Abstract
Anti-IgLON5 (IgLON5-IgG)-associated disease is a newly defined clinical entity. This literature review aims to evaluate its pathogenesis, which remains a pivotal question. Features that favour a primary neurodegenerative mechanism include the non-inflammatory tauopathy neuropathological signature and overrepresentation of microtubule-associated protein tau (MAPT) H1/H1 genotype as seen in other sporadic tauopathies. In contrast, the cell-surface localisation of IgLON5, capability of anti-IgLON5 antibodies to exert direct in vitro pathogenicity and disrupt IgLON5 interactions with its binding partners, human leukocyte antigen (HLA)-DRB1*10:01 and HLA-DQB1*05:01 allele preponderance with high affinity binding of IgLON5 peptides, and responsiveness to immunotherapy favour a primary autoimmune process. The presentation and course of anti-IgLON5-associated disease is heterogenous; hence, we hypothesise that a multitude of immune mechanisms are likely simultaneously operational in this disease cohort.
1. Introduction
Anti-IgLON5 antibodies (IgLON5-IgG) from serum and cerebrospinal fluid (CSF) were first described clinically in a cohort of patients with sleep apnoea, non-rapid eye movement (non-REM) and REM parasomnias, and stridor in 2014 [1]. In the years following this seminal study, there has been increasing research into this disease, albeit largely limited to case reports and case series given its estimated incidence of 1 in 150,000 [2]. The IgLON family is a group of five cell adhesion molecules (IgLON1-5) each with three immunoglobulin-like domains, which mediate a multitude of cellular interactions, particularly at the blood–brain barrier (BBB) [3,4,5,6]. An investigation of the molecular evolution of the IgLON family suggests a role in regulating neural growth and complexity by possessing several motifs that may influence cellular migration and proliferation as well as BBB permeability [7]. While BBB dysfunction, characterized by an increased albumin quotient (CSF albumin/serum albumin ratio) and total CSF protein, appears to be a feature in approximately half of patients with IgLON5-IgG disease, the physiologic role of IgLON5 in BBB integrity is incompletely characterized [8,9]. It is unclear whether IgLON5-IgG is synthesized intrathecally, peripherally, or a combination of both [10,11]. The pathogenesis of IgLON5-IgG disease remains a pivotal question. To date, the literature has focused on whether IgLON5-IgG disease is a primary neurodegenerative process with a secondary inflammatory response against IgLON5 or a primary autoimmune process driven by pathogenic IgLON5-IgG with subsequent neurodegeneration. The aim of this review article is to synthesise the existing literature and evaluate this question. We hypothesise that a multitude of immune mechanisms are likely simultaneously operational in IgLON5-IgG disease as evidenced by the heterogeneity of the patient cohort and, thus, we caution against relying on a dichotomous classification of the pathogenesis.
2. Evidence for a Primary Neurodegenerative Mechanism
The following evidence favours a primary neurodegenerative mechanism underlying IgLON5-IgG disease. Firstly, the neuropathological signature of IgLON5-IgG disease as first described by Sabater et al. and formalised in the recently proposed IgLON5-IgG disease neuropathological criteria involves three key features: (1) subcortical distribution of the tau pathology predominantly affecting the hypothalamus, brainstem tegmentum, and upper spinal cord; (2) tau pathology, nearly exclusively neuronal, with little or no glial and white matter involvement; and (3) disease-associated tau composed of both 3R and 4R isoforms, which are defined by the presence of three and four microtubule-binding repeats, respectively [1,12]. The topographical distribution of the tau pathology is similar to that seen in another tauopathy, progressive supra-nuclear palsy (PSP); however, the absence of glial pathology and sparse supratentorial and basal ganglia involvement in IgLON5-IgG disease is distinct [12,13]. Fearnley et al. examined the spatiotemporal expression of all IgLON family members in the developing murine nervous system at prenatal and postnatal stages [14]. IgLON5 expression was detected prenatally in the eye and olfactory system with subsequent postnatal downregulation at both sites, throughout development in the cerebral cortex, hippocampus, entorhinal region, habenula, and various nuclei of the thalamus and postnatally in the cerebellum and spinal cord [14]. This contrasts against postmortem pathology-mapping, which is largely based on end-stage pathology such as neuronal loss; partial overlap with the regions of interest identified by murine modelling of IgLON5 expression is noted and may explain at least part of the regional vulnerability observed in IgLON5-IgG disease [1,14,15]. The tau filaments seen in IgLON5-IgG disease are of similar structure to those in Alzheimer’s disease (AD), appearing ultrastructurally as paired helical filaments, being composed of both 3R and 4R tau isoforms [12,16].
Secondly, the microtubule-associated protein tau (MAPT) H1/H1 genotype is significantly overrepresented in patients with IgLON5-IgG disease (81.5%, 22/27) compared to healthy controls (46.5%, 54/116) (OR 5.05, 95% CI 1.79–14.25, p = 0.0007) [17]. The MAPT locus is divided into two haplotypes (H1 and H2) by a common inversion polymorphism [18,19,20]. It is well evidenced that H1 homozygosity is associated with an increased risk for tauopathies like PSP, corticobasal degeneration (CBD), and AD [19,21,22,23,24,25,26]. The underlying mechanism linking the H1 haplotype with neurodegeneration is likely related to altered expression levels, altered splicing, or a combination of both [25]. Indeed, the H1 haplotype has been associated with an increased expression of MAPT overall, as well as increased expression of the key exon 10, which, through alternative splicing, generates six tau isoforms (three 3R isoforms and three 4R isoforms) whose expression ratio tightly directs microtubule dynamics [25,26,27,28,29,30]. It is important to note that these findings have not yet been evaluated in an IgLON5-IgG disease cohort.
Finally, poor response to immunotherapy was initially reported and suggested a primary neurodegenerative aetiology of IgLON5-IgG disease [1,31]; however, more recent evidence is discordant with this [2,8,32]. This can likely be attributed to earlier studies having limited case numbers as well as cohort selection bias given that patients were recruited based on a narrow clinical phenotype (the originally described sleep disorder). Subsequent studies have had larger cohorts and selection based on serological screening of stored samples, thus revealing a more diverse spectrum of clinical phenotypes. Further studies of treatment efficacy in populous and clinically diverse cohorts are essential to comprehensively understand IgLON5-IgG disease.
3. Evidence for a Primary Autoimmune Mechanism
The following key principles favour a primary autoimmune aetiology of IgLON5-IgG disease. The cell-surface localisation and role of IgLON5 in the dynamic interactions that occur at the BBB implicate it as a biologically plausible and accessible central nervous system (CNS) autoantigen [1,33,34]. Furthermore, IgLON5-IgG in patient sera exerts a directly pathogenic effect on cultured neurons by causing an irreversible decrease in the cell-surface density of IgLON5 through internalisation, reduction in electrical neuronal activity, and increased frequency of degenerative changes in neurons, such as axonal blebbing and fragmentation [34,35]. Purification of IgG subclasses revealed that this effect was specifically attributable to IgG1 antibodies and not IgG4 antibodies [34]. Interestingly, however, the IgG4 subclass was predominant in the majority of patients from this study, accounting for, on average, 64% of total IgLON5-IgG isolated from patient sera compared to IgG1 antibodies accounting for, on average, 33% of total IgLON5-IgG [34]. It is well established that IgG1 antibodies engage C1q and FcγR more efficiently compared to the reduced binding affinity shown by IgG4 antibodies, and thus, IgG1 antibodies are more potent triggers of pro-inflammatory effector mechanisms; however, IgG4 antibodies are also capable of exerting a pathogenic effect as exemplified in Myasthenia gravis where IgG4 antibodies mediate receptor inactivation [36,37]. In keeping with this, Landa, et al. identified another potential mechanism by which IgLON5-IgG isolated from patient sera may exert a pathogenic effect [38]. Firstly, they demonstrated that physiologic IgLON5 undergoes spontaneous ectodomain shedding and interacts with other members of the IgLON family and, secondly, that both IgLON5-IgG1 and IgG4 subclasses disrupt this interaction [38]. A recent autopsy series published by Berger-Sieczkowski et al. identified two patients with short disease duration without the typical anti-IgLON5-related tauopathy who had extensive neuropil deposition of IgLON5-IgG4 in the brainstem tegmentum, olivary nucleus, and cerebellar cortex [15]. These IgG4 deposits were accompanied by lesser amounts of IgG1 [15]. We postulate that both IgLON5-IgG1 and IgLON5-IgG4 are operational through different mechanisms, thus, potentially explaining a degree of the clinical heterogeneity observed in this disease. There are currently no studies published that investigate complement activation in IgLON5-IgG disease; however, this would be useful in further distinguishing the IgG1- and IgG4-mediated processes in this condition.
Human leukocyte antigen (HLA) genotyping of patients with IgLON5-IgG disease highlighted a genetic susceptibility for autoimmune disease. Initial works identified a robust association with the HLA-DRB1*10:01 and HLA-DQB1*05:01 alleles, reported at a frequency of 57.1–100% of patients [1,17,32]. In addition, IgLON5 peptides exhibit high affinity for the HLA-DRB1 molecules (DRB1*01:01, DRB1*10:01, and DRB1*09:01) isolated from patients [17]. A recent study by Yogeshwar et al. was the largest HLA-association analysis in IgLON5-IgG disease to date, and their findings strongly supported that HLA-DQ, and not HLA-DR, is the actual determinant for disease risk [39]. HLA class II molecules are known to play an integral role in antigen presentation to activate the adaptive immune response, including B-cells and T-cells [17]. The weighting of these responses, based on genetic variation such as HLA, seems likely to impact the individual presentation of IgLON5-IgG disease.
Current evidence supports IgLON5-IgG disease responsiveness to immunotherapy [8,32]. A systematic review by Cabezudo-García et al. reported promising responses and sustained response rates with azathioprine (AZA) (100% [5/5]) and mycophenolate mofetil (MMF) (75% [3/4]), albeit always administered in combination with other agents like corticosteroids (CS) (CS alone had a response rate of 34.2% [12/35]) [32]. A large 2023 retrospective cohort study of 53 patients by Grüter et al. reported favourable response of IgLON5-IgG disease to periodic intravenous immunoglobulin (IVIg) (86.7% [13/15]), AZA (85.7% [6/7]), repetitive plasma exchange (PLEX) (75% [3/4]), and rituximab (RIX) (72.2% [13/18]).
Finally, a notable mention is made of several patients’ case reports with clinical and serological evidence of IgLON5-IgG disease without degenerative features like hyperphosphorylated tau in brain tissue, which may shed light on the sequence of pathogenesis of this disease entity, specifically, that tau accumulation occurs later in the disease course and is a consequence of antibody-mediated neuronal dysfunction [15,40,41,42,43].
4. Heterogeneity of the IgLON5-IgG Disease Cohort
The presentation and progress of IgLON5-IgG disease is diverse. Subgroups appear to diverge in terms of disease time-course and symptomatology [2,8,44]. Grüter et al. reported 28% (15/53) of patients had subacute disease onset (≤4 weeks) while 72% (38/53) had slow progressive disease onset (>4 weeks) with (32%, 12/38) or without (68%, 26/38) overlapping relapse-like exacerbations [8]. Interestingly, patients with subacute disease onset were significantly more likely to manifest psychosis and/or hallucinations as part of their clinical phenotype, have an inflammatory CSF characterised by pleocytosis (albeit mild), and exhibit a more pronounced response to immunotherapy [8]. An important confounding factor is that patients with subacute disease onset, more in keeping with a traditional autoimmune picture, received significantly earlier diagnosis leading to earlier initiation of treatment: an independent predictor of a favourable prognostic outcome [8]. Similar observations were also reported by Gaig et al. who noted that 24% (17/72) of their IgLON5-IgG disease cohort had subacute disease onset (<4 months) and by Honorat et al. who noted that 25% (5/20) of their cohort evolved symptoms in a subacute manner (<4 weeks) [2,44]. Neither clinical characteristics nor CSF examination stratified specifically by disease time-course were available in either of these studies. Furthermore, the IgLON5-IgG disease cohort may be stratified by HLA-DRB1*10:01 and HLA-DQB1*05:01 positivity. Grüter et al. identified that HLA-DRB1*10:01- and HLA-DQB1*05:01-positive patients were significantly younger at disease onset, more frequently exhibited characteristic sleep disorders, and had a higher IgLON5-IgG titre compared to HLA-DRB1*non-10:01 and HLA-DQB1*non-05:01 patients [8]. Similarly, Gaig et al. reported that HLA-DRB1*10:01-positive patients were younger (median 64.5 years [range 46–77]) at diagnosis and more frequently presented with sleep or bulbar clinical phenotypes compared to HLA-DRB1*non-10:01 patients who were older (median 71 years [range 61–83]) at diagnosis and tended to present with PSP-like or cognitive impairment phenotypes; all of these findings reached statistical significance [17]. As described earlier, we further postulate that IgG1 or IgG4 subclass antibodies may influence whether that patient’s disease is more pro-inflammatory and subacute or non-inflammatory and chronic, respectively. Ultimately, the model we propose is that patients with IgLON5-IgG disease may exhibit features of either a primarily neurodegenerative process, a primarily autoimmune process, or a combination of both. For example, a chronic onset process of neurodegeneration presenting with a specific clinical phenotype may then be augmented by exposure to an inflammatory insult, such as a virus infection, triggering an autoimmune process and altering disease course. The heterogeneity of IgLON5-IgG disease is complex and further observational cohort studies are essential.
5. An Atypical Case and Future Directions
Finally, we revisit a previously published atypical case assessed at our centre, initially reported in 2018 [42]. A 49-year-old man presented with a two-year history of cold intolerance followed by the development of involuntary jerking movements, impaired sleep, gait ataxia, dysarthria, and cognitive decline. He was systemically well throughout this chronic course and had no significant past medical history. He was investigated extensively. Prior to the result of his serum and CSF positivity for IgLON5-IgG becoming available, he underwent a stereotactic brain biopsy. This demonstrated cuffing of the white matter blood vessels with CD3+ T lymphocytes as well as mild leptomeningeal chronic inflammation with cortical and white matter gliosis and microglial activation (Figure 1) [42]. Neurodegenerative stains (tau, α-synuclein, β-amyloid, phosphorylated-TDP43, and P62) were all negative; however, it is worth noting that the superficial areas biopsied (cortex and cerebellum) usually do not demonstrate tauopathy in this disease.
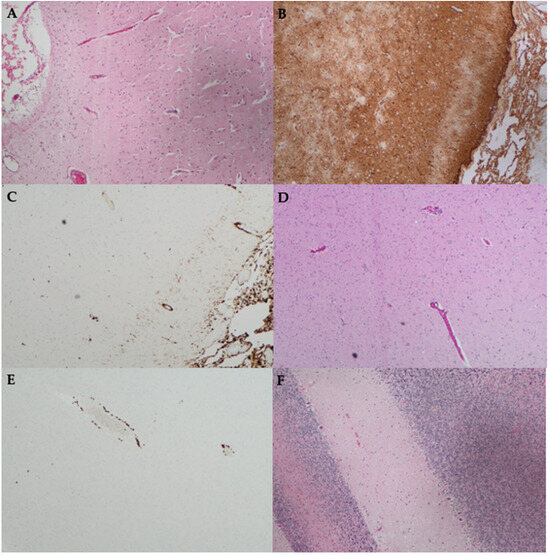
Figure 1.
Histopathology findings from the stereotactic brain biopsy of a patient with IgLON5-IgG disease, which was previously published as a case report [42]. Biopsy of the right frontal cortex stained with (A) Hematoxylin and Eosin (H&E) stain shows leptomeningeal oedema; (B) Glial fibrillary acidic protein (GFAP) shows gliosis; (C) CD163 shows increased histiocytes in the leptomeninges and very mild increase in the underlying cortex; (D) H&E stain shows subtle perivascular lymphocytic infiltrate; and (E) CD3 immunohistochemistry shows CD3+ T lymphocyte cuffing of blood vessels. (F) Biopsy of the left cerebellum stained with H&E shows a reduction in Purkinje cells. Magnification ×100 for all.
The initial treatment regimen consisted of RIX, alemtuzumab (ALEM), PLEX, IVIg, and CS in close succession. In addition to the course of ALEM and RIX, he received two separate courses of intravenous cyclophosphamide (CP) (each course consisting of 500 mg fortnightly for six doses) as per the EuroLupus protocol [45]. He remains on monthly PLEX and IVIg as well as MMF. ALEM was selected based on the presence of CD3+ T lymphocytes on brain biopsy (Figure 1), and to the best of our knowledge, this is the only report of its use to treat IgLON5-IgG disease [42]. ALEM, a monoclonal antibody that targets the CD52 antigen abundantly expressed on T-lymphocytes, has proven useful in the treatment of T-cell-driven pathologies like haematologic malignancies [46]. It was previously thought that T lymphocytes do not have a role in the pathogenesis of IgLON5-IgG disease [17]. However, the usefulness of alemtuzumab in this case and similar reports of perivascular and parenchymal CD3+ and CD8+ T lymphocytes in IgLON5-IgG disease autopsy specimens may contradict this view and support the consideration of a more personalised approach to diagnosis and treatment of rare diseases such as IgLON5-IgG disease [15,42].
Careful phenotyping of the individual patient’s likely underlying pathogenesis, as demonstrated in this atypical case, is essential to instituting the most appropriate treatment regimen. A crucial future direction of this field will be further phenotyping of the IgLON5-IgG disease cohort, in particular, further characterisation of the subsets of patients who present with a more traditional autoimmune picture and those with a more gradual onset of disease, perhaps more in keeping with a neurodegenerative phenotype. The former is indicated by evidence of inflammation and a more acute to subacute presentation with a propensity for the characteristic sleep disorder phenotype. The subset of patients in keeping with a neurodegenerative picture generally present in a more non-inflammatory and chronic manner with a propensity for the PSP-like and cognitive impairment phenotypes [8,17]. While this carries a clear clinical implication for accurate and timely diagnosis, at this stage, the treatment pathway remains equivalent between the subgroups. Immunotherapy is the cornerstone for addressing the inflammatory component of IgLON5-IgG disease as there are currently no effective treatments for the neurodegenerative component though putative therapeutic targets have been identified in the context of other tauopathies [47,48]. Interestingly, these have a role in inflammation as well, indicating that future diagnostic approaches such as sequencing/proteomics may help stratify patient therapy [47,48]. Treatments targeting pathological tau are predominantly in the discovery and preclinical stages [48].
6. Conclusions
In conclusion, IgLON5-IgG disease is a relatively newly defined clinical entity. The non-inflammatory tauopathy neuropathological signature and overrepresentation of MAPT H1/H1 genotype as seen in other sporadic tauopathies is consistent with a primary neurodegenerative process. In these cases, it is conceivable that neurodegeneration preceded the development of an antibody response. In contrast, the cell-surface localisation of IgLON5, capability of IgLON5-IgG to exert direct in vitro pathogenicity and disrupt IgLON5 interactions with its binding partners, HLA-DRB1*10:01 and HLA-DQB1*05:01 allele preponderance loaded with high affinity binding of IgLON5 peptides, and responsiveness to immunotherapy favour a primary autoimmune mechanism.
Nonetheless, we caution against relying on this dichotomous classification. We hypothesise that a multitude of immune mechanisms are likely simultaneously operational in response to varied triggering factors as evidenced by the time-course and phenotype heterogeneity of the IgLON5-IgG disease cohort. This heterogeneity appears to be explained, in part, when patients are stratified by HLA-DRB1*10:01 and HLA-DQB1*05:01 positivity. HLA class II molecules are implicated in antigen presentation and the subsequent activation of the adaptive immune response, including B-cells and T-cells. The weighting of these responses, based on genetic variation such as HLA, seems likely to impact the individual presentation of IgLON5-IgG disease. We promote moving toward a personalised approach to diagnosis and treatment, one that encapsulates the factors outlined above to best define the underlying dominant pathogenic factors in the individual. Moreover, careful clinical phenotyping and bio-banking of these rare diseases is encouraged so that further hypotheses may be generated and tested.
Author Contributions
Conceptualization, D.A.B., M.-W.L., B.J., and J.A.; writing—original draft preparation, J.A.; writing—review and editing, J.A., D.A.B., and M.-W.L.; histopathology, W.V. and M.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sabater, L.; Gaig, C.; Gelpi, E.; Bataller, L.; Lewerenz, J.; Torres-Vega, E.; Contreras, A.; Giometto, B.; Compta, Y.; Embid, C.; et al. A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: A case series, characterisation of the antigen, and post-mortem study. Lancet Neurol. 2014, 13, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Honorat, J.A.; Komorowski, L.; Josephs, K.A.; Fechner, K.; St Louis, E.K.; Hinson, S.R.; Lederer, S.; Kumar, N.; Gadoth, A.; Lennon, V.A.; et al. IgLON5 antibody: Neurological accompaniments and outcomes in 20 patients. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e385. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.; Silva, G.; Junior, W.; Gonçalves, M. Anti-Iglon5 Syndrome: What We Know So Far? A Non-Systematic Review. J. Neurol. Neuromed. 2020, 5, 40–44. [Google Scholar] [CrossRef]
- Reed, J.; McNamee, C.; Rackstraw, S.; Jenkins, J.; Moss, D. Diglons are heterodimeric proteins composed of IgLON subunits, and Diglon-CO inhibits neurite outgrowth from cerebellar granule cells. J. Cell Sci. 2004, 117, 3961–3973. [Google Scholar] [CrossRef]
- McNamee, C.J.; Youssef, S.; Moss, D. IgLONs form heterodimeric complexes on forebrain neurons. Cell Biochem. Funct. 2011, 29, 114–119. [Google Scholar] [CrossRef]
- Madetko, N.; Marzec, W.; Kowalska, A.; Przewodowska, D.; Alster, P.; Koziorowski, D. Anti-IgLON5 Disease—The Current State of Knowledge and Further Perspectives. Front. Immunol. 2022, 13, 852215. [Google Scholar] [CrossRef] [PubMed]
- Kubick, N.; Brösamle, D.; Mickael, M.-E. Molecular Evolution and Functional Divergence of the IgLON Family. Evol. Bioinform. 2018, 14, 1176934318775081. [Google Scholar] [CrossRef] [PubMed]
- Grüter, T.; Möllers, F.E.; Tietz, A.; Dargvainiene, J.; Melzer, N.; Heidbreder, A.; Strippel, C.; Kraft, A.; Höftberger, R.; Schöberl, F.; et al. Clinical, serological and genetic predictors of response to immunotherapy in anti-IgLON5 disease. Brain 2023, 146, 600–611. [Google Scholar] [CrossRef]
- Salluzzo, M.; Vianello, C.; Abdullatef, S.; Rimondini, R.; Piccoli, G.; Carboni, L. The Role of IgLON Cell Adhesion Molecules in Neurodegenerative Diseases. Genes 2023, 14, 1886. [Google Scholar] [CrossRef]
- Zhang, Y.-Z.; Ni, Y.; Gao, Y.-N.; Shen, D.-D.; He, L.; Yin, D.; Meng, H.-Y.; Zhou, Q.-M.; Hu, J.; Chen, S. Anti-IgLON5 disease: A novel topic beyond neuroimmunology. Neural Regen. Res. 2023, 18, 1017–1022. [Google Scholar]
- Koneczny, I.; Macher, S.; Hutterer, M.; Seifert-Held, T.; Berger-Sieczkowski, E.; Blaabjerg, M.; Breu, M.; Dreyhaupt, J.; Dutra, L.A.; Erdler, M.; et al. HLA dependency and possible clinical relevance of intrathecally synthesized anti-IgLON5 IgG4 in anti-IgLON5 disease. Front. Immunol. 2024, 15, 1376456. [Google Scholar] [CrossRef] [PubMed]
- Gelpi, E.; Höftberger, R.; Graus, F.; Ling, H.; Holton, J.L.; Dawson, T.; Popovic, M.; Pretnar-Oblak, J.; Högl, B.; Schmutzhard, E.; et al. Neuropathological criteria of anti-IgLON5-related tauopathy. Acta Neuropathol. 2016, 132, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G. Invited review: Neuropathology of tauopathies: Principles and practice. Neuropathol. Appl. Neurobiol. 2015, 41, 3–23. [Google Scholar] [CrossRef]
- Fearnley, S.; Raja, R.; Cloutier, J.-F. Spatiotemporal expression of IgLON family members in the developing mouse nervous system. Sci. Rep. 2021, 11, 19536. [Google Scholar] [CrossRef] [PubMed]
- Berger-Sieczkowski, E.; Endmayr, V.; Haider, C.; Ricken, G.; Jauk, P.; Macher, S.; Pirker, W.; Högl, B.; Heidbreder, A.; Schnider, P.; et al. Analysis of inflammatory markers and tau deposits in an autopsy series of nine patients with anti-IgLON5 disease. Acta Neuropathol. 2023, 146, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Lidov, H.G.; Duchen, L.W.; Thomas, P.K.; Thrush, D.C. Progressive medullary failure associated with neurofibrillary degeneration. J. Neurol. Neurosurg. Psychiatry 1989, 52, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Gaig, C.; Ercilla, G.; Daura, X.; Ezquerra, M.; Fernández-Santiago, R.; Palou, E.; Sabater, L.; Höftberger, R.; Heidbreder, A.; Högl, B.; et al. HLA and microtubule-associated protein tau H1 haplotype associations in anti-IgLON5 disease. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e605. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef]
- Baker, M.; Litvan, I.; Houlden, H.; Adamson, J.; Dickson, D.; Perez-Tur, J.; Hardy, J.; Lynch, T.; Bigio, E.; Hutton, M. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum. Mol. Genet. 1999, 8, 711–715. [Google Scholar] [CrossRef]
- Stefansson, H.; Helgason, A.; Thorleifsson, G.; Steinthorsdottir, V.; Masson, G.; Barnard, J.; Baker, A.; Jonasdottir, A.; Ingason, A.; Gudnadottir, V.G.; et al. A common inversion under selection in Europeans. Nat. Genet. 2005, 37, 129–137. [Google Scholar] [CrossRef]
- Ghetti, B.; Oblak, A.L.; Boeve, B.F.; Johnson, K.A.; Dickerson, B.C.; Goedert, M. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: A chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 2015, 41, 24–46. [Google Scholar] [CrossRef] [PubMed]
- Pastor, P.; Ezquerra, M.; Tolosa, E.; Muñoz, E.; Martí, M.J.; Valldeoriola, F.; Molinuevo, J.L.; Calopa, M.; Oliva, R. Further extension of the H1 haplotype associated with progressive supranuclear palsy. Mov. Disord. 2002, 17, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Höglinger, G.U.; Melhem, N.M.; Dickson, D.W.; Sleiman, P.M.; Wang, L.S.; Klei, L.; Rademakers, R.; de Silva, R.; Litvan, I.; Riley, D.E.; et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat. Genet. 2011, 43, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Houlden, H.; Baker, M.; Morris, H.R.; MacDonald, N.; Pickering-Brown, S.; Adamson, J.; Lees, A.J.; Rossor, M.N.; Quinn, N.P.; Kertesz, A.; et al. Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology 2001, 56, 1702–1706. [Google Scholar] [CrossRef] [PubMed]
- Myers, A.J.; Pittman, A.M.; Zhao, A.S.; Rohrer, K.; Kaleem, M.; Marlowe, L.; Lees, A.; Leung, D.; McKeith, I.G.; Perry, R.H.; et al. The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol. Dis. 2007, 25, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Kwok, J.B.; Teber, E.T.; Loy, C.; Hallupp, M.; Nicholson, G.; Mellick, G.D.; Buchanan, D.D.; Silburn, P.A.; Schofield, P.R. Tau haplotypes regulate transcription and are associated with Parkinson’s disease. Ann. Neurol. 2004, 55, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, T.M.; Joachim, C.; Paracchini, S.; Esiri, M.M.; Wade-Martins, R. Haplotype-specific expression of exon 10 at the human MAPT locus. Hum. Mol. Genet. 2006, 15, 3529–3537. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.F.; LeBoeuf, A.C.; Massie, M.R.; Jordan, M.A.; Wilson, L.; Feinstein, S.C. Three-and four-repeat tau regulate the dynamic instability of two distinct microtubule subpopulations in qualitatively different manners: Implications for neurodegeneration. J. Biol. Chem. 2005, 280, 13520–13528. [Google Scholar] [CrossRef]
- de Jong, S.; Chepelev, I.; Janson, E.; Strengman, E.; van den Berg, L.H.; Veldink, J.H.; Ophoff, R.A. Common inversion polymorphism at 17q21.31 affects expression of multiple genes in tissue-specific manner. BMC Genom. 2012, 13, 458. [Google Scholar] [CrossRef]
- Gaig, C.; Graus, F.; Compta, Y.; Högl, B.; Bataller, L.; Brüggemann, N.; Giordana, C.; Heidbreder, A.; Kotschet, K.; Lewerenz, J.; et al. Clinical manifestations of the anti-IgLON5 disease. Neurology 2017, 88, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Cabezudo-García, P.; Mena-Vázquez, N.; Estivill Torrús, G.; Serrano-Castro, P. Response to immunotherapy in anti-IgLON5 disease: A systematic review. Acta Neurol. Scand. 2020, 141, 263–270. [Google Scholar] [CrossRef]
- Landa, J.; Gaig, C.; Plagumà, J.; Saiz, A.; Antonell, A.; Sanchez-Valle, R.; Dalmau, J.; Graus, F.; Sabater, L. Effects of IgLON5 Antibodies on Neuronal Cytoskeleton: A Link between Autoimmunity and Neurodegeneration. Ann. Neurol. 2020, 88, 1023–1027. [Google Scholar] [CrossRef]
- Sabater, L.; Planaguma, J.; Dalmau, J.; Graus, F. Cellular investigations with human antibodies associated with the anti-IgLON5 syndrome. J. Neuroinflammation 2016, 13, 226. [Google Scholar] [CrossRef]
- Gamre, M.; Ryding, M.; Nissen, M.S.; Nilsson, A.C.; Meyer, M.; Blaabjerg, M. Investigation of anti-IgLON5-induced neurodegenerative changes in human neurons. bioRxiv 2020, preprint. [Google Scholar]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef]
- Huijbers, M.G.; Zhang, W.; Klooster, R.; Niks, E.H.; Friese, M.B.; Straasheijm, K.R.; Thijssen, P.E.; Vrolijk, H.; Plomp, J.J.; Vogels, P.; et al. MuSK IgG4 autoantibodies cause myasthenia gravis by inhibiting binding between MuSK and Lrp4. Proc. Natl. Acad. Sci. USA 2013, 110, 20783–20788. [Google Scholar] [CrossRef] [PubMed]
- Landa, J.; Serafim, A.B.; Gaig, C.; Saiz, A.; Koneczny, I.; Hoftberger, R.; Santamaria, J.; Dalmau, J.; Graus, F.; Sabater, L. Patients’ IgLON5 autoantibodies interfere with IgLON5-protein interactions. Front. Immunol. 2023, 14, 1151574. [Google Scholar] [CrossRef]
- Yogeshwar, S.M.; Muñiz-Castrillo, S.; Sabater, L.; Peris-Sempere, V.; Mallajosyula, V.; Luo, G.; Yan, H.; Yu, E.; Zhang, J.; Lin, L.; et al. HLA-DQB105 subtypes and not DRB110:01 mediates risk in anti-IgLON5 disease. Brain 2024, 147, 2579–2592. [Google Scholar] [CrossRef]
- Erro, M.E.; Sabater, L.; Martínez, L.; Herrera, M.; Ostolaza, A.; García de Gurtubay, I.; Tuñón, T.; Graus, F.; Gelpi, E. Anti-IGLON5 disease: A new case without neuropathologic evidence of brainstem tauopathy. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e651. [Google Scholar] [CrossRef]
- Cagnin, A.; Mariotto, S.; Fiorini, M.; Gaule, M.; Bonetto, N.; Tagliapietra, M.; Buratti, E.; Zanusso, G.; Ferrari, S.; Monaco, S. Microglial and Neuronal TDP-43 Pathology in Anti-IgLON5-Related Tauopathy. J. Alzheimer’s Dis. 2017, 59, 13–20. [Google Scholar] [CrossRef]
- Morales-Briceño, H.; Cruse, B.; Fois, A.F.; Lin, M.-W.; Jiang, J.; Banerjee, D.; Grunstein, R.; Varikatt, W.; Rodriguez, M.; Shepherd, C.; et al. IgLON5-mediated neurodegeneration is a differential diagnosis of CNS Whipple disease. Neurology 2018, 90, 1113–1115. [Google Scholar] [CrossRef] [PubMed]
- Montagna, M.; Amir, R.; De Volder, I.; Lammens, M.; Huyskens, J.; Willekens, B. IgLON5-Associated Encephalitis With Atypical Brain Magnetic Resonance Imaging and Cerebrospinal Fluid Changes. Front. Neurol. 2018, 9, 329. [Google Scholar] [CrossRef]
- Gaig, C.; Compta, Y.; Heidbreder, A.; Marti, M.J.; Titulaer, M.J.; Crijnen, Y.; Högl, B.; Lewerenz, J.; Erro, M.E.; García-Moncó, J.C.; et al. Frequency and Characterization of Movement Disorders in Anti-IgLON5 Disease. Neurology 2021, 97, e1367–e1381. [Google Scholar] [CrossRef] [PubMed]
- Houssiau, F.A.; Vasconcelos, C.; D’Cruz, D.; Sebastiani, G.D.; Garrido Ed Ede, R.; Danieli, M.G.; Abramovicz, D.; Blockmans, D.; Mathieu, A.; Direskeneli, H.; et al. Immunosuppressive therapy in lupus nephritis: The Euro-Lupus Nephritis Trial, a randomized trial of low-dose versus high-dose intravenous cyclophosphamide. Arthritis Rheum. 2002, 46, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Dearden, C.E.; Matutes, E.; Catovsky, D. Alemtuzumab in T-cell malignancies. Med. Oncol. 2002, 19, S27–S32. [Google Scholar] [CrossRef]
- Góral, I.; Wichur, T.; Sługocka, E.; Godyń, J.; Szałaj, N.; Zaręba, P.; Głuch-Lutwin, M.; Mordyl, B.; Panek, D.; Więckowska, A. Connecting GSK-3β Inhibitory Activity with IKK-β or ROCK-1 Inhibition to Target Tau Aggregation and Neuroinflammation in Alzheimer’s Disease-Discovery, In Vitro and In Cellulo Activity of Thiazole-Based Inhibitors. Molecules 2024, 29, 2616. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Gonzalez, M.I.; Pritchard, M.C.; May, P.C.; Toledo-Sherman, L.M.; Harris, G.A. The therapeutic landscape of tauopathies: Challenges and prospects. Alzheimer’s Res. Ther. 2023, 15, 168. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).