.A.; Aschner, M.; Bowman, A.B. From the Cover: Manganese and Rotenone-Induced Oxidative Stress Signatures Differ in iPSC-Derived Human Dopamine Neurons. Toxicol. Sci. 2017, 159, 366–379. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, S.; Shi, N.; Lian, S.; Lin, W. Nrf2/Ho-1 Pathway Activation by Manganese Is Associated with Reactive Oxygen Species and Ubiquitin–proteasome Pathway, Not MAPKs Signaling. J. Appl. Toxicol. 2011, 31, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Cheng, H.; Su, C.; Chen, P.; Yang, X. PI3K/Akt Signaling Pathway Ameliorates Oxidative Stress-Induced Apoptosis upon Manganese Exposure in PC12 Cells. Biol. Trace Elem. Res. 2022, 200, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Barber, D.S.; Zhang, P.; Liu, B. Complex II of the Mitochondrial Respiratory Chain Is the Key Mediator of Divalent Manganese-Induced Hydrogen Peroxide Production in Microglia. Toxicol. Sci. 2013, 132, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta BBA Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef] [PubMed]
- Rhee, J.W.; Yi, H.; Thomas, D.; Lam, C.K.; Belbachir, N.; Tian, L.; Qin, X.; Malisa, J.; Lau, E.; Paik, D.T.; et al. Modeling Secondary Iron Overload Cardiomyopathy with Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Cell Rep. 2020, 32, 107886. [Google Scholar] [CrossRef] [PubMed]
- Cherrak, S.A.; Mokhtari-Soulimane, N.; Berroukeche, F.; Bensenane, B.; Cherbonnel, A.; Merzouk, H.; Elhabiri, M. In Vitro Antioxidant versus Metal Ion Chelating Properties of Flavonoids: A Structure-Activity Investigation. PLoS ONE 2016, 11, e0165575. [Google Scholar] [CrossRef] [PubMed]
- Kejík, Z.; Kaplánek, R.; Masařík, M.; Babula, P.; Matkowski, A.; Filipenský, P.; Veselá, K.; Gburek, J.; Sýkora, D.; Martásek, P.; et al. Iron Complexes of Flavonoids-Antioxidant Capacity and Beyond. Int. J. Mol. Sci. 2021, 22, 646. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhong, Y.; Gao, C.; Li, J. Myricetin ameliorates scopolamine-induced memory impairment in mice via inhibiting acetylcholinesterase and down-regulating brain iron. Biochem. Biophys. Res. Commun. 2017, 490, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Mu, M.; An, P.; Wu, Q.; Shen, X.; Shao, D.; Wang, H.; Zhang, Y.; Zhang, S.; Yao, H.; Min, J.; et al. The dietary flavonoid myricetin regulates iron homeostasis by suppressing hepcidin expression. J. Nutr. Biochem. 2016, 30, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Liu, S.; Pan, D.; Jia, Y.; Ma, Z.-G. Myricetin reduces cytotoxicity by suppressing hepcidin expression in MES23.5 cells. Neural Regen. Res. 2021, 16, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, N.; Gadde, R.; Betharia, S. Dithiolethiones Protect Against Iron Overload-Induced Cytotoxicity and Serve as Ferroptosis Inhibitors in U-87 MG Cells. Neurochem. Res. 2023, 48, 2542–2551. [Google Scholar] [CrossRef] [PubMed]
- Rosiak, N.; Cielecka-Piontek, J.; Skibiński, R.; Lewandowska, K.; Bednarski, W.; Zalewski, P. Do Rutin and Quercetin Retain Their Structure and Radical Scavenging Activity after Exposure to Radiation? Molecules 2023, 28, 2713. [Google Scholar] [CrossRef] [PubMed]
- Hamad, R.S. Rutin, a Flavonoid Compound Derived from Garlic, as a Potential Immunomodulatory and Anti-Inflammatory Agent against Murine Schistosomiasis mansoni. Nutrients 2023, 15, 1206. [Google Scholar] [CrossRef] [PubMed]
- Pervin, M.; Unno, K.; Ohishi, T.; Tanabe, H.; Miyoshi, N.; Nakamura, Y. Beneficial Effects of Green Tea Catechins on Neurodegenerative Diseases. Molecules 2018, 23, 1297. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.-L.; Chen, H.-H.; Xu, J.; Huang, M.-Y.; Wang, J.-F.; Shen, H.-J.; Shen, S.-X.; Gao, C.-X.; Qian, C.-D. Myricetin Acts as an Inhibitor of Type II NADH Dehydrogenase from Staphylococcus aureus. Molecules 2024, 29, 2354. [Google Scholar] [CrossRef] [PubMed]
- Verdura, S.; Encinar, J.A.; Teixidor, E.; Segura-Carretero, A.; Micol, V.; Cuyàs, E.; Bosch-Barrera, J.; Menendez, J.A. Silibinin Overcomes EMT-Driven Lung Cancer Resistance to New-Generation ALK Inhibitors. Cancers 2022, 14, 6101. [Google Scholar] [CrossRef] [PubMed]
- Espíndola, K.M.M.; Varela, E.L.P.; Albuquerque, R.d.F.V.d.; Figueiredo, R.A.; dos Santos, S.M.; Malcher, N.S.; Seabra, P.S.d.S.; Fonseca, A.D.N.; Sousa, K.M.d.A.; de Oliveira, S.B.B.; et al. Alpha-Lipoic Acid and Its Enantiomers Prevent Methemoglobin Formation and DNA Damage Induced by Dapsone Hydroxylamine: Molecular Mechanism and Antioxidant Action. Int. J. Mol. Sci. 2023, 24, 57. [Google Scholar] [CrossRef] [PubMed]
- Kukoc-Modun, L.; Kraljevic, T.; Tsikas, D.; Spassov, T.G.; Kolev, S.D. Determination of N-Acetyl-L-cysteine Ethyl Ester (NACET) by Sequential Injection Analysis. Sensors 2024, 24, 312. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Zhou, J.; Zhao, C.-N.; Gan, R.-Y.; Li, H.-B. Health Benefits and Molecular Mechanisms of Resveratrol: A Narrative Review. Foods 2020, 9, 340. [Google Scholar] [CrossRef] [PubMed]
- Janus, E.; Pinheiro, L.R.; Nowak, A.; Kucharska, E.; Świątek, E.; Podolak, N.; Perużyńska, M.; Piotrowska, K.; Duchnik, W.; Kucharski, Ł.; et al. New Ferulic Acid and Amino Acid Derivatives with Increased Cosmeceutical and Pharmaceutical Potential. Pharmaceutics 2023, 15, 117. [Google Scholar] [CrossRef] [PubMed]
- Bouhlel, J.; Caffin, F.; Gros-Désormeaux, F.; Douki, T.; Benoist, J.-F.; Castelli, F.A.; Chu-Van, E.; Piérard, C.; Junot, C.; Fenaille, F. Metabolomics Analysis of Rabbit Plasma after Ocular Exposure to Vapors of Sulfur Mustard. Metabolites 2024, 14, 349. [Google Scholar] [CrossRef]
- Nukala, U.; Thakkar, S.; Krager, K.J.; Breen, P.J.; Compadre, C.M.; Aykin-Burns, N. Antioxidant Tocols as Radiation Countermeasures (Challenges to be Addressed to Use Tocols as Radiation Countermeasures in Humans). Antioxidants 2018, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Giordano, M.E.; Caricato, R.; Lionetto, M.G. Concentration Dependence of the Antioxidant and Prooxidant Activity of Trolox in HeLa Cells: Involvement in the Induction of Apoptotic Volume Decrease. Antioxidants 2020, 9, 1058. [Google Scholar] [CrossRef]
- Liang, X.; Pacuła-Miszewska, A.J.; Vartak, R.; Prajapati, M.; Zheng, H.; Zhao, C.; Mao, G.; Patel, K.; Fedosova, N.U.; Ścianowski, J.; et al. N-3-Methylbutyl-benzisoselenazol-3(2H)-one Exerts Antifungal Activity In Vitro and in a Mouse Model of Vulvovaginal Candidiasis. Curr. Issues Mol. Biol. 2024, 46, 2480–2496. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Kansakar, U.; Varzideh, F.; Mone, P.; Jankauskas, S.S.; Lombardi, A. Functional Role of Taurine in Aging and Cardiovascular Health: An Updated Overview. Nutrients 2023, 15, 4236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, D.; Yi, B.; Liao, Z.; Tang, L.; Yin, D.; He, M. Taurine supplementation reduces oxidative stress and protects the liver in an iron-overload murine model. Mol. Med. Rep. 2014, 10, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, M.; Xu, Y.; Yu, X.; Xiong, T.; Du, M.; Sun, J.; Liu, L.; Tang, Y.; Yao, P. Iron-Mediated Lysosomal Membrane Permeabilization in Ethanol-Induced Hepatic Oxidative Damage and Apoptosis: Protective Effects of Quercetin. Oxidative Med. Cell. Longev. 2016, 2016, 4147610. [Google Scholar] [CrossRef] [PubMed]
- Hezaveh, Z.S.; Azarkeivan, A.; Janani, L.; Hosseini, S.; Shidfar, F. The effect of quercetin on iron overload and inflammation in β-thalassemia major patients: A double-blind randomized clinical trial. Complement. Ther. Med. 2019, 46, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Zhang, G.; Chen, B.; He, Q.; Mai, J.; Chen, W.; Pan, Z.; Yang, J.; Li, J.; Ma, Y.; et al. Quercetin protects against iron overload-induced osteoporosis through activating the Nrf2/HO-1 pathway. Life Sci. 2023, 322, 121326. [Google Scholar] [CrossRef] [PubMed]
- Moayedi, B.; Gharagozloo, M.; Esmaeil, N.; Maracy, M.R.; Hoorfar, H.; Jalaeikar, M. A randomized double-blind, placebo-controlled study of therapeutic effects of silymarin in β-thalassemia major patients receiving desferrioxamine. Eur. J. Haematol. 2013, 90, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S.; Bao, B. Molecular Mechanisms of Zinc as a Pro-Antioxidant Mediator: Clinical Therapeutic Implications. Antioxidants 2019, 8, 164. [Google Scholar] [CrossRef] [PubMed]
- Deore, M.S.; Keerthana, S.; Naqvi, S.; Kumar, A.; Flora, S.J.S. Alpha-Lipoic Acid Protects Co-Exposure to Lead and Zinc Oxide Nanoparticles Induced Neuro, Immuno and Male Reproductive Toxicity in Rats. Front. Pharmacol. 2021, 12, 626238. [Google Scholar] [CrossRef] [PubMed]
- Amer, M.G.; Karam, R.A. Morphological and Biochemical Features of Cerebellar Cortex After Exposure to Zinc Oxide Nanoparticles: Possible Protective Role of Curcumin. Anat. Record 2018, 301, 1454–1466. [Google Scholar] [CrossRef] [PubMed]
- Heidai-Moghadam, A.; Khorsandi, L.; Jozi, Z. Curcumin attenuates nephrotoxicity induced by zinc oxide nanoparticles in rats. Environ. Sci. Pollut. Res. 2019, 26, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Khorsandi, L.; Mansouri, E.; Orazizadeh, M.; Jozi, Z. Curcumin Attenuates Hepatotoxicity Induced by Zinc Oxide Nanoparticles in Rats. Balk. Med. J. 2016, 33, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Nagar, V.; Singh, T.; Tiwari, Y.; Aseri, V.; Pandit, P.P.; Chopade, R.L.; Pandey, K.; Lodha, P.; Awasthi, G. ZnO Nanoparticles: Exposure, toxicity mechanism and assessment. Mater. Today Proc. 2022, 69, 56–63. [Google Scholar] [CrossRef]
- San Miguel, S.M.; Opperman, L.A.; Allen, E.P.; Zielinski, J.E.; Svoboda, K.K. Antioxidant combinations protect oral fibroblasts against metal-induced toxicity. Arch. Oral Biol. 2013, 58, 299–310. [Google Scholar] [CrossRef]
- Tamagno, W.A.; Santini, W.; Dos Santos, A.; Alves, C.; Bilibio, D.; Sutorillo, N.T.; Zamberlan, D.C.; Kaizer, R.R.; Barcellos, L.J.G. Pitaya Fruit Extract Ameliorates the Healthspan on Copper-induced Toxicity of Caenorhabditis elegans. J. Food Biochem. 2022, 46, e14050. [Google Scholar] [CrossRef] [PubMed]
- Tamagno, W.A.; Santini, W.; Alves, C.; Vanin, A.P.; Pompermaier, A.; Bilibio, D.; Sutorillo, N.T.; Kaizer, R.R.; Gil Barcellos, L.J. Neuroprotective and antioxidant effects of pitaya fruit on Cu-induced stress in adult zebrafish. J. Food Biochem. 2022, 46, e14147. [Google Scholar] [CrossRef]
- Azeez, O.; Braimah, S. Mitigating effect of vitamin-E on copper sulphate-induced toxicity in African Catfish (Clarias gariepinus). Eur. J. Med. Health Sci. 2020, 2, 4. [Google Scholar] [CrossRef]
- Maryam, T.; Rana, N.F.; Alshahrani, S.M.; Batool, F.; Fatima, M.; Tanweer, T.; Alrdahe, S.S.; Alanazi, Y.F.; Alsharif, I.; Alaryani, F.S.; et al. Silymarin Encapsulated Liposomal Formulation: An Effective Treatment Modality against Copper Toxicity Associated Liver Dysfunction and Neurobehavioral Abnormalities in Wistar Rats. Molecules 2023, 28, 1514. [Google Scholar] [CrossRef] [PubMed]
- Shibata, T.; Nagayasu, H.; Kawano, T.; Kitajo, H.; Hamada, J.; Moriuchi, T.; Okada, F.; Watanabe, S.; Yasuda, S.; Okuyama, H.; et al. Unsaturated fatty acid feeding prevents the development of acute hepatitis in Long-Evans cinnamon (LEC) rats. Anticancer Res. 1999, 19, 5169–5174. [Google Scholar] [PubMed]
- Yamamoto, H.; Watanabe, T.; Mizuno, H.; Endo, K.; Fukushige, J.; Hosokawa, T.; Kazusaka, A.; Fujita, S. The antioxidant effect of DL-alpha-lipoic acid on copper-induced acute hepatitis in Long-Evans Cinnamon (LEC) rats. Free Radic. Res. 2001, 34, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Nishikawa, A.; Nakamura, H.; Furukawa, F.; Imazawa, T.; Umemura, T.; Uchida, K.; Hirose, M. Effects of N-acetylcysteine, quercetin, and phytic acid on spontaneous hepatic and renal lesions in LEC rats. Toxicol. Pathol. 2005, 33, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Peres, T.V.; Schettinger, M.R.C.; Chen, P.; Carvalho, F.; Avila, D.S.; Bowman, A.B.; Aschner, M. Manganese-induced neurotoxicity: A review of its behavioral consequences and neuroprotective strategies. BMC Pharmacol. Toxicol. 2016, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Chtourou, Y.; Garoui, E.M.; Boudawara, T.; Zeghal, N. Protective role of silymarin against manganese-induced nephrotoxicity and oxidative stress in rat. Environ. Toxicol. 2014, 29, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- da Silva Santos, V.; Bisen-Hersh, E.; Yu, Y.; Cabral, I.S.R.; Nardini, V.; Culbreth, M.; da Rocha, J.B.T.; Barbosa, F.; Aschner, M. Anthocyanin-Rich Açaí (Euterpe oleracea Mart.) Extract Attenuates Manganese-Induced Oxidative Stress in Rat Primary Astrocyte Cultures. J. Toxicol. Environ. Health Part A 2014, 77, 390–404. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, A.P.; Schneider, J.A.; Nelson, B.C.; Atha, D.H.; Jain, A.; Soliman, K.F.; Aschner, M.; Mazzio, E.; Reams, R.R. Manganese-induced oxidative DNA damage in neuronal SH-SY5Y cells: Attenuation of thymine base lesions by glutathione and N-acetylcysteine. Toxicol. Lett. 2013, 218, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Adedara, I.A.; Ego, V.C.; Subair, T.I.; Oyediran, O.; Farombi, E.O. Quercetin Improves Neurobehavioral Performance Through Restoration of Brain Antioxidant Status and Acetylcholinesterase Activity in Manganese-Treated Rats. Neurochem. Res. 2017, 42, 1219–1229. [Google Scholar] [CrossRef] [PubMed]
- Bahar, E.; Kim, J.-Y.; Yoon, H. Quercetin Attenuates Manganese-Induced Neuroinflammation by Alleviating Oxidative Stress through Regulation of Apoptosis, iNOS/NF-κB and HO-1/Nrf2 Pathways. Int. J. Mol. Sci. 2017, 18, 1989. [Google Scholar] [CrossRef] [PubMed]
- Lebda, M.A.; El-Neweshy, M.S.; El-Sayed, Y.S. Neurohepatic toxicity of subacute manganese chloride exposure and potential chemoprotective effects of lycopene. Neurotoxicology 2012, 33, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Cordova, F.M.; Aguiar, A.S.; Peres, T.V.; Lopes, M.W.; Gonçalves, F.M.; Pedro, D.Z.; Lopes, S.C.; Pilati, C.; Prediger, R.D.S.; Farina, M.; et al. Manganese-exposed developing rats display motor deficits and striatal oxidative stress that are reversed by Trolox. Arch. Toxicol. 2013, 87, 1231–1244. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, A.M.; Santos, D.; Au, C.; Milatovic, D.; Aschner, M.; Batoréu, M.C.C. Antioxidants prevent the cytotoxicity of manganese in RBE4 cells. Brain Res. 2008, 1236, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Milatovic, D.; Gupta, R.C.; Yu, Y.; Zaja-Milatovic, S.; Aschner, M. Protective effects of antioxidants and anti-inflammatory agents against manganese-induced oxidative damage and neuronal injury. Toxicol. Appl. Pharmacol. 2011, 256, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Ky, S.Q.; Deng, H.S.; Xie, P.Y.; Hu, W. A report of two cases of chronic serious manganese poisoning treated with sodium para-aminosalicylic acid. Br. J. Ind. Med. 1992, 49, 66–69. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Iron | Copper | Manganese | |||
|---|---|---|---|---|---|
| Compound | Effects | Compound | Effects | Compound | Effects |
| Ebselen (in vitro) | ↓ Fe-uptake ↓ ROS | Resveratrol, Ferulic acid, Phloretin Tetrahydro curcuminoids (in vitro) | ↑ Cell viability and proliferation ↓ ROS | Silymarin (in vitro, co-treat) | ↓ H2O2 and ROS levels ↑ MnSOD, SOD, GPx, CAT activity |
| Quercetin Catechin Rutin (in vitro) | ↓ Hemolyzed GSH ↓ MDA levels, CAT activity | Pitaya-extract (in vivo) | ↓ AChE activity ↓ Lipid peroxidation ↓ MDA, CAT levels | Açaí extract (anthocyanins) (in vitro) | ↑ GSH/GSSG ratio ↑ Glutamate uptake ↓ Lipid peroxidation ↓ Nrf2 activation |
| α-Lipoic acid + vitamin D (in vitro) | ↓ Fe-content ↓ ROS ↓ p53, APP and SOD content | Pitaya-extract (in vivo, co-treatment) | ↓ GST, CAT activity (brain) ↑ SOD activity (brain) ↑ GST activity (gut) ↓ CAT activity (gut) ↑ AChE and δ-aminolevulinate dehydratase activity (brain) ↓ cortisol levels | N-acetylcysteine/GSH (in vitro, pre-treatment) | ↑ Cell viability ↓ DNA damage |
| Myricetin (in vitro, in vivo) | ↓ Fe-uptake ↓ TfR1 ↓ Lipid peroxidation ↓ DNA oxidation products | Vitamin E (in vivo, co-treatment) | ↑ GSH, GPx levels ↓ H2O2, MDA levels | Quercetin (in vivo) | ↓ H2O2 levels ↓ Lipid peroxidation ↑ SOD, CAT activity ↓ AChE activity ↑ Locomotor impairment |
| Taurine (in vivo) | ↓ Lipid peroxidation ↓ Loss of GSH levels | Silymarin (in vivo, encapsulated in a liposomal formulation) | ↑ Spatial memory ↓ Liver damage (AST, ALT, Total-Bilirubin) | Quercetin (in vitro, pre-treatment) | ↑ Cell viability ↓ ROS and MDA levels ↑ SOD, CAT, GSH activity ↓ Loss of MMP ↓ TNF-α, IL-1β, IL-6 protein content ↓ NF-kB, iNOS mRNA levels ↑ HO-1, Nrf2 mRNA levels ↓ Bax, Cyt c, caspase 3, PARP-1 levels |
| Quercetin (in vivo, human studies) | ↑ Fe-depletion ↓ Oxidative stress ↓ C-reactive protein levels ↓ Ferritin content ↓ Transferrin saturation ↑ Transferrin levels | α-Lipoic acid (in vivo) | ↓ Liver damage ↑ GPx, GR, SOD activity ↓ Lipid peroxidation | Lycopene (in vivo, pre-treatment) | ↓ ALT, AST, AChE, glucose levels (serum) ↓ AChE levels (brain) |
| Silymarin (human studies) | ↓ Fe-content ↓ Ferritin content ↓ Fe-binding capacity ↓ Hepcidin and transferrin levels | N-acetylcysteine (in vivo) | ↓ Cu/Zn ratio ↓ Liver and kidney damage | Trolox (in vivo, co-treatment) | ↓ Caspase activity ↓ Oxidative stress ↑ Motor coordination deficits |
| Trolox (in vitro/in vivo, pre-treatment) | ↓ Oxidative stress ↓ ATP depletion | ||||
| ↑ increase, ↓ decrease | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fontes, A.; Jauch, A.T.; Sailer, J.; Engler, J.; Azul, A.M.; Zischka, H. Metabolic Derangement of Essential Transition Metals and Potential Antioxidant Therapies. Int. J. Mol. Sci. 2024, 25, 7880. https://doi.org/10.3390/ijms25147880
Fontes A, Jauch AT, Sailer J, Engler J, Azul AM, Zischka H. Metabolic Derangement of Essential Transition Metals and Potential Antioxidant Therapies. International Journal of Molecular Sciences. 2024; 25(14):7880. https://doi.org/10.3390/ijms25147880
Chicago/Turabian StyleFontes, Adriana, Adrian T. Jauch, Judith Sailer, Jonas Engler, Anabela Marisa Azul, and Hans Zischka. 2024. "Metabolic Derangement of Essential Transition Metals and Potential Antioxidant Therapies" International Journal of Molecular Sciences 25, no. 14: 7880. https://doi.org/10.3390/ijms25147880