Abstract
T-cell malignancies, including T-cell acute lymphoblastic leukemia (T-ALL) and T-cell lymphoblastic lymphoma (T-LBL), present significant challenges to treatment due to their aggressive nature and chemoresistance. Chemotherapies remain a mainstay for their management, but the aggressiveness of these cancers and their associated toxicities pose limitations. Immunepotent CRP (ICRP), a bovine dialyzable leukocyte extract, has shown promise in inducing cytotoxicity against various cancer types, including hematological cancers. In this study, we investigated the combined effect of ICRP with a panel of chemotherapies on cell line models of T-ALL and T-LBL (CEM and L5178Y-R cells, respectively) and its impact on immune system cells (peripheral blood mononuclear cells, splenic and bone marrow cells). Our findings demonstrate that combining ICRP with chemotherapies enhances cytotoxicity against tumoral T-cell lymphoblasts. ICRP + Cyclophosphamide (CTX) cytotoxicity is induced through a caspase-, reactive oxygen species (ROS)-, and calcium-dependent mechanism involving the loss of mitochondrial membrane potential, an increase in ROS production, and caspase activation. Low doses of ICRP in combination with CTX spare non-tumoral immune cells, overcome the bone marrow-induced resistance to CTX cell death, and improves the CTX antitumor effect in vivo in syngeneic Balb/c mice challenged with L5178Y-R. This led to a reduction in tumor volume and a decrease in Ki-67 proliferation marker expression and the granulocyte/lymphocyte ratio. These results set the basis for further research into the clinical application of ICRP in combination with chemotherapeutic regimens for improving outcomes in T-cell malignancies.
1. Introduction
T-cell malignancies comprise a group of neoplasms that arise from the expansion of dysfunctional T-cells at different stages of development. T-cell acute lymphoblastic leukemia (T-ALL) is the most common T-cell cancer in children. In contrast, T-cell lymphoblastic lymphoma (T-LBL) accounts for 20% of the non-Hodgkin lymphoma cases in children. Studies have lent strength to the theory that T-LBL and T-ALL may evolve from a common malignant precursor cell [1,2]; moreover, both diseases are aggressive forms of hematological cancers since T-cell’s overall prognosis is poorer than B-cell malignancies [3,4]. Chemotherapies, such as cyclophosphamide (CTX), etoposide (ETO), and anthracyclines such as doxorubicin (DOX) and epirubicin (EPI) remain a potential strategy for T-ALL/T-LBL [5,6,7,8,9]. Several chemotherapies act primarily through the induction of apoptosis beyond distinct targets for these agents in susceptible cancer cells [10]. Also, in high doses, they cause severe secondary effects, such as bone marrow suppression, spleen toxicity [5,6,7,8,9], cognitive impairment, and microglial death [7].
Managing treatment during disease recurrence remains challenging due to chemoresistance, which arises from various mechanisms, including the inherent sensitivity of cancer cells to evade cell death [5,11,12]. Therefore, efforts to overcome resistance have pointed out the use of multi-targeted agents through the assessment of drug combinations, guided by an understanding of the molecular mechanisms underlying cell death. In this regard, recent studies highlighted the advances and the growing relevance of simultaneously blocking multiple pathogenic pathways in B-cell malignancies and lymphoma [13,14]. The multiple targets can belong to the same or different pathways of cell death that converge at a pathway site, resulting in an enhanced effect. Combination therapy works in a synergistic, additive, or antagonistic manner depending on the amount of the drug combination effect, which can be quantified by several models [15]. A substantial amount of evidence uses the combination index (CI) analysis proposed by Chou-Talay, which mitigates uncertainties in identifying effective combination treatments by enabling the scoring of synergistic drug effects [16,17]. Multiple reports provide evidence of combining chemotherapies and immunotherapies [18], which enables a reduction in the toxic effects on healthy cells and enhances efficacy against cancer cells at lower dosages, potentially overcoming chemo-resistance [19].
Immunepotent CRP (ICRP), a bovine dialyzable leukocyte extract, is an immunotherapy reported to exhibit immunomodulatory properties and cytotoxicity against several cancer cell lines [20,21]. The combinational therapy of ICRP with DOX and CTX modified the tumor microenvironment in a murine breast cancer model [22]. Furthermore, the combination of ICRP and oxaliplatin (OXP), induced immunogenic cell death (ICD) in murine melanoma [23]. ICRP was also reported to improve the clinical parameters of breast cancer patients receiving standard chemotherapy [24]. Therefore, ICRP shows potential when combined with various chemotherapies, including CTX, a major chemotherapy used for hematologic malignancies. Thus, in the present study, we investigated the combinatorial effects of a panel of chemotherapies and ICRP treatment on two T-cell malignancies, T-ALL and T-LBL c, chosen for their aggressive nature, poor prognosis, response to therapy, and chemoresistance, focusing on the mechanism of the CTX-ICRP combination and its in vivo effects.
2. Results
2.1. ICRP, CTX, DOX, EPI, and ETO Induce Tumoral T-Cell Lymphoblasts Cell Death
CEM and L5178Y-R death was analyzed after ICRP (dark gray) or chemotherapy (light gray) treatment. Results showed that all treatments augment tumoral T-cell lymphoblast cell death as treatment concentration increases (Figure 1A–E). Data show cell death of 20% of the cells (CC20) at 0.2 and 0.15 U/mL of ICRP for CEM and L5178Y-R cells, respectively, meanwhile 50% of the cells were dead (CC50) at 0.6 and 0.3 U/mL of ICRP (Figure 1A), respectively. On the other hand, CTX CC20 was 15 mM for both cell lines while 20 mM CTX was required to induce cell death in 50% of the cell population for both cell lines (Figure 1B). Likewise, DOX CC20 was shown at 5 μM for CEM and 10 μM for L5178Y-R, whereas DOX CC50 was shown at 15 μM for both cell lines (Figure 1C). Furthermore, EPI CC20 was 30 μM for CEM and 3 μM for L5178Y-R, whereas EPI CC50 was obtained at 40 μM for CEM and 12 μM for L5178Y-R (Figure 1D). Additionally, 20 μM and 40 μM ETO were the CC20, while 100 μM and 200 μM ETO were the CC50 of CEM and L5178Y-R, respectively (Figure 1E). CC20 and CC50 cytotoxic concentrations were found and the sublethal concentration was taken as the highest concentration of each treatment that does not induce notable cell death, for each cell line and treatment. These concentrations are summarized in the table shown in Figure 1F.
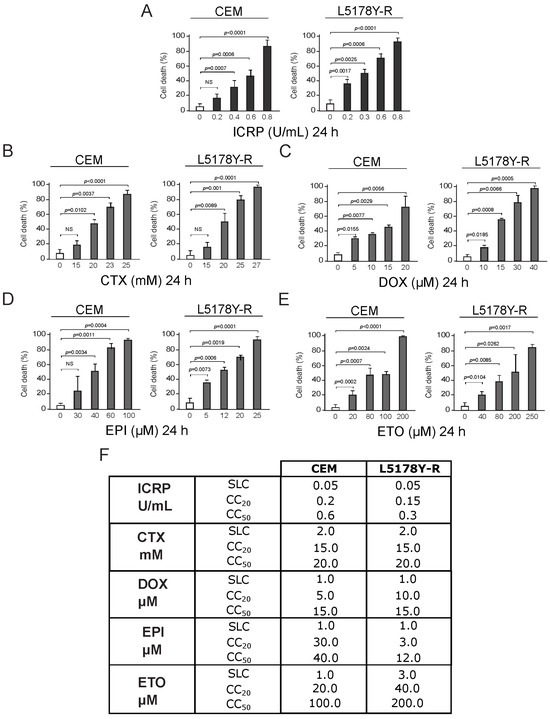
Figure 1.
ICRP, CTX, DOX, EPI, and ETO induce cell death in tumoral T-cell lymphoblasts. CEM and L5178Y-R cell lines were treated for 24 h, and biochemical features of cell death were assessed and expressed in percentage (%). Cell death was analyzed by Annexin V/PI staining for (A) ICRP-, (B) CTX-, and (E) ETO-treated cells or only AnnV for (C) DOX and (D) EPI treatments. (F) Sublethal concentration (SLC), cytotoxic concentration that induced cell death of 20% of the cells (CC20) and cytotoxic concentration that induced cell death of 50% of the cells (CC50) found for IMMUNEPOTENT CRP (ICRP), Cyclophosphamide (CTX), Doxorubicin (DOX), Epirubicin (EPI) and Etoposide (ETO) are summarized for CEM and L5178Y-R cell lines. Graphs are the means ± SD of triplicates from at least three independent experiments. NS was assigned to p > 0.05.
Although chemotherapies have different mechanisms of action, we proposed that a potentiated cytotoxic effect could be achieved by combining them with ICRP.
2.2. The Combination of ICRP and Chemotherapies Potentiates Cell Death against Tumoral T-Cell Lymphoblasts
Different combination ratios were designed for investigating the effect of several concentrations of ICRP on chemotherapies’ cytotoxicity. The chemotherapies for combination studies were chosen from a panel of chemotherapies (with different mechanisms of action such as alkylating agents and topoisomerase inhibitors) that were able to directly induce cell death as monotherapies in the cell lines tested. In contrast, we discarded the antimetabolites Ara-C and Methotrexate as they were unable to induce 50% cell death in L5178Y cells (Figure S1). First, we used a non-cytotoxic concentration (SLC, sublethal) of ICRP, in combination with the CTX, DOX, EPI, and ETO − CC50 of each tumoral T-cell lymphoblasts cell line. To investigate whether chemotherapies affect ICRP cell death, we tested the combination of CC50 ICRP with SLC CTX, DOX, EPI, and ETO. To examine the combined effect of equipotent concentrations of both treatments, we tested the combination of CC20 of ICRP and CTX or the combination of CC50 of both treatments. Moreover, to investigate whether ICRP at a low dose affects chemotherapies’ cell death, we treated cells with CC20 ICRP + CC50 CTX, DOX, EPI, and ETO.
As Figure 2A shows, a significant increase in CEM and L5178Y-R cell death compared to single agents was observed, reaching 85% and 96%, respectively, when combining SLC ICRP + CC50 CTX. Results showed a non-significant cell death increase in CEM with the combination of CC50 ICRP + SLC CTX, whereas this combination induced a significant increase in L5178Y-R death reaching 69% and 77% cell death, respectively. Cell death assessment showed that CC20 ICRP + CC20 CTX reached 91% cell death in CEM and L5178Y-R. Likewise, CC50 ICRP + CC50 CTX showed a significant increase in cell death compared to single treatments, reaching 98% and 95% in CEM and L5178Y-R, respectively, and the combination using CC20 ICRP + CC50 CTX demonstrated 98% cell death in the two cell lines tested. Furthermore, Figure 2B shows that SLC ICRP + CC50 DOX demonstrated a significant increase in CEM cell death compared to single treatments, reaching 93%, whereas L5178Y-R showed no significant increase, reaching 59% cell death. The combination using CC50 ICRP + SLC DOX induced 50% cell death in CEM and L5178Y-R. A significant increase in CEM and L5178Y-R cell death compared to single agents was observed, reaching 40% and 43%, respectively, when combining CC20 ICRP + CC20 DOX. When the combination of CC50 ICRP + CC50 DOX was used we observed a significant cell death increase in CEM, reaching 94%, whereas this combination reached 58% in L5178Y-R cells. The assessment revealed a significant increase in cell death to 97% in CEM when combining CC20 ICRP + CC50 DOX. Conversely, this combination showed a non-significant increase in cell death in L5178Y-R cells, with 52%.
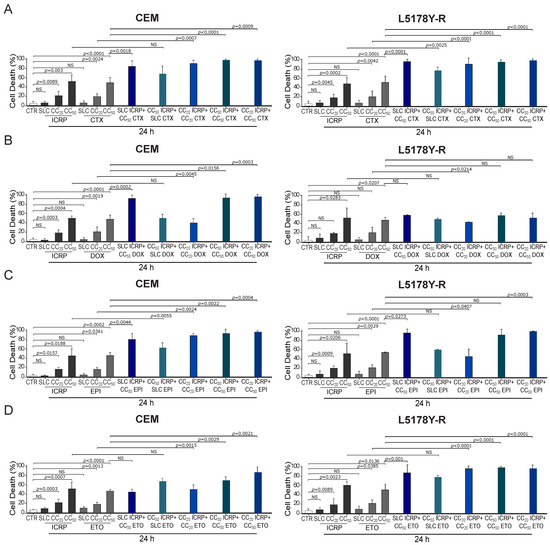
Figure 2.
ICRP + chemotherapy-induced cell death in tumoral T-cell lymphoblasts. (A–D) CEM and L5178Y-R were treated for 24 and analyzed by flow cytometry using Ann/PI staining or Ann alone for DOX and EPI. Cell death induced by (A) ICRP, CTX, and its combination, (B) ICRP, DOX, and its combination, (C) ICRP, EPI, and its combination, and (D) ICRP, ETO, and its combination. Graphs are the means ± SD of triplicates from at least three independent experiments. NS was assigned to p > 0.05.
Moreover, as shown in Figure 2C, a significant increase in cell death occurs when combining SLC ICRP + CC50 EPI, demonstrating 81% and 96% cell death in CEM and L5178Y-R, respectively. Results showed a significant cell death augmentation in CEM with the combination of CC50 ICRP + SLC EPI reaching 59%, while this combination in L5178Y-R reached 60%. Cell death assessment induced by CC20 ICRP + CC20 EPI showed a significant increase compared to single treatments, reaching 89% and 46% in CEM and L5178Y-R, respectively. Likewise, CC50 ICRP + CC50 EPI showed 87% and 91% cell death in CEM and L5178Y-R, respectively, and the combination using CC20 ICRP + CC50 EPI demonstrated 97% cell death in CEM and 99% in L5178Y-R.
Additionally, Figure 2D shows that SLC ICRP + CC50 ETO showed a non-significant increase in CEM cell death compared to ETO alone, reaching 45%, whereas L5178Y-R showed a significant increase, reaching 88% cell death. The combination using CC50 ICRP + SLC ETO demonstrated an increase in cell death with 68% and 82% values in CEM and L5178Y-R, respectively. When combining CC20 ICRP + CC20 ETO, a significant increase in CEM and L5178Y-R cell death was observed compared to single agents, reaching 51% and 95%, respectively. Results showed a significant cell death augmentation in the two cell lines tested when the combination of CC50 ICRP + CC50 ETO was used, reaching 70% in CEM and 93% in L5178Y-R. Finally, the assessment showed a significant increase in cell death to 87% in CEM and 96% in L5178Y-R when combining CC20 ICRP + CC50 ETO.
2.3. The Combination of ICRP with Chemotherapy Induces a Synergistic Cytotoxic Effect Allowing a Reduction in Chemotherapy Doses
To correctly define whether the combined effect is superior to the single drugs, we used the combination index (CI) to quantify the drug interaction effect induced by ICRP in combination with each chemotherapy by the software Compusyn. Table 1 shows the CI values obtained from all the tested combinations, revealing a synergistic effect (CI < 1) by all the chemotherapies and ratios tested. Nevertheless, the highest synergic effect, according to the CI values shown in both cell lines, was obtained from the combinations of ICRP with CTX.

Table 1.
CI values compilation from the combinations of ICRP with chemotherapies in tumoral T-cell lymphoblasts.
Furthermore, when looking for a decreasing toxicity in single drugs, as the combined effect is higher than monotherapy, we calculated the degree of chemotherapy dosage reduction by drug reduction index (DRI). All the chemotherapies tested showed DRI values above 1 reaching up to 1724.07, indicating a favorable dose reduction. DRI values are summarized in Table 2.
Table 2.
DRI values compilation from the combinations of ICRP with chemotherapies in tumoral T-cell lymphoblasts.
Considering CTX demonstrated the greatest synergistic effect across both cell lines and a favorable reduction in DRI values, combinations involving SLC ICRP + CC50 CTX, CC50 ICRP + CC50 CTX and CC20 ICRP + CC50 CTX combinations were chosen to further determine several biochemical features of ICRP + CTX cell death, assessing the main characteristics elicited by each monotherapy.
2.4. The Combination of ICRP with CTX Induces Mitochondrial Alterations in Tumoral T-Cell Lymphoblasts
The right panel of Figure 3A shows a significant increase in the loss of mitochondrial membrane potential assessment by SLC ICRP + CC50 CTX in CEM and L5178Y-R reaching 75% and 82%, respectively, whereas L5178Y-R also showed a significant increase with CC50 ICRP + CC50 CTX and CC20 ICRP + CC50 CTX-treatment (86–88%) compared to CTX monotherapy. Likewise, CEM CC20 ICRP + CC50 CTX-treated cells showed 55% ROS production, and L5178Y-R at all the combination ratios showed a significant increase in ROS production compared to CTX alone, demonstrated by up to 82% HE+ cells (Figure 3B). Additionally, a significant increase in caspase activation was observed after SLC ICRP + CC50 CTX, CC50 ICRP + CC50 CTX, and CC20 ICRP + CC50 CTX treatment in CEM and L5178Y-R, compared to single agents (Figure 3C).
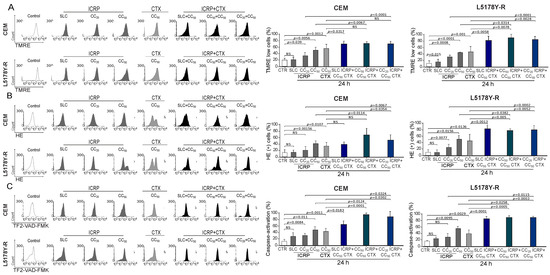
Figure 3.
ICRP + CTX cell death-induced mitochondrial alterations in tumoral T-cell lymphoblasts. Cells were treated with ICRP + CTX in distinct ratios for 24 h and analyzed by flow cytometry. Representative histograms and graphs from (A) loss of mitochondrial membrane potential, (B) ROS production, and (C) caspase activation measured using TMRE, HE, and TF2-VAD-FMK staining, respectively, in CEM and L5178Y-R. Graphs are the means ± SD of triplicates from at least three independent experiments. NS was assigned to p > 0.05.
2.5. The Combination of ICRP with CTX Induces Cell Death Involving Caspases, ROS Production, and Calcium Augmentation in Tumoral T-Cell Lymphoblasts
We aimed to investigate the effectors of ICRP + CTX cell death. For this, we analyzed the caspase dependence using the pan-caspase inhibitor QVD. We found that QVD diminished cell death induced by CC50 ICRP and CC50 CTX in the two cell lines; also in CEM, QVD diminished the cell death in the different combination ratios tested. Whereas, in L5178Y-R, QVD inhibited cell death when cells were treated with SLC ICRP + CC50 CTX and CC50 ICRP + CC50 CTX, but not with the combination CC20 ICRP + CC50 CTX (Figure 4A). Furthermore, after using the antioxidant NAC, cell death diminished significantly when cells were treated with all the combination ratios tested in both cell lines (Figure 4B). Additionally, pre-treatment with the extracellular calcium chelator BAPTA decreased cell death induced by CC50 ICRP and CC50 CTX, as well as all the combination ratios tested in both cell lines (Figure 4C).
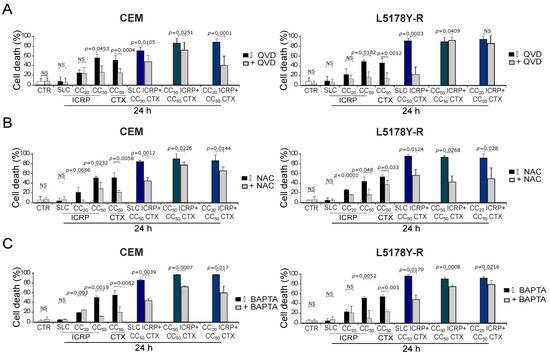
Figure 4.
ICRP + CTX cell death effectors in tumoral T-cell lymphoblasts. Cells were treated with (A) QVD, (B) NAC, or (C) BAPTA for 30 min before treatment with ICRP + CTX in distinct ratios for 24 h, and cell death was analyzed by flow cytometry. Graphs from AnnV/PI measurement of CEM (left panel) and L5178Y-R (right panel). Bars are the means ± SD of triplicates from at least three independent experiments. NS was assigned to p > 0.05.
2.6. The Combination of ICRP with CTX Does Not Potentiate CTX Cell Death in Non-Tumoral Immune System Cells and Protects Bone Marrow Cells from CTX Cell Death
To evaluate if the combination of ICRP with CTX could also potentiate the cytotoxicity of non-tumoral immune system cells, we chose the highest cytotoxic concentration used in the tumoral cells to investigate the cytotoxicity of this combination in peripheral blood mononuclear cells (PBMC), splenocytes, and bone marrow cells. As Figure 5 shows, ICRP is not cytotoxic to PBMC (Figure 5A), spleen (Figure 5B), and bone marrow cells (Figure 5C) as only a low relative cell viability reduction was observed at CC50 ICRP of CEM (0.6 U/mL, 17% reduction). In contrast, CC50 CTX (20 mM) induced a strong reduction in cell viability in all the non-tumoral immune system cells, ranging from 57% to 94% reduction. Interestingly, any of the combination ratios tested increased this reduction in cell viability. Importantly, we observed a significant increase in the relative cell viability of bone marrow cells when treated with all the combination ratios tested, compared to CTX alone. This indicates that ICRP protects against cell death in bone marrow cells.
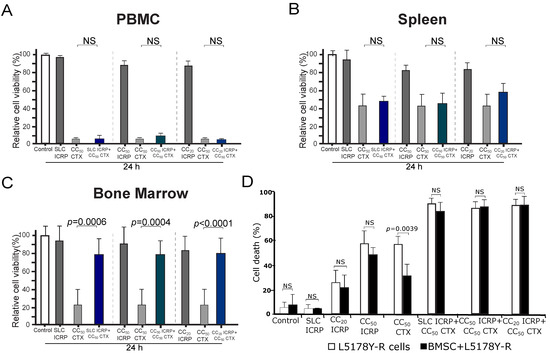
Figure 5.
ICRP + CTX cell death in non-tumoral immune system cells and tumoral cells in the presence of BMSC environment. PBMC (A), spleen (B), and bone marrow cells (C) were treated for 24 h with ICRP, CTX, and their combination, analyzed by flow cytometry using Ann/PI staining, and expressed as relative cell viability by the exclusion of Ann V/PI positive cells considering control cells as 100% cell viability. (D) Cell death induced by ICRP, CTX, and its combination in L5178Y-R co-cultivated with bone marrow stromal cells (BMSC) and analyzed by flow cytometry. NS was assigned to p > 0.05.
2.7. The Combination of ICRP with CTX Overcomes Cell Death Resistance Induced by Bone Marrow Stromal Cells
Next, we assessed whether ICRP + CTX-induced cell death could be protected by the survival stimuli provided by the bone marrow microenvironment [25]. In Figure 5D, while cell death induced by CC20 and CC50 ICRP persisted even when L5178Y-R were cocultured with BMSC, the presence of BMSC inhibited cell death in CTX-treated cells. In contrast, the cell death induced by the combination of ICRP + CTX remained unchanged even when using SL concentrations of ICRP (Figure 5D).
2.8. The Combination of ICRP with CTX Has an Antitumor Effect against T-Cell Lymphoma
As Figure 6A shows, female L5178Y-R-bearing mice were treated with a low dose of ICRP, CTX, and their combination. Treatment with two units of ICRP every two days led to a moderate decrease in tumor volume, whereas weekly injections of 125 mg/kg CTX resulted in a significant decrease in tumor volume compared to the control (vehicle-treated group). However, when the low dose of ICRP was combined with CTX, tumor volume significantly diminished compared to CTX monotherapy. This reduction in tumor volume is consistent with the tumor size shown in Figure 6B.
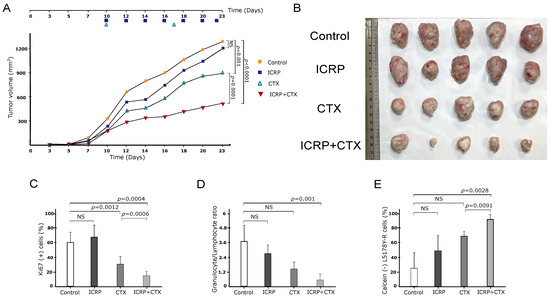
Figure 6.
ICRP + CTX induces an antitumor effect against tumoral T-cell lymphoblasts. Female BALB/c mice (n = 5 per group) were inoculated s.c. with 1 × 106 L5178Y-R viable cells. When the tumor reached 100–120 mm3 after inoculation, mice were treated with 2 U/mL i.p. ICRP (purple squares) every two days, 125 mg/kg CTX i.p, weekly (green triangles), or the combination of ICRP + CTX (inverted red triangles). Control mice (yellow circles) were treated with 100 μL sterile water for injection. Data are shown in (A) graph of tumor volume, (B) tumor size photograph, (C) Ki67 in tumor cells analyzed by flow cytometry, (D) granulocyte/lymphocyte ratio obtained from hematic biometry and (E) splenocytes cytotoxicity of mice treated with ICRP, CTX, or its combination against L5178Y-R stained with calcein-AM and analyzed by flow cytometry. NS was assigned to p > 0.05.
Additionally, as shown in Figure 6C, tumor cells from the control and ICRP groups exhibited a high percentage of the Ki-67 proliferation marker. In contrast, the CTX group showed a decrease in the percentage of Ki-67, which was further reduced in the ICRP + CTX group compared to CTX monotherapy.
A hematic biometry was conducted after treatment, and the granulocyte/lymphocyte ratio was determined. It was observed that this ratio remained unchanged in the peripheral blood of ICRP- and CTX-treated mice compared to control mice. However, the granulocyte/lymphocyte ratio was significantly decreased only in the ICRP + CTX-treated group (Figure 6D). Furthermore, to analyze the specific cytotoxicity of immune cells against cancer cells after treatment, we assessed the cytotoxicity of splenocytes to L5178Y cells. We observed that only splenocytes obtained from ICRP + CTX-treated mice induced a significant increase in L5178Y-R cell cytotoxicity, as evidenced by the loss of calcein staining (Figure 6E).
3. Discussion
Chemotherapies are well-known apoptosis inducers and exhibit significant immunosuppressive effects on various organs, including bone marrow, spleen, and the central nervous system [5,6,7,8,9,10]. Immunepotent CRP (ICRP), a bovine dialyzable leukocyte extract, displays selective cytotoxicity against several solid and hematologic cancers by inducing ROS-dependent apoptosis in T-cell acute lymphoblastic leukemia (T-ALL) cells, leading to nuclear and mitochondrial damage [20,26]. This study reported the first use of ICRP in conjunction with chemotherapy to enhance cytotoxicity against T-ALL and T-LBL, which are often resistant to conventional treatments [27,28]. Our findings revealed that combining ICRP with chemotherapy significantly boosts cytotoxicity in T-cell lymphoblasts, showing potential for enhanced antitumor effects. The concept of combination therapy was pioneered by Frei, Holland, and Freireich, who developed the first chemotherapy regimen for ALL [29]. In subsequent studies, there were combined doses of CC50 cisplatin after 4-hydroperoxycyclophosphamide treatment, achieving up to 85% inhibition of leukemic cell viability [30], similar to our results of 84–96% cell death using sublethal doses of various chemotherapies combined with ICRP.
Our study indicated that synergistic cytotoxic effects are enhanced by combining ICRP with chemotherapy. There have been reported synergistic cytotoxic effects induced by combinations of low doses of chemotherapies with other treatments, such as combinations of CC10 nutlin-3a with CC20 doxorubicin (DOX), CC25 chlorambucil (CLB), or CC15 fludarabine (FLU), which showed 50% to 65% cell death in B-cell chronic lymphocytic leukemia patient’s samples [31]. These results are similar to our findings when using sublethal doses of CTX, DOX, EPI, or ETO, combined with CC50 ICRP, where cell death reached 50% to 82%. On the other hand, using a sublethal inhibitory concentration of nelarabine (nela) in combination with the inhibitory concentration 15 (IC15) of ZSTK-474, induced a 25% cell viability inhibition of T-ALL patient’s samples [32]. These results are different from the ones observed when we combined suboptimal (CC20) concentrations of ICRP and CTX, DOX, EPI, or ETO as these combinations reached up to 95% cell death, demonstrating a synergistic effect of CI values lower than 1.0. Furthermore, improved efficacy in terms of cytotoxicity was obtained by treatment using CC20 nela plus CC50 ZSTK-474, inducing 60% cell viability inhibition. Remarkably, CC20 ICRP plus CC50 CTX, DOX, EPI, or ETO improved the cell death induced by monotherapies, showing 87% to 98%. These data underline the potential of ICRP in potentiating chemotherapy-induced cell death, even when used at non-lethal or suboptimal concentrations.
Combinations of several agents such as BV6, a bivalent SMAC mimetic, and nela, with chemotherapies at ratios using equipotent concentrations of both treatments, revealed higher cytotoxicity induced by ICRP plus CTX, DOX, EPI, or ETO. For instance, IC50 BV6 combined with IC50 CTX showed a decrease in cell viability to 20% in primary ALL cells [12]. On the other hand, a combination that included CC40 nela and CC40 ZSTK-474 against ALL cells reached 70% inhibition of cell viability. When we combined CC50 ICRP+ CC50 of each chemotherapy, our results produced up to 98% cell death, leading to CI values representing a synergistic cytotoxic effect [32].
Combination therapy with synergistic or additive effects may produce a more potent cytotoxic effect in lower doses of each monotherapy. We observed CI values reaching 0.00745–0.99682, showing a stronger synergism as well as more favorable DRI values (1.59690–1724.07) than shown previously by Hosseini M. and colleagues which combined different ratios of carfilzomib (cfz) and dexamethasone (Dex) against MOLT-4, a T-ALL cell line, and obtained 0.983–0.749 in CI values and 2.243–41.951 [33]. Furthermore, our results regarding the CI and DRI values are also different from the ones reported by Hassani S et al., who combined azidothymidine (AZT) and arsenic trioxide (ATO) in different ratios and found a reduction in the ATO cytotoxicity, showing an antagonistic effect with CI values of 1.21–5.54 and non-favorable or non-dose-reduction for ATO with 0.46–1.32 DRI values [34]. These data emphasize the potential of ICRP in boosting the effectiveness of existing chemotherapy protocols in T-ALL and T-LBL, particularly at suboptimal concentrations that are less toxic to healthy cells.
A synergistic effect could be triggered by actions on multiple targets that reside in the same or different pathways, negative regulation of counteractive actions, facilitating actions, or due to complementary actions [15]. Our data showed that both ICRP and CTX induce the loss of mitochondrial membrane potential, an increase in ROS production, and caspase activation. These effects were significantly augmented when the treatments were combined, compared to each treatment alone, in most of the combination ratios tested in both cell lines. Therefore, it seems that the increased cytotoxic effects of ICRP + CTX could be at least in some part due to the enhancement of mitochondrial alterations which can initiate cell death, similar to the results previously reported by combining Cfz + Dex which showed a significant increase in caspase 3, BAX and BCL2 gene expression in a T-ALL cell line compared to monotherapy [33]. Moreover, we further identified the role of caspase activation, ROS production, and intracellular calcium overload during cell death. As previously reported, ICRP and CTX cell death rely on caspase activation and ROS production, whereas ICRP cell death also depends on the increase in intracellular calcium levels in T-ALL [5,6,20,35]. Yet, here we first reported the relevance of an increase in the intracellular calcium for CTX-mediated cell death as it was previously described for cardiomyocyte toxicity [36]. ICRP + CTX showed mostly caspase-dependent, ROS-dependent, and Calcium-dependent cell death. However, we could note that even if ICRP alone induces caspase-dependent cell death, caspases were dispensable when using CC20 ICRP alone, such independence was maintained in the combination CTX + ICRP CC20 in L5178Y-R cells. We previously demonstrated that in breast cancer cell lines (MCF-7, MDA-MB231, and 4T1 cells) ICRP induces caspase-independent cell death, and the combination of ICRP + CTX maintains such caspase-independent cell death; however, ROS dependence was not assessed [37]. Other ROS- and caspases-dependent cell death modalities have been shown by the combination of bortezomib with PCI-24781 (an HDAC inhibitor) synergized against a Hodgkin and a non-Hodkin lymphoma cell line [38]. The combination of phytosphingosine and ionizing radiation in a T-cell lymphoma cell line resistant to radiation also involved the loss of mitochondrial membrane potential and resulted in a caspase-independent mechanism [39].
Conventional chemotherapies can be toxic to healthy cells, leading to multiple side effects, including a reduction in the immune system by affecting lymphoid organs such as bone marrow and the spleen [5,6,7,8,9]. Although combination therapy can be toxic, the low therapeutic dosage required of each drug may prevent the toxic effects on healthy cells, while potentiating the cytotoxic effects on cancer cells. This may occur if one drug in the combination regimen is non-cytotoxic to healthy cells [19], as is the case in several immunotherapies, which show immunomodulatory activities but also present cytotoxic activities against cancer cells [40]. Although CTX induced variable cytotoxic effects in non-tumoral immune system cells, ICRP was not toxic. When combining both treatments using the concentrations and combination ratios tested in tumoral cells, ICRP + CTX did not demonstrate an increase in the cytotoxic effect of CTX in PBMC and spleen cells, but also, ICRP inhibited the CTX toxicity induced in bone marrow cells. This cytoprotection observed in bone marrow cells is in accordance with previous reports of our research group, where it was demonstrated that ICRP was able to induce in vivo bone marrow cell protection after 5-Fluorouracil treatment by reducing ROS production [41]. Other naturally derived products, such as a mixture of honeybee compounds, showed the in vivo amelioration of the cytotoxic effects of CTX in bone marrow cells, sperm, and the liver when used in combination with CTX [42]. However, the Janus-like effect of ICRP, where on one hand it is cytotoxic to cancerous cells and cytoprotective to bone marrow cells, could be related to its capacity to induce ER stress. This was demonstrated in T-ALL, where it induces ER stress through ER-Ca2+ mobilization and prosurvival autophagosome formation [35]. It has been demonstrated that depending on the duration and intensity of the stress, ER stress can switch from protection to cell death induction [43], and even in the presence of autophagy, the same molecular cascades that initially support the cytoprotection shift to a cytotoxic mode and ultimately promote cell death [44]. Here, we observed that Ca2+ mobilization in CTX + ICRP treatment is important for cell death induction, and it has been demonstrated that T-ALL cells upregulate the machinery and signaling molecules associated with ER stress and autophagy [43,45,46]. On the other hand, autophagosomes usually serve as a cell antioxidant pathway [47], which can be linked to the antioxidant activity previously observed in bone marrow cells of mice treated with ICRP. Thus, it is plausible that the mechanism induced by ICRP is in the tightrope between cytoprotective effects in bone marrow cells and the cytotoxic effect observed in cancer cells. This overexpressed ER stress machinery in leukemic cells, which usually promotes prosurvival mechanisms when activated by ICRP treatment, could trigger perturbations that exceed cellular repair capacities leading to cell death. However, further studies on the precise role of ICRP in cytoprotection and the comparison between non-tumor and tumor cells must be performed to better understand this Janus-like role.
Bone marrow niches support stem cells and their progeny, protecting malignant cells from chemotherapy and ultimately contributing to the recurrence of hematological malignancies [25]. Our results revealed that CTX cell death is modulated by BMSC; in contrast, ICRP-induced cell death remained unchanged under these conditions. Also, ICRP + CTX overcame this CTX resistance, even when the combination included SL concentrations of ICRP. Similar results were reported by the peptide RCP168 which partially inhibited stroma-mediated resistance of Jurkat cells (T-ALL) to cytarabine (Ara-C) cell death [48]. Further analysis should be performed to identify the molecular mechanism by which ICRP + CTX overcomes the BMSC-mediated CTX resistance.
The combination strategies are based on sequential or concurrent therapy [49]. Our results show that concurrent therapy, initiating the administration of ICRP when beginning chemotherapy treatment, improved the tumor volume and the proliferation marker reduction induced by CTX alone in T-cell lymphoblastic lymphoma-bearing mice. A previous clinical trial in non-small cell lung cancer ICRP was administered on the third day after chemotherapy and cisplatin treatment. In this study, no changes in tumor size were observed when ICRP was administered, with respect to conventional treatment alone, although ICRP showed a beneficial effect in lymphocyte numbers and improved the Karnofsky score in patients [50]. Later, a clinical trial in breast cancer patients was performed using ICRP starting with 1-week administration prior to chemotherapy, with continued administration during the chemotherapy cycle and up to 1 month after the completion of chemotherapy. ICRP also showed a beneficial effect in lymphocyte numbers and improved Karnofsky score, but this schema also achieved better complete response percentages in stage III and IV patients, and the regression of metastatic lesions was obtained in less time than in the control group [24]. These results point out that administering ICRP at the same time as or before chemotherapy could be the best option in a conventional treatment for T-ALL or T-LBL. However, clinical trials must be performed to confirm this.
In previous research, when CTX was combined with Interferon type I (IFN-I) in vivo, it delayed tumor development and prevented 60% of mice bearing two types of T-cell lymphoma, whereas CTX or IFN alone did not prevent tumor-bearing mice [51]. Furthermore, mice surviving after IFN + CTX could generate immunologic memory, as hypothesized by our results as splenocytes from mice treated with ICRP + CTX showed cytotoxic capacity against the T-LBL cell line. Additionally, the significant decrease in the granulocyte/lymphocyte ratio shown by ICRP + CTX indicates a better anti-tumor efficiency as an elevated ratio seems to be associated with tumor progression and metastasis, perhaps because granulocytes compromise the natural antitumor function of lymphocytes [52].
Overall, throughout this study, we demonstrated that combining ICRP with chemotherapy synergically enhances cytotoxicity against T-cell lymphoblasts even when ICRP was used at non-lethal or suboptimal concentrations, whereas ICRP + CTX overcomes the bone marrow-induced resistance to CTX cell death. Furthermore, ICRP improves the CTX antitumor effect in vivo and promotes cancer cell killing by splenocytes ex vivo (Figure 7). These results set the basis for further research into the clinical application of ICRP in combination with chemotherapeutic regimens for improving outcomes in T-cell malignancies.
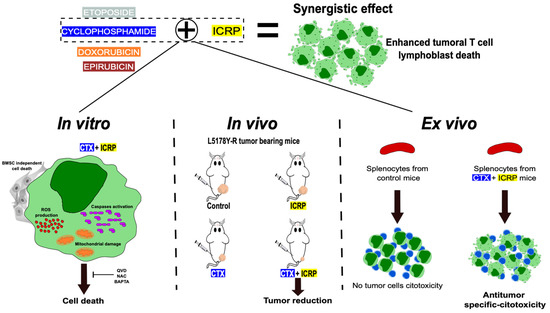
Figure 7.
Immunepotent CRP synergistic enhances chemotherapy-induced cell death against tumoral T-cell lymphoblasts. When Immunepotent CRP (ICRP) is combined with Cyclophosphamide (CTX) it enhances ROS production, caspase activation, mitochondrial damage and induces cell death even in the presence of protecting bone marrow stromal cells. The cell death induced depends on caspases, ROS, and calcium. In vivo, the combination of ICRP and Cyclophosphamide enhance the reduction in tumor volume, leading ex vivo to the specific antitumor cytotoxicity induced by splenocytes of the treated mice.
4. Materials and Methods
4.1. Cytotoxic Agents, Cell Culture Mediums, and Inhibitors
Cells were cultured in RPMI-1640 supplemented with heat-inactivated-10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (GIBCO by Life Technologies, Grand Island, NY, USA) referred to now as complete RPMI. The Laboratory of Immunology and Virology from the School of Biological Sciences produced IMMUNEPOTENT CRP (ICRP). One unit of ICRP contains 24 mg of peptides obtained from 15 × 108 leukocytes. The general characterization of ICRP was previously reported [53,54,55], where physical, bromatological, chemical, and in silico analyses were reported. ICRP and Cyclophosphamide (Cryofaxol from Cryopharma; Tlajomulco de Zuñiga, Jalisco, Mexico) were dissolved in complete RPMI. Doxorubicin (DOX), Epirubicin (EPI) (Farmorubicin RD®, purchased from Pfizer, Mexico City, Mexico), and Etoposide (ETO, Cavep®. from Accord Farma, Mexico City, Mexico) were dissolved in sterile water for injection as appropriate. The antioxidant, N-acetyl-L-cysteine (NAC), was dissolved in water to a final concentration of 500 mM. The pan-caspase inhibitor, QVD.opH (QVD, 1 mM), and the extracellular calcium chelator, BAPTA (50 μM), were dissolved in dimethyl sulfoxide (DMSO) and were incubated for 30 min before treatment. All the solutions were wrapped in foil and stored according to the manufacturer’s instructions.
4.2. Cell Culture
The CEM cell line, female human T-cell acute lymphoblastic leukemia (ATCC CCL-119), and L5178Y-R, murine T-cell lymphoblasts (ATCC CRL-1722), were obtained from the American Type Culture Collection (ATCC) and maintained according to its standards in a humidified incubator at 37 °C and 5% CO2. Cells were maintained in 25 cm3 cell culture flasks (CORNING Enterprises, Corning, NY, USA) containing complete RPMI.
4.3. Ethical Consideration
All experiments were reviewed and approved by the Ethical Committee (CEIBA) of the College of Biological Sciences at the UANL: CEIBA-2020-015. For animal samples, all experiments were performed following the Mexican regulation NOM-062-ZOO-1999 and were designed according to the Arrive guidelines for animal care and protection [56]. The procedures in our study involving human samples were conducted in accordance with the Helsinki Declaration.
4.4. Animals
The animal house at the Universidad Autónoma de Nuevo León, Mexico, supplied female BALB/c mice (eight-to-ten-week-old; 25 ± 5 g weight). Mice were housed in plastic cages in groups of five, and seven days were given to acclimate to the housing facility. Animals were maintained at 21 ± 3 °C, 55% ±10% humidity, and 12 h light/dark cycle. Mice were provided with rodent maintenance food (LabDiet, St. Louis, MO, USA) and water ad libitum, and health status was monitored daily. Mice were randomly assigned to different groups for all the studies.
4.5. Lymphoid Cell Isolation
Male mice (n = 4) were anesthetized using 100 mg/kg sodium pentobarbital (CHEMINOVA, Mexico City, Mexico) and sacrificed by cervical dislocation. Then, the spleen, femur, and tibia were obtained. The spleen was filtered through a cell strainer (70 μM) with PBS. Bone marrow cells were obtained by flushing the femur and tibia into complete RPMI. All cells were maintained at 2 × 105 per well in complete RPMI at 37 °C in a 5% CO2 atmosphere.
4.6. Peripheral Blood Mononuclear Cells (PBMC) Isolation
After obtaining written informed consent, human PBMC isolation from healthy donors was performed by gradient centrifugation using Ficoll-Paque™ PLUS (GE Healthcare, Chicago, IL, USA). Cell layers were obtained from which the population corresponding to PBMC was taken. Cells were maintained in complete RPMI at 2 × 105 cells per well at 37 °C in a 5% CO2 atmosphere.
4.7. Cell Death Analysis
Cells (5 × 105 cells/mL) were exposed to ICRP (0.2–0.8 U/mL), CTX (15–27 mM), DOX (5–40 μM), EPI (5–100 μM), ETO (20–250 μM), and the cytotoxic concentrations (CC) used for the combination treatment were obtained. For the following assays, different combination ratios of ICRP + CTX, ICRP + DOX, ICRP + EPI, and ICRP + ETO were used to treat cells for 24 h in 96-well dishes (Life Sciences, Darmstadt, Germany). After incubation, cells were collected and washed with PBS and suspended in 100 μL of binding buffer (10 mM HEPES/NaOH pH 7.4, 140 mM NaCl, 2.5 mM CaCl2) containing Annexin-V-APC (AnnV, 1 μg/mL, BD Pharmingen, San Jose, CA, USA) and propidium iodide staining (PI, 0.5 μg/mL, MilliporeSigma, Eugene, OR, USA) to measure cell death with BD Accuri c6 flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) and analyzed using FlowJo 10.7.2 Software (BD Biosciences, Ashland, OR, USA).
4.8. Pharmacological Inhibition of Cell Death Analysis
Before treatment with ICRP + CTX, cells were treated for 30 min with or without 1.5 µM QVD, 0.25 mM NAC, or 50 μM BAPTA for cell death inhibition. After 24 h, cells were obtained and washed with PBS twice, and suspended in 100 μL of binding buffer (10 mM HEPES/NaOH pH 7.4, 140 mM NaCl, 2.5 mM CaCl2) containing Annexin-V-APC (1 μg/mL, BD Pharmingen, San Jose, CA, USA) and 0.5 μg/mL propidium iodide (PI, MilliporeSigma, Eugene, OR, USA) to determine cell death using a BD Accury c6 flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). FlowJo Software was used to analyze data (BD Biosciences).
4.9. Stromal Bone Marrow Cells’ Protection Analysis
Bone marrow cells were obtained as mentioned above and plated in a flat plate for 48 h. Adherent cells were taken as stromal cells. L5178Y-R was then incubated with the bone marrow stromal cells (BMSC) and its supernatant at a 1:10 ratio (tumor to BMSC) prior to ICRP, CTX, and ICRP + CTX treatment as mentioned above. Cell death was then measured as described previously.
4.10. ROS Production Analysis
Quantification of ROS production was performed using 2.5 μM Hydroetidine (HE) staining (Invitrogen, St. Louis, MO, USA). Cells (5 × 105 cells/mL) were exposed to ICRP, CTX, and their combination in 96-well dishes (CORNING) for 24 h. Cells were then harvested and washed with PBS before staining incubation. HE was incubated for 30 min at 37 °C and then washed with PBS for assessment by flow cytometry and analyzed as described above.
4.11. Mitochondrial Membrane Potential Analysis
In 5 × 105 cells/mL plated in 96-well dishes (CORNING), treated as mentioned before, and then collected, we performed tetramethyl rhodamine ethyl ester staining analysis (TMRE, 125 nM, Sigma-Aldrich, St. Louis, MO, USA) which was incubated at 37 °C for 30 min to determine loss of mitochondrial membrane potential. Then, cells were washed with PBS to measure the loss of TMRE-fluorescence by flow cytometry as described above.
4.12. Caspase Activity Assay
TF2-VAD-FMK, the Generic Caspase Activity FMK staining kit staining (Abcam, Cambridge, UK) was used to assess caspase activity in cells (5 × 105 cells/mL) that were treated with ICRP, CTX, and ICRP + CTX-combinations for 24 h, according to manufacturer’s instructions. Analyses were performed by flow cytometry as described above.
4.13. Tumor Establishment and Treatment
L5178Y-R cells (1 × 106) were suspended in 100 μL PBS and injected into the female mice left hind s.c. Three times per week, the tumor volume and mice weight were measured using a caliper (Digimatic Caliper Mitutoyo Corporation, Kanagawa, Japan) and a digital scale (American Weigh Scale-600-BLK, Atlanta, GA, USA). When the tumor reached 100–120 mm3 after inoculation, mice (n = 5 per group, assigned randomly) were injected with 2 U i.p. every two days, 125 mg/kg CTX i.p., weekly, or the combination of ICRP + CTX. Control mice were treated with 100 μL sterile water for injection. All treatments were dissolved in sterile water for injection. The following formula was used to determine tumor volume: tumor volume (mm3) = (Length × width2)/2. Twenty-three days after inoculation of tumor cells, mice were anesthetized as mentioned above, blood was obtained by cardiac puncture for hematic biometry, from which the granulocyte/lymphocyte ratio was determined, and mice were then euthanized by cervical dislocation. Tumor and spleen were obtained and weighed.
4.14. Splenocytes + L5178Y-R Co-Culture
L5178Y-R was stained with 0.1 mg/mL Calcein-AM (BD biosciences, San José, CA, USA) for 30 min at 37 °C and 5% CO2. Cells were then washed twice with PBS. Thus, splenocytes (obtained as previously described) were added in a 44:1 (splenocytes to tumor) ratio. Co-culture was maintained at 37 °C and 5% CO2 for 24 h and calcein-negative L5178Y-R cells were measured by flow cytometry.
4.15. Ki67 Analysis
Dissected tumors were macerated and filtered through a cell strainer (70 μM) with PBS and tumor cells (1 × 106) were fixed then in ethanol dropwise gradient (50% to 70%) while vortexing and incubated at −20 °C overnight. Cells were washed twice and analyzed using Ki-67 (Alexa Fluor 647 anti-human Ki-67 Antibody, BioLegend, San Diego, CA, USA).
4.16. Statistical Analysis
Triplicate determinations from at least three independent experiments were presented as means ± SD in graphs. Results were analyzed by GraphPad Prism software (San Diego, CA, USA), using paired Student’s t-tests for in vitro studies, and two-tailed unpaired Student’s-t-tests and Mann–Whitney tests for the ex vivo and in vivo studies, considering statistical significance as p < 0.05.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/ijms25147938/s1.
Author Contributions
A.L.R.-L., K.M.C.-R., M.I.-R. and A.C.M.-T.: planned experiments; A.L.R.-L., K.M.C.-R. and M.I.-R.: performed experiments; A.L.R.-L., K.M.C.-R., M.I.-R., J.M.V.-G., A.C.M.-T. and C.R.-P.: analyzed and interpreted data; A.C.M.-T. and C.R.-P.: contributed with reagents or other essential material; A.L.R.-L. and A.C.M.-T.: wrote the paper; A.C.M.-T.: conceived, designed, and supervised the work; A.L.R.-L., K.M.C.-R., M.I.-R., J.M.V.-G., A.C.M.-T. and C.R.-P.: reviewed the work; A.L.R.-L., K.M.C.-R., M.I.-R., J.M.V.-G., A.C.M.-T. and C.R.-P.: revised and approved the final version. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
All experiments involving animals were reviewed and approved by the Ethical Committee (CEIBA) of the School of Biological Sciences at the UANL: CEIBA-2020-015. For animal samples, all experiments were performed following the Mexican regulation NOM-062-ZOO-1999 and were designed according to the Arrive guidelines for animal care and protection [56]. The procedures in our study involving human samples were conducted in accordance with the Helsinki Declaration.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data is contained within the article or Supplementary Material.
Acknowledgments
Authors thank Alejandra Arreola-Triana for english language editing, and the Laboratory of Immunology and Virology at the UANL for the infrastructure provided to achieve this work. A.L.R.-L., M.I.-R., and K.M.C.-R. are thankful for the scholarship provided by CONAHCYT. A.L.R.-L., K.M.C.-R., A.C.M.-T., and C.R.-P. thank ECOS-NORD, ANUIES CONAHCYT.
Conflicts of Interest
All authors have read the journal’s policy on disclosure of potential conflicts of interest. The authors declare no conflicts of interest. C.R.P. is an employee of LONGEVEDEN SA de CV; the company had no role in the design of the study, in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Network, H.M.R. (n.d.). Classification of Haematological Malignancies. Network, Haematological Malignancy Research. Available online: https://hmrn.org/about/classification (accessed on 15 July 2024).
- Intermesoli, T.; Weber, A.; Leoncin, M.; Frison, L.; Skert, C.; Bassan, R. Lymphoblastic Lymphoma: A Concise Review. Curr. Oncol. Rep. 2022, 24, 1–12. [Google Scholar] [CrossRef]
- Fleischer, L.C.; Spencer, H.T.; Raikar, S.S. Targeting T cell malignancies using CAR-based immunotherapy: Challenges and potential solutions. J. Hematol. Oncol. 2019, 12, 141. [Google Scholar] [CrossRef]
- Kroeze, E.; Loeffen, J.L.C.; Poort, V.M.; Meijerink, J.P.P. T-cell lymphoblastic lymphoma and leukemia: Different diseases from a common premalignant progenitor? Blood Adv. 2020, 4, 3466–3473. [Google Scholar] [CrossRef]
- Emadi, A.; Jones, R.J.; Brodsky, R.A. Cyclophosphamide and cancer: Golden anniversary. Nat. Reviews. Clin. Oncol. 2009, 6, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Pan, T.; Liu, C.; Shan, X.; Xu, Z.; Hong, H.; Lin, H.; Chen, J.; Sun, H. Cyclophosphamide induced physiological and biochemical changes in mice with an emphasis on sensitivity analysis. Ecotoxicol. Environ. Saf. 2021, 211, 111889. [Google Scholar] [CrossRef] [PubMed]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef]
- Khasraw, M.; Bell, R.; Dang, C. Epirubicin: Is it like doxorubicin in breast cancer? A clinical review. Breast 2012, 21, 142–149. [Google Scholar] [CrossRef]
- Wood, L.J.; Nail, L.M.; Perrin, N.A.; Elsea, C.R.; Fischer, A.; Druker, B.J. The cancer chemotherapy drug etoposide (VP-16) induces proinflammatory cytokine production and sickness behavior-like symptoms in a mouse model of cancer chemotherapy-related symptoms. Biol. Res. Nurs. 2006, 8, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A. Apoptosis and the dilemma of cancer chemotherapy. Blood 1997, 89, 1845–1853. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Sadeghi, M.; Hamdi Hajibaba, H.; Valizadeh, Y.; Movasaghpour Akbari, A.A.; Hosseinpour Feizi, A.A.; Aghebati-Maleki, L.; Jadidi-Niaragh, F. Combinational therapy of acute lymphoblastic leukemia with cyclophosphamide and BV6 synergistically induces apoptosis in leukemic cells. ImmunoAnalysis 2022, 2, 9. [Google Scholar] [CrossRef]
- Smith, A.L.; Pal, D.; Martinez-Rico, R.; Eiken, A.P.; Durden, D.L.; Kutateladze, T.G.; El-Gamal, D. Preclinical activity of a novel multi-axis inhibitor in aggressive and indolent B-cell malignancies. Leuk. Lymphoma 2023, 64, 2333–2337. [Google Scholar] [CrossRef] [PubMed]
- Barreca, M.; Lang, N.; Tarantelli, C.; Spriano, F.; Barraja, P.; Bertoni, F. Antibody-drug conjugates for lymphoma patients: Preclinical and clinical evidences. Explor. Target. Anti-Tumor Ther. 2022, 3, 763–794. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhu, F.; Ma, X.; Cao, Z.; Cao, Z.W.; Li, Y.; Li, Y.X.; Chen, Y.Z. Mechanisms of drug combinations: Interaction and network perspectives. Nat. Rev. Drug Discov. 2009, 8, 111–128. [Google Scholar] [CrossRef]
- Pemovska, T.; Bigenzahn, J.W.; Superti-Furga, G. Recent advances in combinatorial drug screening and synergy scoring. Curr. Opin. Pharmacol. 2018, 42, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.; Talalay, P. Analysis of combined drug effects: A new look at a very old problem. Trends Pharmacol. Sci. 1983, 4, 450–454. [Google Scholar] [CrossRef]
- Correia, A.S.; Gärtner, F.; Vale, N. Drug combination and repurposing for cancer therapy: The example of breast cancer. Heliyon 2021, 7, e05948. [Google Scholar] [CrossRef] [PubMed]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Anota, H.Y.; Martínez-Torres, A.C.; Scott-Algara, D.; Tamez-Guerra, R.S.; Rodríguez-Padilla, C. Bovine Dialyzable Leukocyte Extract IMMUNEPOTENT-CRP Induces Selective ROS-Dependent Apoptosis in T-Acute Lymphoblastic Leukemia Cell Lines. J. Oncol. 2020, 2020, 1598503. [Google Scholar] [CrossRef]
- Lorenzo-Anota, H.Y.; Martínez-Loria, A.B.; Tamez-Guerra, R.S.; Scott-Algara, D.; Martínez-Torres, A.C.; Rodríguez-Padilla, C. Changes in the natural killer cell repertoire and function induced by the cancer immune adjuvant candidate IMMUNEPOTENT-CRP. Cell. Immunol. 2022, 374, 104511. [Google Scholar] [CrossRef]
- Santana-Krímskaya, S.E.; Franco-Molina, M.A.; Zárate-Triviño, D.G.; Prado-García, H.; Zapata-Benavides, P.; Torres-Del-Muro, F.; Rodríguez-Padilla, C. IMMUNEPOTENT CRP plus doxorubicin/cyclophosphamide chemotherapy remodel the tumor microenvironment in an air pouch triple-negative breast cancer murine model. Biomed. Pharmacother. 2020, 126, 110062. [Google Scholar] [CrossRef]
- Rodríguez-Salazar, M.D.C.; Franco-Molina, M.A.; Mendoza-Gamboa, E.; Martínez-Torres, A.C.; Zapata-Benavides, P.; López-González, J.S.; Coronado-Cerda, E.E.; Alcocer-González, J.M.; Tamez-Guerra, R.S.; Rodríguez-Padilla, C. The novel immunomodulator IMMUNEPOTENT CRP combined with chemotherapy agent increased the rate of immunogenic cell death and prevented melanoma growth. Oncol. Lett. 2017, 14, 844–852. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lara, H.H.; Turrent, L.I.; Garza-Treviño, E.N.; Tamez-Guerra, R.; Rodriguez-Padilla, C. Clinical and immunological assessment in breast cancer patients receiving anticancer therapy and bovine dialyzable leukocyte extract as an adjuvant. Exp. Ther. Med. 2010, 1, 425–431. [Google Scholar] [CrossRef]
- Méndez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef]
- Reyes-Ruiz, A.; Calvillo-Rodriguez, K.M.; Martínez-Torres, A.C.; Rodríguez-Padilla, C. The bovine dialysable leukocyte extract IMMUNEPOTENT CRP induces immunogenic cell death in breast cancer cells leading to long-term antitumour memory. Br. J. Cancer 2021, 124, 1398–1410. [Google Scholar] [CrossRef]
- Follini, E.; Marchesini, M.; Roti, G. Strategies to Overcome Resistance Mechanisms in T-Cell Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2019, 20, 3021. [Google Scholar] [CrossRef]
- Savage, P. Chemotherapy Curability in Leukemia, Lymphoma, Germ Cell Tumors and Gestational Malignancies: A Reflection of the Unique Physiology of Their Cells of Origin. Front. Genet. 2020, 11, 426. [Google Scholar] [CrossRef] [PubMed]
- Frei, E., 3rd; Karon, M.; Levin, R.H.; Freireich, E.J.; Taylor, R.J.; Hananian, J.; Selawry, O.; Holland, J.F.; Hoogstraten, B.; Wolman, I.J.; et al. The effectiveness of combinations of antileukemic agents in inducing and maintaining remission in children with acute leukemia. Blood 1965, 26, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Peters, R.H.; Stuart, R.K. Synergism between 4-hydroperoxycyclophosphamide and cisplatin: Importance of incubation sequence and measurement of cisplatin accumulation. Biochem. Pharmacol. 1990, 39, 607–609. [Google Scholar] [CrossRef]
- Coll-Mulet, L.; Iglesias-Serret, D.; Santidrián, A.F.; Cosialls, A.M.; de Frias, M.; Castaño, E.; Campàs, C.; Barragán, M.; de Sevilla, A.F.; Domingo, A.; et al. MDM2 antagonists activate p53 and synergize with genotoxic drugs in B-cell chronic lymphocytic leukemia cells. Blood 2006, 107, 4109–4114. [Google Scholar] [CrossRef]
- Lonetti, A.; Cappellini, A.; Bertaina, A.; Locatelli, F.; Pession, A.; Buontempo, F.; Evangelisti, C.; Evangelisti, C.; Orsini, E.; Zambonin, L.; et al. Improving nelarabine efficacy in T cell acute lymphoblastic leukemia by targeting aberrant PI3K/AKT/mTOR signaling pathway. J. Hematol. Oncol. 2016, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, M.S.; Mohammadi, M.H.; Vahabpour Roudsari, R.; Jafari, L.; Mashati, P.; Gharehbaghian, A. Proteasome Inhibition by Carfilzomib Induced Apotosis and Autophagy in a T-cell Acute Lymphoblastic Leukemia Cell Line. Iran. J. Pharm. Res. 2019, 18 (Suppl. 1), 132–145. [Google Scholar] [CrossRef]
- Hassani, S.; Ghaffari, S.H.; Zaker, F.; Mirzaee, R.; Mardani, H.; Bashash, D.; Zekri, A.; Yousefi, M.; Zaghal, A.; Alimoghaddam, K.; et al. Azidothymidine hinders arsenic trioxide-induced apoptosis in acute promyelocytic leukemia cells by induction of p21 and attenuation of G2/M arrest. Ann. Hematol. 2013, 92, 1207–1220. [Google Scholar] [CrossRef]
- Lorenzo-Anota, H.Y.; Reyes-Ruiz, A.; Calvillo-Rodríguez, K.M.; Mendoza-Reveles, R.; Urdaneta-Peinado, A.P.; Alvarez-Valadez, K.M.; Martínez-Torres, A.C.; Rodríguez-Padilla, C. IMMUNEPOTENT CRP increases intracellular calcium through ER-calcium channels, leading to ROS production and cell death in breast cancer and leukemic cell lines. EXCLI J. 2023, 22, 352–366. [Google Scholar] [CrossRef]
- Iqubal, A.; Iqubal, M.K.; Sharma, S.; Ansari, M.A.; Najmi, A.K.; Ali, S.M.; Ali, J.; Haque, S.E. Molecular mechanism involved in cyclophosphamide-induced cardiotoxicity: Old drug with a new vision. Life Sci. 2019, 218, 112–131. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Lazarín, A.L.; Martínez-Torres, A.C.; de la Hoz-Camacho, R.; Guzmán-Aguillón, O.L.; Franco-Molinaa, M.A.; Rodríguez-Padilla, C. The bovine dialyzable leukocyte extract, immunepotent CRP, synergically enhances cyclophosphamide-induced breast cancer cell death, through a caspase-independent mechanism. EXCLI J. 2023, 22, 131–145. [Google Scholar] [CrossRef]
- Bhalla, S.; Balasubramanian, S.; David, K.; Sirisawad, M.; Buggy, J.; Mauro, L.; Prachand, S.; Miller, R.; Gordon, L.I.; Evens, A.M. PCI-24781 induces caspase and reactive oxygen species-dependent apoptosis through NF-kappaB mechanisms and is synergistic with bortezomib in lymphoma cells. Clin. Cancer Res. 2009, 15, 3354–3365. [Google Scholar] [CrossRef] [PubMed]
- Park, M.T.; Kim, M.J.; Kang, Y.H.; Choi, S.Y.; Lee, J.H.; Choi, J.A.; Kang, C.M.; Cho, C.K.; Kang, S.; Bae, S.; et al. Phytosphingosine in combination with ionizing radiation enhances apoptotic cell death in radiation-resistant cancer cells through ROS-dependent and -independent AIF release. Blood 2005, 105, 1724–1733. [Google Scholar] [CrossRef]
- Calvillo-Rodríguez, K.M.; Lorenzo-Anota, H.Y.; Rodríguez-Padilla, C.; Martínez-Torres, A.C.; Scott-Algara, D. Immunotherapies inducing immunogenic cell death in cancer: Insight of the innate immune system. Front. Immunol. 2023, 14, 1294434. [Google Scholar] [CrossRef]
- Coronado-Cerda, E.E.; Franco-Molina, M.A.; Mendoza-Gamboa, E.; Prado-García, H.; Rivera-Morales, L.G.; Zapata-Benavides, P.; Rodríguez-Salazar Mdel, C.; Caballero-Hernandez, D.; Tamez-Guerra, R.S.; Rodríguez-Padilla, C. In Vivo Chemoprotective Activity of Bovine Dialyzable Leukocyte Extract in Mouse Bone Marrow Cells against Damage Induced by 5-Fluorouracil. J. Immunol. Res. 2016, 2016, 6942321. [Google Scholar] [CrossRef]
- Fahmy, M.; Hassan, N.; El-Fiky, S.; Elalfy, H. A mixture of honey bee products ameliorates the genotoxic side effects of cyclophosphamide. Asian Pac. J. Trop. Dis. 2015, 5, 638–644. [Google Scholar] [CrossRef]
- Féral, K.; Jaud, M.; Philippe, C.; Di Bella, D.; Pyronnet, S.; Rouault-Pierre, K.; Mazzolini, L.; Touriol, C. ER Stress and Unfolded Protein Response in Leukemia: Friend, Foe, or Both? Biomolecules 2021, 11, 199. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galassi, C.; Zitvogel, L.; Galluzzi, L. Immunogenic cell stress and death. Nat. Immunol. 2022, 23, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.L.; Yu, S.J.; Li, C.L. Role of Autophagy and Apoptosis in Acute Lymphoblastic Leukemia. Cancer Control 2021, 28, 10732748211019138. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.Z.; Garcia-Prat, L.; Voisin, V.; Ferrari, R.; Gan, O.I.; Wagenblast, E.; Kaufmann, K.B.; Zeng, A.G.X.; Takayanagi, S.I.; Patel, I.; et al. Sphingolipid Modulation Activates Proteostasis Programs to Govern Human Hematopoietic Stem Cell Self-Renewal. Cell Stem Cell 2019, 25, 639–653. [Google Scholar] [CrossRef]
- Li, D.; Ding, Z.; Du, K.; Ye, X.; Cheng, S. Reactive Oxygen Species as a Link between Antioxidant Pathways and Autophagy. Oxidative Med. Cell. Longev. 2021, 2021, 5583215. [Google Scholar] [CrossRef]
- Zeng, Z.; Samudio, I.J.; Munsell, M.; An, J.; Huang, Z.; Estey, E.; Andreeff, M.; Konopleva, M. Inhibition of CXCR4 with the novel RCP168 peptide overcomes stroma-mediated chemoresistance in chronic and acute leukemias. Mol. Cancer Ther. 2006, 5, 3113–3121. [Google Scholar] [CrossRef]
- Kwon, M.; Jung, H.; Nam, G.H.; Kim, I.S. The right Timing, right combination, right sequence, and right delivery for Cancer immunotherapy. J. Control. Release 2021, 331, 321–334. [Google Scholar] [CrossRef]
- Franco-Molina, M.A.; Mendoza-Gamboa, E.; Zapata-Benavides, P.; Vera-García, M.E.; Castillo-Tello, P.; García de la Fuente, A.; Mendoza, R.D.; Garza, R.G.; Tamez-Guerra, R.S.; Rodríguez-Padilla, C. IMMUNEPOTENT CRP (bovine dialyzable leukocyte extract) adjuvant immunotherapy: A phase I study in non-small cell lung cancer patients. Cytotherapy 2008, 10, 490–496. [Google Scholar] [CrossRef]
- Schiavoni, G.; Sistigu, A.; Valentini, M.; Mattei, F.; Sestili, P.; Spadaro, F.; Sanchez, M.; Lorenzi, S.; D’Urso, M.T.; Belardelli, F.; et al. Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis. Cancer Res. 2011, 71, 768–778. [Google Scholar] [CrossRef]
- Liu, H.; Tabuchi, T.; Takemura, A.; Kasuga, T.; Motohashi, G.; Hiraishi, K.; Katano, M.; Nakada, I.; Ubukata, H.; Tabuchi, T. The granulocyte/lymphocyte ratio as an independent predictor of tumour growth, metastasis and progression: Its clinical applications. Mol. Med. Rep. 2008, 1, 699–704. [Google Scholar] [CrossRef] [PubMed]
- García Coronado, P.L.; Franco Molina, M.A.; Zárate Triviño, D.G.; Menchaca Arredondo, J.L.; Zapata Benavides, P.; Rodriguez Padilla, C. Putative Wound Healing Induction Functions of Exosomes Isolated from IMMUNEPOTENT CRP. Int. J. Mol. Sci. 2023, 24, 8971. [Google Scholar] [CrossRef] [PubMed]
- Franco-Molina, M.A.; Santana-Krímskaya, S.E.; Zarate-Triviño, D.G.; Zapata-Benavides, P.; Hernández-Martínez, S.P.; Cervantes-Wong, F.; Rodríguez-Padilla, C. Bovine Dialyzable Leukocyte Extract IMMUNEPOTENT CRP: Evaluation of Biological Activity of the Modified Product. Appl. Sci. 2021, 11, 3505. [Google Scholar] [CrossRef]
- Franco-Molina, M.A.; Santana-Krímskaya, S.E.; Coronado-Cerda, E.E.; Hernández-Luna, C.E.; Zarate-Triviño, D.G.; Zapata-Benavides, P.; Rodríguez-Padilla, C. Increase of the antitumour efficacy of the biocompound IMMUNEPOTENT CRP by enzymatic treatment. Biotechnol. Biotechnol. Equip. 2018, 32, 1028–1035. [Google Scholar] [CrossRef]
- Percie du Sert, N.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; Emerson, M.; et al. Reporting animal research: Explanation and elaboration for the ARRIVE guidelines 2.0. PLoS Biol. 2020, 18, e3000411. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).