
Navigating Lipodystrophy: Insights from Laminopathies and Beyond

Abstract
:1. Introduction
2. Lamin A in Lipodystrophies
3. Functional Overview of the Human Adipose Tissue
3.1. White Adipose Tissue
3.2. Brown and Beige Adipose Tissue
4. Lipodystrophies—Genes, Pathways and Phenotypes
4.1. Hutchison Gilford Progeria Syndrome

{kind=link}
{kind=link}
| Disease | Disease Type | OMIM # | Gen Association | Chromosomal Location of Mutation | Prevalence (PV) | Clinical Phenotype | Inheritance | Lipodystrophy Specification | Symptomatic Onset (Maximum Life Expectancy) | References |
|---|---|---|---|---|---|---|---|---|---|---|
| HGPS | 176,670 | LMNA | 1q22 | 132 described patients (PV: 1:8,000,000) | -lipodystrophy -micrognathia -hair loss -neoplasm -short stature -accelerated aging -failure to thrive -alopecia -late loss of primary teeth -nail dystrophy -early atherosclerosis -sclerodermatous skin -joint stiffness -muscular abnormalities | autosomal dominant | generalized loss of subcutaneous fat tissue [19] | 6 months (14.5 years) | [65,67,81,82] | |
| MAD | A | 248,370 | LMNA | 1q21 | ~40 described cases (PV unknown) | -lipodystrophy -skin pigmentation -osteoporosis -osteolysis -accelerated aging -retrognathia -joint stiffness -musculoskeletal abnormalities -insulin resistance -hypertriglyceridemia | autosomal recessive | partial loss of SAT in extremities | 2–4 years (-) | [83,84,85,86] |
| B | 608,612 | ZMPSTE24 | 1p34 | 20+ cases (PV unknown) | -lipodystrophy -skin pigmentation -osteoporosis -osteolysis -accelerated aging -retrognathia -joint stiffness -musculoskeletal abnormalities -insulin reistance -hypertriglyceridemia -loss of hair | autosomal recessive | generelized loss of SAT | 2–4 years (-) | [85,87] | |
| FPLD | 1 (Köbberling Type) | 608,600 | polygenic | - | (PV: 1:1,000,000) 13+ described | -lipodystrophy -insulin resistance -coronary artery disease -hypertension -hyperglycemia -hypertriglyceridemia -impaired glucose tolerance | polygenic | SAT loss at extremities Normal/increased fat distribution in face, neck and trunk | Onset in childhood (-) | [2,88,89] |
| 2 (Dunningan type) | 151,660 | LMNA | 1q21 | >500 cases | -lipodystrophy -acanthosis nigricans -female hirtuism -female mens- -trual abnormalities -increased heart disease risk -dyslipidemia -pancreatitis -diabetis mellitus -insulin resistance | autosomal dominant | Partial loss of SAT at limbs, torso, buttocks, hips fat increase in face, neck and buffalo hump formation | Onset in puberty () | [83,90,91] | |
| 3 | 604,367 | PPARG | 3q25 | 20 | -lipodystrophy -hypertension -hypertriglyceridemia -insulin resistance -diabetes mellitus -pancreatitis | autosomal dominant | Partial fat loss in extremities – fat loss stronger in forearms and calves than upper arms and neck | ? (-) | [83,92] | |
| 4 | 613,877 | PLIN1 | 15q26 | 4 families | -lipodystrophy -muscular hypertrhorpy -cushingoid appearance -insulin-resistant -diabetes mellitus -acanthosis nigricans -hypertriglyceridemia -hypertension, -hepatic steatosis -ovarian dysfunction -increased fibrosis -increased macrophage infiltration | autosomal dominant | Partial loss of SAT in gluteal region and lower limbs | Childhood/young adult (-) | [83,93] | |
| 5 | 615,238 | CIDEC | 3p25 | 1 patient | -lipodystrophy -abnormal menstrual cycle -insulin resistance -diabetes mellitus -hepatomegaly -hepatic steatosis -small lipid droplets -increased mitochondrial density -leptin/adiponectin decreased -acanthosis nigricans | autosomal recessive | Partial loss of SAT in lower limbs, normal fat in neck, face and axillary region | Childhood (-) | [83,94] | |
| 6 | 615,980 | LIPE | 19q13 | 2 families | -lipodystrophy -menstrual irregularity -diabetes mellitus -hypertriglyceridemia -hepatic steatosis -low level of HLP | autosomal recessive | abnormal fat increase in neck, abdomen, clavicular regions, axillae, labia majora, back, and area below the triceps fat loss in lower limbs | Adult onset (-) | [83,95,96] | |
| 7 | 606,721 | CAV1 | 7q31.2 | 3 patients | -hyperlipoproteinemia -pancreatitis -acanthosis nigricans | autosomal dominant | subcutaneous fat loss upper body, subcutaneous fat los face | Adult onset | [97] | |
| AKT2 related | - | AKT2 | 19q13.2 | 1 family | -diabetes mellitus -insulin resistance | autosomal dominat | partial lipoathrophy, diabetes mellitus, severe insulin resistance | Adult onset | [98] | |
| Progeroid Syndroms | 614,008 | BANF1 | 11q13 | 3 families | -light brown skin spots on thorax -severe osteoporosis -osteolysis -micrognathia -scoliosis -pulmonary hypertension -mitral regurgitation | autosomal recessive | generalized lipoathrophy | [99] | ||
| Atypical Progeroid Sydromes | LMNA c.1045 C > T (R349W) | LMNA | 1q22 | 10 patients | -lipodystrophy -progeroid syndromes (see HGPS) -cardiomyopathy -hearing impairment | autosomal domiant | generalized lipoathrophy | childhood onset | [74] | |
| Aquired Lipodystrophy (ART therapy) | - | (ZMPSTE24) | - | 40% of HAART patients (up to 70% in long term treatment) | -decreased mortality due to HIV -development of metabolic syndrome -lipodystrophy -diabetes mellitus -hypertension -mammary hypertrophy -lipomas | - | -Buffalo hump development -peripheral lipoathrophy in face, buttocks, arms and legs -abdominal lipohypertrophy | Treatment dependent (-) | [100,101,102] |
4.2. Mandibuloacral Dysplasia
4.3. Familial Partial Lipodystrophy Disease
4.4. Acquired Lipodystrophies (Antiretroviral HIV Drugs)
5. Adipocyte Homeostasis—Essential for Glucose Metabolism and Cardiovascular Health
6. Amelioration of Lipodystrophy in Novel Therapies
6.1. Leptin/Metreleptin
6.2. LDL Lowering Drugs
| Possible/Current Syndroms of Application | State of Research (Preclinical Cell Culture = 1, Preclinical Mouse Studies = 2, Clinical Studies in Human = 3 ) | Benefits | Side Effects | Refernece | ||
|---|---|---|---|---|---|---|
| Leptin/Metreleptin | FPLD | 3 | -overall satiety -reduced hunger -reduced TG, HbA1c, LDL-cholesterol levels -increased insulin sensitivity | -proteinuria -development of neutralizing antibodies -headache -lymphoma -hypersensitivity -decreased weight -abdominal pain -dizziness -fatigue | [210,212] | |
| Gemcabene | FPLD | 3 | -reduced dyslipidaemia -reduces CRP levels -reduced LDL-cholesterol | -nausea/vomiting -anaemia -fever -rash -thrombocytopenia -oedema -increased AST, ALT and ALP | [219,220] | |
| RNA based approaches | Evinacumab | HGPS, MAD, FPLD* | 1 (in lipodystrophies) 3 (in hypercholisterinaemia) | -reduced fasting TGs -reduced LDL levels | -increased ALT and AST -flu like symptoms | [221,222] |
| Volanesorsen | HGPS.FPLD, MAD* | 1 (lipodystrophy) 3 (familial cyclomicroneamia) | -reduction of blood level cyclomicrons -70–80% reduction of TGs -reduced pancreatitis -reduced insulin resistance -increased well being in FCS | -serum sickness -dehydration -thrombocytopenia -injection side swelling, itching and bruising -rush -hypersensitivity -blood in urine -headache | [147,223] | |
| AKCEA-APOCIII-LRx | HGPS, MAD, FPLD* | /(lipodystrophy) 3 (CVD and Hypertriglyceridemia) | -reduced TG levels -artherosclerotic proteins reduced -84% reduced apoC3 levels and 30% apoB reduced -HDL levels increased | -no sever side effects | [224] | |
| Viral vectors (AAV mediated FGF21 therapy) | HGPS, MAD, FPLD* | 2 (obesity & metabolic disease) | -inhibited gluconeogenesis -increased adipose thermogenesis -incraesed liver fatty acid oxidation -reduced inflammation in pancreatic cells -improved energy homeostasis -reduced body weight -reduced hepatic steatosis -reduced fibrosis | -no severe side effects observed in animals | [225,226,227] | |
| Gene Therapy | Splicing site silencing | HGPS | 1 | ○ / | ○ / | [228,229,230] |
6.3. Anti-Inflammatory Drugs
6.4. RNA-Based Approaches: ASOs (Evinacumab-Like Mechanism)
6.5. Gene Therapies
6.6. Posttranscriptional Modifications
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Araújo-Vilar, D.; Santini, F. Diagnosis and treatment of lipodystrophy: A step-by-step approach. J. Endocrinol. Investig. 2019, 42, 61–73. [Google Scholar] [CrossRef]
- Herbst, K.L.; Tannock, L.R.; Deeb, S.S.; Purnell, J.Q.; Brunzell, J.D.; Chait, A. Köbberling type of familial partial lipodystrophy: An underrecognized syndrome. Diabetes Care 2003, 26, 1819–1824. [Google Scholar] [CrossRef] [PubMed]
- Garg, A. Acquired and Inherited Lipodystrophies. N. Engl. J. Med. 2004, 350, 1220–1234. [Google Scholar] [CrossRef]
- Rochlani, Y.; Pothineni, N.V.; Kovelamudi, S.; Mehta, J.L. Metabolic syndrome: Pathophysiology, management, and modulation by natural compounds. Ther. Adv. Cardiovasc. Dis. 2017, 11, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.M. Role of Insulin Resistance in Human Disease (Syndrome X): An Expanded Definition. Annu. Rev. Med. 1993, 44, 121–131. [Google Scholar] [CrossRef]
- Brown, R.J.; Araujo-Vilar, D.; Cheung, P.T.; Dunger, D.; Garg, A.; Jack, M.; Mungai, L.; Oral, E.A.; Patni, N.; Rother, K.I.; et al. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J. Clin. Endocrinol. Metab. 2016, 101, 4500–4511. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.; Samaras, K.; Burton, S.; Law, M.; Freund, J.; Chisholm, D.J.; Cooper, D.A. A syndrome of peripheral lipodystrophy, hyperlipidaemia and insulin resistance in patients receiving HIV protease inhibitors. AIDS 1998, 12, F51–F58. [Google Scholar] [CrossRef]
- Lin, F.; Worman, H.J. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 1993, 268, 16321–16326. [Google Scholar] [CrossRef]
- Turgay, Y.; Medalia, O. The structure of lamin filaments in somatic cells as revealed by cryo-electron tomography. Nucleus 2017, 8, 475–481. [Google Scholar] [CrossRef]
- van Steensel, B.; Belmont, A.S. Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef]
- Meuleman, W.; Peric-Hupkes, D.; Kind, J.; Beaudry, J.B.; Pagie, L.; Kellis, M.; Reinders, M.; Wessels, L.; van Steensel, B. Constitutive nuclear lamina-genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res. 2013, 23, 270–280. [Google Scholar] [CrossRef]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008, 453, 948–951. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, T.; Zhang, S.; Yang, T.; Gong, Y.; Deng, H.W.; Bai, D.; Tian, W.; Chen, Y. Discovery and functional assessment of a novel adipocyte population driven by intracellular Wnt/β-catenin signaling in mammals. Elife 2022, 11, e77740. [Google Scholar] [CrossRef]
- Cenni, V.; Capanni, C.; Mattioli, E.; Schena, E.; Squarzoni, S.; Bacalini, M.G.; Garagnani, P.; Salvioli, S.; Franceschi, C.; Lattanzi, G. Lamin A involvement in ageing processes. Ageing Res. Rev. 2020, 62, 101073. [Google Scholar] [CrossRef] [PubMed]
- Solovei, I.; Wang, A.S.; Thanisch, K.; Schmidt, C.S.; Krebs, S.; Zwerger, M.; Cohen, T.V.; Devys, D.; Foisner, R.; Peichl, L.; et al. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 2013, 152, 584–598. [Google Scholar] [CrossRef] [PubMed]
- Bronshtein, I.; Kepten, E.; Kanter, I.; Berezin, S.; Lindner, M.; Redwood, A.B.; Mai, S.; Gonzalo, S.; Foisner, R.; Shav-Tal, Y.; et al. Loss of lamin A function increases chromatin dynamics in the nuclear interior. Nat. Commun. 2015, 6, 8044. [Google Scholar] [CrossRef] [PubMed]
- Siiteri, P.K. Adipose tissue as a source of hormones. Am. J. Clin. Nutr. 1987, 45, 277–282. [Google Scholar] [CrossRef]
- Kershaw, E.E.; Flier, J.S. Adipose Tissue as an Endocrine Organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef]
- Sakaguchi, K.; Takeda, K.; Maeda, M.; Ogawa, W.; Sato, T.; Okada, S.; Ohnishi, Y.; Nakajima, H.; Kashiwagi, A. Glucose area under the curve during oral glucose tolerance test as an index of glucose intolerance. Diabetol. Int. 2015, 7, 53–58. [Google Scholar] [CrossRef]
- Wu, J.; Boström, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.-H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef]
- Pond, C.M. An evolutionary and functional view of mammalian adipose tissue. Proc. Nutr. Soc. 1992, 51, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Saely, C.H.; Geiger, K.; Drexel, H. Brown versus White Adipose Tissue: A Mini-Review. Gerontology 2012, 58, 15–23. [Google Scholar] [CrossRef]
- Roberts, L.D.; West, J.A.; Vidal-Puig, A.; Griffin, J.L. Chapter Twelve—Methods for Performing Lipidomics in White Adipose Tissue. In Methods in Enzymology; MacDougald, O.A., Ed.; Academic Press: Cambridge, MA, USA, 2014; Volume 538, pp. 211–231. [Google Scholar]
- Gesta, S.; Tseng, Y.-H.; Kahn, C.R. Developmental Origin of Fat: Tracking Obesity to Its Source. Cell 2007, 131, 242–256. [Google Scholar] [CrossRef]
- Kissebah, A.H.; Krakower, G.R. Regional adiposity and morbidity. Physiol. Rev. 1994, 74, 761–811. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Red Eagle, A.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Neeland, I.J.; Ayers, C.R.; Rohatgi, A.K.; Turer, A.T.; Berry, J.D.; Das, S.R.; Vega, G.L.; Khera, A.; McGuire, D.K.; Grundy, S.M.; et al. Associations of visceral and abdominal subcutaneous adipose tissue with markers of cardiac and metabolic risk in obese adults. Obesity 2013, 21, E439–E447. [Google Scholar] [CrossRef]
- Frayn, K. Adipose tissue as a buffer for daily lipid flux. Diabetologia 2002, 45, 1201–1210. [Google Scholar] [CrossRef]
- Virtue, S.; Vidal-Puig, A. Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome—An allostatic perspective. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2010, 1801, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Chawla, A. Pleiotropic Actions of Insulin Resistance and Inflammation in Metabolic Homeostasis. Science 2013, 339, 172. [Google Scholar] [CrossRef]
- Schenk, S.; Saberi, M.; Olefsky, J.M. Insulin sensitivity: Modulation by nutrients and inflammation. J. Clin. Investig. 2008, 118, 2992–3002. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Herrero, L.; Naaz, A. Obesity, Inflammation, and Insulin Resistance. Gastroenterology 2007, 132, 2169–2180. [Google Scholar] [CrossRef] [PubMed]
- Plomgaard, P.; Bouzakri, K.; Krogh-Madsen, R.; Mittendorfer, B.; Zierath, J.R.; Pedersen, B.K. Tumor necrosis factor-alpha induces skeletal muscle insulin resistance in healthy human subjects via inhibition of Akt substrate 160 phosphorylation. Diabetes 2005, 54, 2939–2945. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; McNeill, J.; Goon, K.; Little, D.; Kimmerling, K.; Huebner, J.; Kraus, V.; Guilak, F. Conditional Macrophage Depletion Increases Inflammation and Does Not Inhibit the Development of Osteoarthritis in Obese Macrophage Fas-Induced Apoptosis-Transgenic Mice. Arthritis Rheumatol. 2017, 69, 1772–1783. [Google Scholar] [CrossRef] [PubMed]
- van Loo, G.; Bertrand, M.J.M. Death by TNF: A road to inflammation. Nat. Rev. Immunol. 2023, 23, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Murano, I.; Barbatelli, G.; Parisani, V.; Latini, C.; Muzzonigro, G.; Castellucci, M.; Cinti, S. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J. Lipid Res. 2008, 49, 1562–1568. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Cawthorn, W.P.; Heyd, F.; Hegyi, K.; Sethi, J.K. Tumour necrosis factor-alpha inhibits adipogenesis via a beta-catenin/TCF4(TCF7L2)-dependent pathway. Cell Death Differ. 2007, 14, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Schutz, Y. Macronutrients and energy balance in obesity. Metab.—Clin. Exp. 1995, 44, 7–11. [Google Scholar] [CrossRef]
- Porter, C. Quantification of UCP1 function in human brown adipose tissue. Adipocyte 2017, 6, 167–174. [Google Scholar] [CrossRef]
- Klingenberg, M. Uncoupling protein--a useful energy dissipator. J. Bioenerg. Biomembr. 1999, 31, 419–430. [Google Scholar] [CrossRef]
- Cannon, B.; Nedergaard, J.A.N. Brown Adipose Tissue: Function and Physiological Significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Fernández, L.; Fernández-Galilea, M.; Felix-Soriano, E.; Escoté, X.; González-Muniesa, P.; Moreno-Aliaga, M.J. Chapter 4—Inflammation and Oxidative Stress in Adipose Tissue: Nutritional Regulation. In Obesity; del Moral, A.M., Aguilera García, C.M., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 63–92. [Google Scholar] [CrossRef]
- Wu, J.; Cohen, P.; Spiegelman, B.M. Adaptive thermogenesis in adipocytes: Is beige the new brown? Genes Dev. 2013, 27, 234–250. [Google Scholar] [CrossRef] [PubMed]
- El Hadi, H.; Di Vincenzo, A.; Vettor, R.; Rossato, M. Food Ingredients Involved in White-to-Brown Adipose Tissue Conversion and in Calorie Burning. Front. Physiol. 2019, 9, 1954. [Google Scholar] [CrossRef] [PubMed]
- Ferré, P.; Burnol, A.F.; Leturque, A.; Terretaz, J.; Penicaud, L.; Jeanrenaud, B.; Girard, J. Glucose utilization in vivo and insulin-sensitivity of rat brown adipose tissue in various physiological and pathological conditions. Biochem. J. 1986, 233, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Wang, X.; Berleth, N.; Deitersen, J.; Wallot-Hieke, N.; Böhler, P.; Schlütermann, D.; Stuhldreier, F.; Cox, J.; Schmitz, K.; et al. The Autophagy-Initiating Kinase ULK1 Controls RIPK1-Mediated Cell Death. Cell Rep. 2020, 31, 107547. [Google Scholar] [CrossRef] [PubMed]
- Valverde, A.M.; Teruel, T.; Navarro, P.; Benito, M.; Lorenzo, M. Tumor necrosis factor-alpha causes insulin receptor substrate-2-mediated insulin resistance and inhibits insulin-induced adipogenesis in fetal brown adipocytes. Endocrinology 1998, 139, 1229–1238. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, M.; Fernández-Veledo, S.; Vila-Bedmar, R.; Garcia-Guerra, L.; De Alvaro, C.; Nieto-Vazquez, I. Insulin resistance induced by tumor necrosis factor-alpha in myocytes and brown adipocytes. J. Anim. Sci. 2008, 86, E94–E104. [Google Scholar] [CrossRef]
- Sakamoto, T.; Nitta, T.; Maruno, K.; Yeh, Y.S.; Kuwata, H.; Tomita, K.; Goto, T.; Takahashi, N.; Kawada, T. Macrophage infiltration into obese adipose tissues suppresses the induction of UCP1 level in mice. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E676–E687. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.; Ricciardi, C.J.; Esposito, D.; Komarnytsky, S.; Hu, P.; Curry, B.J.; Brown, P.L.; Gao, Z.; Biggerstaff, J.P.; Chen, J.; et al. Activation of pattern recognition receptors in brown adipocytes induces inflammation and suppresses uncoupling protein 1 expression and mitochondrial respiration. Am. J. Physiol. Cell Physiol. 2014, 306, C918–C930. [Google Scholar] [CrossRef]
- Okla, M.; Zaher, W.; Alfayez, M.; Chung, S. Inhibitory Effects of Toll-Like Receptor 4, NLRP3 Inflammasome, and Interleukin-1β on White Adipocyte Browning. Inflammation 2018, 41, 626–642. [Google Scholar] [CrossRef]
- Nøhr, M.K.; Bobba, N.; Richelsen, B.; Lund, S.; Pedersen, S.B. Inflammation Downregulates UCP1 Expression in Brown Adipocytes Potentially via SIRT1 and DBC1 Interaction. Int. J. Mol. Sci. 2017, 18, 1006. [Google Scholar] [CrossRef] [PubMed]
- Valladares, A.; Roncero, C.; Benito, M.; Porras, A. TNF-alpha inhibits UCP-1 expression in brown adipocytes via ERKs. Opposite effect of p38MAPK. FEBS Lett. 2001, 493, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Naknukool, S.; Yoshitake, R.; Hanafusa, Y.; Tokiwa, S.; Li, Y.; Sakamoto, T.; Nitta, T.; Kim, M.; Takahashi, N.; et al. Proinflammatory cytokine interleukin-1β suppresses cold-induced thermogenesis in adipocytes. Cytokine 2016, 77, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Infantes, D.; White, U.A.; Elks, C.M.; Morrison, R.F.; Gimble, J.M.; Considine, R.V.; Ferrante, A.W.; Ravussin, E.; Stephens, J.M. Oncostatin m is produced in adipose tissue and is regulated in conditions of obesity and type 2 diabetes. J. Clin. Endocrinol. Metab. 2014, 99, E217–E225. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Infantes, D.; Cereijo, R.; Peyrou, M.; Piquer-Garcia, I.; Stephens, J.M.; Villarroya, F. Oncostatin m impairs brown adipose tissue thermogenic function and the browning of subcutaneous white adipose tissue. Obesity 2017, 25, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Polyák, Á.; Winkler, Z.; Kuti, D.; Ferenczi, S.; Kovács, K.J. Brown adipose tissue in obesity: Fractalkine-receptor dependent immune cell recruitment affects metabolic-related gene expression. Biochim. Biophys. Acta 2016, 1861, 1614–1622. [Google Scholar] [CrossRef] [PubMed]
- Pirzgalska, R.M.; Seixas, E.; Seidman, J.S.; Link, V.M.; Sánchez, N.M.; Mahú, I.; Mendes, R.; Gres, V.; Kubasova, N.; Morris, I.; et al. Sympathetic neuron-associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nat. Med. 2017, 23, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Camell, C.D.; Sander, J.; Spadaro, O.; Lee, A.; Nguyen, K.Y.; Wing, A.; Goldberg, E.L.; Youm, Y.H.; Brown, C.W.; Elsworth, J.; et al. Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature 2017, 550, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Hussain, I.; Garg, A. Lipodystrophy Syndromes. Endocrinol. Metab. Clin. N. Am. 2016, 45, 783–797. [Google Scholar] [CrossRef]
- Vigouroux, C.; Capeau, J. A-type lamin-linked lipodystrophies. Novartis Found. Symp. 2005, 264, 166–177, discussion 177–182, 227–130. [Google Scholar]
- Ullrich, N.J.; Gordon, L.B. Hutchinson-Gilford progeria syndrome. Handb. Clin. Neurol. 2015, 132, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Hennekam, R.C.M. Hutchinson–Gilford progeria syndrome: Review of the phenotype. Am. J. Med. Genet. Part A 2006, 140, 2603–2624. [Google Scholar] [CrossRef] [PubMed]
- DeBusk, F.L. The Hutchinson-Gilford progeria syndrome. Report of 4 cases and review of the literature. J. Pediatr. 1972, 80, 697–724. [Google Scholar] [CrossRef] [PubMed]
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N. Engl. J. Med. 2008, 358, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [PubMed]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin A Truncation in Hutchinson-Gilford Progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Lee, L.; Kudlow, B.A.; Dos Santos, H.G.; Sletvold, O.; Shafeghati, Y.; Botha, E.G.; Garg, A.; Hanson, N.B.; Martin, G.M.; et al. LMNA mutations in atypical Werner’s syndrome. Lancet 2003, 362, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Duband-Goulet, I.; Woerner, S.; Gasparini, S.; Attanda, W.; Kondé, E.; Tellier-Lebègue, C.; Craescu, C.T.; Gombault, A.; Roussel, P.; Vadrot, N.; et al. Subcellular localization of SREBP1 depends on its interaction with the C-terminal region of wild-type and disease related A-type lamins. Exp. Cell Res. 2011, 317, 2800–2813. [Google Scholar] [CrossRef] [PubMed]
- Bastard, J.-P.; Caron, M.; Vidal, H.; Jan, V.; Auclair, M.; Vigouroux, C.; Luboinski, J.; Laville, M.; Maachi, M.; Girard, P.-M.; et al. Association between altered expression of adipogenic factor SREBP1 in lipoatrophic adipose tissue from HIV-1-infected patients and abnormal adipocyte differentiation and insulin resistance. Lancet 2002, 359, 1026–1031. [Google Scholar] [CrossRef]
- Kim, J.B.; Sarraf, P.; Wright, M.; Yao, K.M.; Mueller, E.; Solanes, G.; Lowell, B.B.; Spiegelman, B.M. Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J. Clin. Investig. 1998, 101, 1–9. [Google Scholar] [CrossRef]
- Xiong, Z.M.; LaDana, C.; Wu, D.; Cao, K. An inhibitory role of progerin in the gene induction network of adipocyte differentiation from iPS cells. Aging 2013, 5, 288–303. [Google Scholar] [CrossRef]
- Magno, S.; Ceccarini, G.; Pelosini, C.; Ferrari, F.; Prodam, F.; Gilio, D.; Maffei, M.; Sessa, M.R.; Barison, A.; Ciccarone, A.; et al. Atypical Progeroid Syndrome and Partial Lipodystrophy Due to LMNA Gene p.R349W Mutation. J. Endocr. Soc. 2020, 4, bvaa108. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.P.; Savage, D.B. What lipodystrophies teach us about the metabolic syndrome. J. Clin. Investig. 2019, 129, 4009–4021. [Google Scholar] [CrossRef]
- Rochford, J.J. Molecular mechanisms controlling human adipose tissue development: Insights from monogenic lipodystrophies. Expert. Rev. Mol. Med. 2010, 12, e24. [Google Scholar] [CrossRef] [PubMed]
- Vigouroux, C.; Guénantin, A.C.; Vatier, C.; Capel, E.; Le Dour, C.; Afonso, P.; Bidault, G.; Béréziat, V.; Lascols, O.; Capeau, J.; et al. Lipodystrophic syndromes due to LMNA mutations: Recent developments on biomolecular aspects, pathophysiological hypotheses and therapeutic perspectives. Nucleus 2018, 9, 235–248. [Google Scholar] [CrossRef]
- Revêchon, G.; Viceconte, N.; McKenna, T.; Sola Carvajal, A.; Vrtačnik, P.; Stenvinkel, P.; Lundgren, T.; Hultenby, K.; Franco, I.; Eriksson, M. Rare progerin-expressing preadipocytes and adipocytes contribute to tissue depletion over time. Sci. Rep. 2017, 7, 4405. [Google Scholar] [CrossRef]
- Bridger, J.M.; Kill, I.R. Aging of Hutchinson-Gilford progeria syndrome fibroblasts is characterised by hyperproliferation and increased apoptosis. Exp. Gerontol. 2004, 39, 717–724. [Google Scholar] [CrossRef]
- Sagelius, H.; Rosengardten, Y.; Hanif, M.; Erdos, M.R.; Rozell, B.; Collins, F.S.; Eriksson, M. Targeted transgenic expression of the mutation causing Hutchinson-Gilford progeria syndrome leads to proliferative and degenerative epidermal disease. J. Cell Sci. 2008, 121, 969–978. [Google Scholar] [CrossRef]
- Harhouri, K.; Frankel, D.; Bartoli, C.; Roll, P.; De Sandre-Giovannoli, A.; Lévy, N. An overview of treatment strategies for Hutchinson-Gilford Progeria syndrome. Nucleus 2018, 9, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.B.; Brown, W.T.; Collins, F.S. Hutchinson-Gilford Progeria Syndrome. In GeneReviews(®); Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Bagias, C.; Xiarchou, A.; Bargiota, A.; Tigas, S. Familial Partial Lipodystrophy (FPLD): Recent Insights. Diabetes Metab. Syndr. Obes. 2020, 13, 1531–1544. [Google Scholar] [CrossRef]
- Tsoukas, M.A.; Mantzoros, C.S. Chapter 37—Lipodystrophy Syndromes. In Endocrinology: Adult and Pediatric, 7th ed.; Jameson, J.L., De Groot, L.J., de Kretser, D.M., Giudice, L.C., Grossman, A.B., Melmed, S., Potts, J.T., Weir, G.C., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2016; pp. 648–661.e645. [Google Scholar] [CrossRef]
- Cenni, V.; D’Apice, M.R.; Garagnani, P.; Columbaro, M.; Novelli, G.; Franceschi, C.; Lattanzi, G. Mandibuloacral dysplasia: A premature ageing disease with aspects of physiological ageing. Ageing Res. Rev. 2018, 42, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fiorenza, C.G.; Chou, S.H.; Mantzoros, C.S. Lipodystrophy: Pathophysiology and advances in treatment. Nat. Rev. Endocrinol. 2011, 7, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Hitzert, M.M.; van der Crabben, S.N.; Baldewsingh, G.; van Amstel, H.K.P.; van den Wijngaard, A.; van Ravenswaaij-Arts, C.M.A.; Zijlmans, C.W.R. Mandibuloacral dysplasia type B (MADB): A cohort of eight patients from Suriname with a homozygous founder mutation in ZMPSTE24 (FACE1), clinical diagnostic criteria and management guidelines. Orphanet J. Rare Dis. 2019, 14, 294. [Google Scholar] [CrossRef] [PubMed]
- Johansen, K.; Rasmussen, M.H.; Kjems, L.L.; Astrup, A. An unusual type of familial lipodystrophy. J. Clin. Endocrinol. Metab. 1995, 80, 3442–3446. [Google Scholar] [CrossRef]
- Köbberling, J.; Dunnigan, M.G. Familial partial lipodystrophy: Two types of an X linked dominant syndrome, lethal in the hemizygous state. J. Med. Genet. 1986, 23, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Garg, A. Clinical review#: Lipodystrophies: Genetic and acquired body fat disorders. J. Clin. Endocrinol. Metab. 2011, 96, 3313–3325. [Google Scholar] [CrossRef] [PubMed]
- Diker-Cohen, T.; Cochran, E.; Gorden, P.; Brown, R.J. Partial and generalized lipodystrophy: Comparison of baseline characteristics and response to metreleptin. J. Clin. Endocrinol. Metab. 2015, 100, 1802–1810. [Google Scholar] [CrossRef]
- Barroso, I.; Gurnell, M.; Crowley, V.E.; Agostini, M.; Schwabe, J.W.; Soos, M.A.; Maslen, G.L.; Williams, T.D.; Lewis, H.; Schafer, A.J.; et al. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 1999, 402, 880–883. [Google Scholar] [CrossRef] [PubMed]
- Gandotra, S.; Le Dour, C.; Bottomley, W.; Cervera, P.; Giral, P.; Reznik, Y.; Charpentier, G.; Auclair, M.; Delépine, M.; Barroso, I.; et al. Perilipin deficiency and autosomal dominant partial lipodystrophy. N. Engl. J. Med. 2011, 364, 740–748. [Google Scholar] [CrossRef]
- Rubio-Cabezas, O.; Puri, V.; Murano, I.; Saudek, V.; Semple, R.K.; Dash, S.; Hyden, C.S.; Bottomley, W.; Vigouroux, C.; Magré, J.; et al. Partial lipodystrophy and insulin resistant diabetes in a patient with a homozygous nonsense mutation in CIDEC. EMBO Mol. Med. 2009, 1, 280–287. [Google Scholar] [CrossRef]
- Zolotov, S.; Xing, C.; Mahamid, R.; Shalata, A.; Sheikh-Ahmad, M.; Garg, A. Homozygous LIPE mutation in siblings with multiple symmetric lipomatosis, partial lipodystrophy, and myopathy. Am. J. Med. Genet. A 2017, 173, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Carboni, N.; Brancati, F.; Cocco, E.; Solla, E.; D‘Apice, M.R.; Mateddu, A.; McIntyre, A.; Fadda, E.; Mura, M.; Lattanzi, G.; et al. Partial lipodystrophy associated with muscular dystrophy of unknown genetic origin. Muscle Nerve 2014, 49, 928–930. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Alston, L.; Ruschman, J.; Hegele, R.A. Heterozygous CAV1 frameshift mutations (MIM 601047) in patients with atypical partial lipodystrophy and hypertriglyceridemia. Lipids Health Dis 2008, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Rochford, J.J.; Wolfrum, C.; Gray, S.L.; Schinner, S.; Wilson, J.C.; Soos, M.A.; Murgatroyd, P.R.; Williams, R.M.; Acerini, C.L.; et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science 2004, 304, 1325–1328. [Google Scholar] [CrossRef] [PubMed]
- Puente, X.S.; Quesada, V.; Osorio, F.G.; Cabanillas, R.; Cadiñanos, J.; Fraile, J.M.; Ordóñez, G.R.; Puente, D.A.; Gutiérrez-Fernández, A.; Fanjul-Fernández, M.; et al. Exome sequencing and functional analysis identifies BANF1 mutation as the cause of a hereditary progeroid syndrome. Am. J. Hum. Genet. 2011, 88, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Batterham, M.J.; Garsia, R.; Greenop, P. Prevalence and predictors of HIV-associated weight loss in the era of highly active antiretroviral therapy. Int. J. STD AIDS 2002, 13, 744–747. [Google Scholar] [CrossRef]
- Mutimura, E.; Stewart, A.; Rheeder, P.; Crowther, N.J. Metabolic function and the prevalence of lipodystrophy in a population of HIV-infected African subjects receiving highly active antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2007, 46, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Guzman, N.; Vijayan, V. HIV-Associated Lipodystrophy. In StatPearls; StatPearls Publishing Copyright © 2024; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2024. [Google Scholar]
- Campisi, J. The role of cellular senescence in skin aging. J. Investig. Dermatol. Symp. Proc. 1998, 3, 1–5. [Google Scholar]
- Capeau, J.; Magré, J.; Caron-Debarle, M.; Lagathu, C.; Antoine, B.; Béréziat, V.R.; Lascols, O.; Bastard, J.P.; Vigouroux, C. Human lipodystrophies: Genetic and acquired diseases of adipose tissue. Endocr. Dev. 2010, 19, 1–20. [Google Scholar] [CrossRef]
- Suganami, T.; Mieda, T.; Itoh, M.; Shimoda, Y.; Kamei, Y.; Ogawa, Y. Attenuation of obesity-induced adipose tissue inflammation in C3H/HeJ mice carrying a Toll-like receptor 4 mutation. Biochem. Biophys. Res. Commun. 2007, 354, 45–49. [Google Scholar] [CrossRef]
- Najdi, F.; Krüger, P.; Djabali, K. Impact of Progerin Expression on Adipogenesis in Hutchinson-Gilford Progeria Skin-Derived Precursor Cells. Cells 2021, 10, 1598. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat. Cell Biol. 2008, 10, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lian, Q.; Zhu, G.; Zhou, F.; Sui, L.; Tan, C.; Mutalif, R.A.; Navasankari, R.; Zhang, Y.; Tse, H.F.; et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011, 8, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.-F.; Wong, W.-T. Progress and trends in the development of therapies for Hutchinson–Gilford progeria syndrome. Aging Cell 2020, 19, e13175. [Google Scholar] [CrossRef] [PubMed]
- Varela, I.; Pereira, S.; Ugalde, A.P.; Navarro, C.L.; Suárez, M.F.; Cau, P.; Cadiñanos, J.; Osorio, F.G.; Foray, N.; Cobo, J.; et al. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat. Med. 2008, 14, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.B.; Kleinman, M.E.; Massaro, J.; D’Agostino, R.B., Sr.; Shappell, H.; Gerhard-Herman, M.; Smoot, L.B.; Gordon, C.M.; Cleveland, R.H.; Nazarian, A.; et al. Clinical Trial of the Protein Farnesylation Inhibitors Lonafarnib, Pravastatin, and Zoledronic Acid in Children With Hutchinson-Gilford Progeria Syndrome. Circulation 2016, 134, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Beyret, E.; Liao, H.K.; Yamamoto, M.; Hernandez-Benitez, R.; Fu, Y.; Erikson, G.; Reddy, P.; Izpisua Belmonte, J.C. Single-dose CRISPR-Cas9 therapy extends lifespan of mice with Hutchinson-Gilford progeria syndrome. Nat. Med. 2019, 25, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Rajeev, M.; Ratan, C.; Krishnan, K.; Vijayan, M. Hutchinson-Gilford Progeria Syndrome (Hgps) and Application of Gene Therapy Based Crispr/Cas Technology as A Promising Innovative Treatment Approach. Recent. Pat. Biotechnol. 2021, 15, 266–285. [Google Scholar] [CrossRef] [PubMed]
- Erdos, M.R.; Cabral, W.A.; Tavarez, U.L.; Cao, K.; Gvozdenovic-Jeremic, J.; Narisu, N.; Zerfas, P.M.; Crumley, S.; Boku, Y.; Hanson, G.; et al. A targeted antisense therapeutic approach for Hutchinson-Gilford progeria syndrome. Nat. Med. 2021, 27, 536–545. [Google Scholar] [CrossRef]
- Koblan, L.W.; Erdos, M.R.; Wilson, C.; Cabral, W.A.; Levy, J.M.; Xiong, Z.M.; Tavarez, U.L.; Davison, L.M.; Gete, Y.G.; Mao, X.; et al. In vivo base editing rescues Hutchinson-Gilford progeria syndrome in mice. Nature 2021, 589, 608–614. [Google Scholar] [CrossRef]
- Villa-Bellosta, R. ATP-based therapy prevents vascular calcification and extends longevity in a mouse model of Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 23698–23704. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, L.; Cohen, E.; Atalaia, A.; Ben Yaou, R.; Bonne, G.; Bertrand, A.T. Preclinical Advances of Therapies for Laminopathies. J. Clin. Med. 2021, 10, 4834. [Google Scholar] [CrossRef] [PubMed]
- Osorio, F.G.; Navarro, C.L.; Cadiñanos, J.; López-Mejía, I.C.; Quirós, P.M.; Bartoli, C.; Rivera, J.; Tazi, J.; Guzmán, G.; Varela, I.; et al. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci. Transl. Med. 2011, 3, 106ra107. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Nobumori, C.; Tu, Y.; Choi, C.; Yang, S.H.; Jung, H.J.; Vickers, T.A.; Rigo, F.; Bennett, C.F.; Young, S.G.; et al. Modulation of LMNA splicing as a strategy to treat prelamin A diseases. J. Clin. Investig. 2016, 126, 1592–1602. [Google Scholar] [CrossRef] [PubMed]
- Harhouri, K.; Navarro, C.; Baquerre, C.; Da Silva, N.; Bartoli, C.; Casey, F.; Mawuse, G.K.; Doubaj, Y.; Lévy, N.; De Sandre-Giovannoli, A. Antisense-Based Progerin Downregulation in HGPS-Like Patients’ Cells. Cells 2016, 5, 31. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, G.; Bruno, I.G.; Zhang, N.; Sho, S.; Tedone, E.; Lai, T.-P.; Cooke, J.P.; Shay, J.W. Transient introduction of human telomerase mRNA improves hallmarks of progeria cells. Aging Cell 2019, 18, e12979. [Google Scholar] [CrossRef]
- Endisha, H.; Merrill-Schools, J.; Zhao, M.; Bristol, M.; Wang, X.; Kubben, N.; Elmore, L.W. Restoring SIRT6 Expression in Hutchinson-Gilford Progeria Syndrome Cells Impedes Premature Senescence and Formation of Dysmorphic Nuclei. Pathobiology 2015, 82, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K.; Fryns, J.P.; Auchus, R.J.; Garg, A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum. Mol. Genet. 2003, 12, 1995–2001. [Google Scholar] [CrossRef]
- Novelli, G.; Muchir, A.; Sangiuolo, F.; Helbling-Leclerc, A.; D‘Apice, M.R.; Massart, C.; Capon, F.; Sbraccia, P.; Federici, M.; Lauro, R.; et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am. J. Hum. Genet. 2002, 71, 426–431. [Google Scholar] [CrossRef]
- Simha, V.; Garg, A. Body fat distribution and metabolic derangements in patients with familial partial lipodystrophy associated with mandibuloacral dysplasia. J. Clin. Endocrinol. Metab. 2002, 87, 776–785. [Google Scholar] [CrossRef]
- Filesi, I.; Gullotta, F.; Lattanzi, G.; D’Apice, M.R.; Capanni, C.; Nardone, A.M.; Columbaro, M.; Scarano, G.; Mattioli, E.; Sabatelli, P.; et al. Alterations of nuclear envelope and chromatin organization in mandibuloacral dysplasia, a rare form of laminopathy. Physiol. Genom. 2005, 23, 150–158. [Google Scholar] [CrossRef]
- Lloyd, D.J.; Trembath, R.C.; Shackleton, S. A novel interaction between lamin A and SREBP1: Implications for partial lipodystrophy and other laminopathies. Hum. Mol. Genet. 2002, 11, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Vantyghem, M.C.; Faivre-Defrance, F.; Marcelli-Tourvieille, S.; Fermon, C.; Evrard, A.; Bourdelle-Hego, M.F.; Vigouroux, C.; Defebvre, L.; Delemer, B.; Wemeau, J.L. Familial partial lipodystrophy due to the LMNA R482W mutation with multinodular goitre, extrapyramidal syndrome and primary hyperaldosteronism. Clin. Endocrinol. 2007, 67, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, F.; Evangelisti, C.; Cenni, V.; Fazio, A.; Paganelli, F.; Martelli, A.M.; Lattanzi, G. The Cutting Edge: The Role of mTOR Signaling in Laminopathies. Int. J. Mol. Sci. 2019, 20, 847. [Google Scholar] [CrossRef]
- Pellegrini, C.; Columbaro, M.; Schena, E.; Prencipe, S.; Andrenacci, D.; Iozzo, P.; Angela Guzzardi, M.; Capanni, C.; Mattioli, E.; Loi, M.; et al. Altered adipocyte differentiation and unbalanced autophagy in type 2 Familial Partial Lipodystrophy: An in vitro and in vivo study of adipose tissue browning. Exp. Mol. Med. 2019, 51, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hussain, I.; Patni, N.; Garg, A. Lipodystrophies, dyslipidaemias and atherosclerotic cardiovascular disease. Pathology 2019, 51, 202–212. [Google Scholar] [CrossRef]
- Fernandez-Pombo, A.; Diaz-Lopez, E.J.; Castro, A.I.; Sanchez-Iglesias, S.; Cobelo-Gomez, S.; Prado-Moraña, T.; Araujo-Vilar, D. Clinical Spectrum of LMNA-Associated Type 2 Familial Partial Lipodystrophy: A Systematic Review. Cells 2023, 12, 725. [Google Scholar] [CrossRef]
- Guillín-Amarelle, C.; Fernández-Pombo, A.; Sánchez-Iglesias, S.; Araújo-Vilar, D. Lipodystrophic laminopathies: Diagnostic clues. Nucleus 2018, 9, 249–260. [Google Scholar] [CrossRef]
- Guillín-Amarelle, C.; Sánchez-Iglesias, S.; Castro-Pais, A.; Rodriguez-Cañete, L.; Ordóñez-Mayán, L.; Pazos, M.; González-Méndez, B.; Rodríguez-García, S.; Casanueva, F.F.; Fernández-Marmiesse, A.; et al. Type 1 familial partial lipodystrophy: Understanding the Köbberling syndrome. Endocrine 2016, 54, 411–421. [Google Scholar] [CrossRef]
- Garg, A.; Peshock, R.M.; Fleckenstein, J.L. Adipose tissue distribution pattern in patients with familial partial lipodystrophy (Dunnigan variety). J. Clin. Endocrinol. Metab. 1999, 84, 170–174. [Google Scholar] [CrossRef]
- Kozusko, K.; Tsang, V.; Bottomley, W.; Cho, Y.H.; Gandotra, S.; Mimmack, M.L.; Lim, K.; Isaac, I.; Patel, S.; Saudek, V.; et al. Clinical and molecular characterization of a novel PLIN1 frameshift mutation identified in patients with familial partial lipodystrophy. Diabetes 2015, 64, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Visser, M.E.; Kropman, E.; Kranendonk, M.E.; Koppen, A.; Hamers, N.; Stroes, E.S.; Kalkhoven, E.; Monajemi, H. Characterisation of non-obese diabetic patients with marked insulin resistance identifies a novel familial partial lipodystrophy-associated PPARγ mutation (Y151C). Diabetologia 2011, 54, 1639–1644. [Google Scholar] [CrossRef] [PubMed]
- Chiquette, E.; Oral, E.A.; Garg, A.; Araújo-Vilar, D.; Dhankhar, P. Estimating the prevalence of generalized and partial lipodystrophy: Findings and challenges. Diabetes Metab. Syndr. Obes. 2017, 10, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Rutkowska, L.; Salachna, D.; Lewandowski, K.; Lewiński, A.; Gach, A. Familial Partial Lipodystrophy-Literature Review and Report of a Novel Variant in PPARG Expanding the Spectrum of Disease-Causing Alterations in FPLD3. Diagnostics 2022, 12, 1122. [Google Scholar] [CrossRef] [PubMed]
- Haque, W.A.; Shimomura, I.; Matsuzawa, Y.; Garg, A. Serum adiponectin and leptin levels in patients with lipodystrophies. J. Clin. Endocrinol. Metab. 2002, 87, 2395. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.B.; Soos, M.A.; Powlson, A.; O‘Rahilly, S.; McFarlane, I.; Halsall, D.J.; Barroso, I.; Thomas, E.L.; Bell, J.D.; Scobie, I.; et al. Familial partial lipodystrophy associated with compound heterozygosity for novel mutations in the LMNA gene. Diabetologia 2004, 47, 753–756. [Google Scholar] [CrossRef]
- Verstraeten, V.L.; Broers, J.L.; van Steensel, M.A.; Zinn-Justin, S.; Ramaekers, F.C.; Steijlen, P.M.; Kamps, M.; Kuijpers, H.J.; Merckx, D.; Smeets, H.J.; et al. Compound heterozygosity for mutations in LMNA causes a progeria syndrome without prelamin A accumulation. Hum. Mol. Genet. 2006, 15, 2509–2522. [Google Scholar] [CrossRef] [PubMed]
- Arioglu, E.; Duncan-Morin, J.; Sebring, N.; Rother, K.I.; Gottlieb, N.; Lieberman, J.; Herion, D.; Kleiner, D.E.; Reynolds, J.; Premkumar, A.; et al. Efficacy and safety of troglitazone in the treatment of lipodystrophy syndromes. Ann. Intern. Med. 2000, 133, 263–274. [Google Scholar] [CrossRef]
- Oral, E.A.; Simha, V.; Ruiz, E.; Andewelt, A.; Premkumar, A.; Snell, P.; Wagner, A.J.; DePaoli, A.M.; Reitman, M.L.; Taylor, S.I.; et al. Leptin-replacement therapy for lipodystrophy. N. Engl. J. Med. 2002, 346, 570–578. [Google Scholar] [CrossRef]
- Melzer, F.; Geisler, C.; Schulte, D.M.; Laudes, M. Rapid response to leptin therapy in a FPLD patient with a novel PPARG missense variant. Endocrinol. Diabetes Metab. Case Rep. 2021, 2021, EDM210082. [Google Scholar] [CrossRef]
- Witztum, J.L.; Gaudet, D.; Freedman, S.D.; Alexander, V.J.; Digenio, A.; Williams, K.R.; Yang, Q.; Hughes, S.G.; Geary, R.S.; Arca, M.; et al. Volanesorsen and Triglyceride Levels in Familial Chylomicronemia Syndrome. N. Engl. J. Med. 2019, 381, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Esan, O.; Wierzbicki, A.S. Volanesorsen in the Treatment of Familial Chylomicronemia Syndrome or Hypertriglyceridaemia: Design, Development and Place in Therapy. Drug Des. Devel Ther. 2020, 14, 2623–2636. [Google Scholar] [CrossRef] [PubMed]
- Olkkonen, V.M.; Sinisalo, J.; Jauhiainen, M. New medications targeting triglyceride-rich lipoproteins: Can inhibition of ANGPTL3 or apoC-III reduce the residual cardiovascular risk? Atherosclerosis 2018, 272, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Mandema, J.W.; Hermann, D.; Wang, W.; Sheiner, T.; Milad, M.; Bakker-Arkema, R.; Hartman, D. Model-based development of gemcabene, a new lipid-altering agent. AAPS J. 2005, 7, E513–E522. [Google Scholar] [CrossRef] [PubMed]
- Fisher, H.G.; Patni, N.; Scheuerle, A.E. An additional case of Néstor-Guillermo progeria syndrome diagnosed in early childhood. Am. J. Med. Genet. A 2020, 182, 2399–2402. [Google Scholar] [CrossRef] [PubMed]
- Gong, V. Acquired immunodeficiency syndrome (AIDS). Am. J. Emerg. Med. 1984, 2, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Tartaglione, T.A.; Collier, A.C. Development of antiviral agents for the treatment of human immunodeficiency virus infection. Clin. Pharm. 1987, 6, 927–940. [Google Scholar]
- Usach, I.; Melis, V.; Peris, J.-E. Non-nucleoside reverse transcriptase inhibitors: A review on pharmacokinetics, pharmacodynamics, safety and tolerability. J. Int. AIDS Soc. 2013, 16, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hughes, P.J.; Cretton-Scott, E.; Teague, A.; Wensel, T.M. Protease Inhibitors for Patients With HIV-1 Infection: A Comparative Overview. Pharm. Ther. 2011, 36, 332–345. [Google Scholar]
- Eron, J.J., Jr. HIV-1 Protease Inhibitors. Clin. Infect. Dis. 2000, 30, S160–S170. [Google Scholar] [CrossRef]
- Kotler, D.; Heymsfield, S.B. HIV infection: A model chronic illness for studying wasting diseases. Am. J. Clin. Nutr. 1998, 68, 519–520. [Google Scholar] [CrossRef] [PubMed]
- Koethe, J.R.; Lagathu, C.; Lake, J.E.; Domingo, P.; Calmy, A.; Falutz, J.; Brown, T.T.; Capeau, J. HIV and antiretroviral therapy-related fat alterations. Nat. Rev. Dis. Primers 2020, 6, 48. [Google Scholar] [CrossRef] [PubMed]
- Abel, G.; Thompson, L. “I don’t want to look like an AIDS victim”: A New Zealand case study of facial lipoatrophy. Health Soc. Care Community 2018, 26, 41–47. [Google Scholar] [CrossRef] [PubMed]
- de Waal, R.; Cohen, K.; Maartens, G. Systematic review of antiretroviral-associated lipodystrophy: Lipoatrophy, but not central fat gain, is an antiretroviral adverse drug reaction. PLoS ONE 2013, 8, e63623. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Escuredo, J.M.; Del Mar Gutierrez, M.; Diaz-Delfin, J.; Domingo, J.C.; Mateo, M.G.; Domingo, P.; Giralt, M.; Villarroya, F. Differential effects of efavirenz and lopinavir/ritonavir on human adipocyte differentiation, gene expression and release of adipokines and pro-inflammatory cytokines. Curr. HIV Res. 2010, 8, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Albu, J.B.; Engelson, E.S.; Fried, S.K.; Inada, Y.; Ionescu, G.; Kotler, D.P. Increased systemic and adipose tissue cytokines in patients with HIV-associated lipodystrophy. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E261–E271. [Google Scholar] [CrossRef] [PubMed]
- Afonso, P.; Auclair, M.; Boccara, F.; Vantyghem, M.C.; Katlama, C.; Capeau, J.; Vigouroux, C.; Caron-Debarle, M. LMNA mutations resulting in lipodystrophy and HIV protease inhibitors trigger vascular smooth muscle cell senescence and calcification: Role of ZMPSTE24 downregulation. Atherosclerosis 2016, 245, 200–211. [Google Scholar] [CrossRef]
- Domingo, P.; Gutierrez, M.d.M.; Gallego-Escuredo, J.M.; Torres, F.; Mateo, G.M.; Villarroya, J.; de los Santos, I.; Domingo, J.C.; Villarroya, F.; Rio, L.D.; et al. Effects of Switching from Stavudine to Raltegravir on Subcutaneous Adipose Tissue in HIV-Infected Patients with HIV/HAART-Associated Lipodystrophy Syndrome (HALS). A Clinical and Molecular Study. PLoS ONE 2014, 9, e89088. [Google Scholar] [CrossRef] [PubMed]
- Coffinier, C.; Hudon, S.E.; Farber, E.A.; Chang, S.Y.; Hrycyna, C.A.; Young, S.G.; Fong, L.G. HIV protease inhibitors block the zinc metalloproteinase ZMPSTE24 and lead to an accumulation of prelamin A in cells. Proc. Natl. Acad. Sci. USA 2007, 104, 13432–13437. [Google Scholar] [CrossRef]
- Dean, J.M.; He, A.; Tan, M.; Wang, J.; Lu, D.; Razani, B.; Lodhi, I.J. MED19 Regulates Adipogenesis and Maintenance of White Adipose Tissue Mass by Mediating PPARγ-Dependent Gene Expression. Cell Rep. 2020, 33, 108228. [Google Scholar] [CrossRef]
- Klok, M.D.; Jakobsdottir, S.; Drent, M.L. The role of leptin and ghrelin in the regulation of food intake and body weight in humans: A review. Obes. Rev. 2007, 8, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Bjørbaek, C.; Kahn, B.B. Leptin signaling in the central nervous system and the periphery. Recent. Prog. Horm. Res. 2004, 59, 305–331. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Katagiri, H.; Ishigaki, Y.; Ogihara, T.; Imai, J.; Uno, K.; Hasegawa, Y.; Gao, J.; Ishihara, H.; Niijima, A.; et al. Signals from intra-abdominal fat modulate insulin and leptin sensitivity through different mechanisms: Neuronal involvement in food-intake regulation. Cell Metab. 2006, 3, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.Y.; Qu, S.L.; Xiong, W.H.; Rom, O.; Chang, L.; Jiang, Z.S. Perivascular adipose tissue (PVAT) in atherosclerosis: A double-edged sword. Cardiovasc. Diabetol. 2018, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.K.; Zhou, Z.; Zhang, J.; Zeng, R.; Wu, J.; Eitzman, D.T.; Chen, Y.E.; Chang, L. Perivascular adipose tissue in vascular function and disease: A review of current research and animal models. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1621–1630. [Google Scholar] [CrossRef] [PubMed]
- van Dam, A.D.; Boon, M.R.; Berbée, J.F.P.; Rensen, P.C.N.; van Harmelen, V. Targeting white, brown and perivascular adipose tissue in atherosclerosis development. Eur. J. Pharmacol. 2017, 816, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Li, R.M.; Chen, S.Q.; Zeng, N.X.; Zheng, S.H.; Guan, L.; Liu, H.M.; Zhou, L.Q.; Xu, J.W. Browning of Abdominal Aorta Perivascular Adipose Tissue Inhibits Adipose Tissue Inflammation. Metab. Syndr. Relat. Disord. 2017, 15, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Reynés, B.; Palou, M.; Palou, A. Gene expression modulation of lipid and central energetic metabolism related genes by high-fat diet intake in the main homeostatic tissues. Food Funct. 2017, 8, 629–650. [Google Scholar] [CrossRef]
- Tian, X.Y.; Ganeshan, K.; Hong, C.; Nguyen, K.D.; Qiu, Y.; Kim, J.; Tangirala, R.K.; Tontonoz, P.; Chawla, A. Thermoneutral Housing Accelerates Metabolic Inflammation to Potentiate Atherosclerosis but Not Insulin Resistance. Cell Metab. 2016, 23, 165–178. [Google Scholar] [CrossRef]
- Chang, L.; Villacorta, L.; Li, R.; Hamblin, M.; Xu, W.; Dou, C.; Zhang, J.; Wu, J.; Zeng, R.; Chen, Y.E. Loss of perivascular adipose tissue on peroxisome proliferator-activated receptor-γ deletion in smooth muscle cells impairs intravascular thermoregulation and enhances atherosclerosis. Circulation 2012, 126, 1067–1078. [Google Scholar] [CrossRef]
- Friederich-Persson, M.; Nguyen Dinh Cat, A.; Persson, P.; Montezano, A.C.; Touyz, R.M. Brown Adipose Tissue Regulates Small Artery Function Through NADPH Oxidase 4-Derived Hydrogen Peroxide and Redox-Sensitive Protein Kinase G-1α. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Lopez, N.; Thompson, J.M.; Watts, S.W. Perivascular Adipose Tissue’s Impact on Norepinephrine-Induced Contraction of Mesenteric Resistance Arteries. Front. Physiol. 2017, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Piché, M.E.; Vasan, S.K.; Hodson, L.; Karpe, F. Relevance of human fat distribution on lipid and lipoprotein metabolism and cardiovascular disease risk. Curr. Opin. Lipidol. 2018, 29, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulos, A.S.; Antoniades, C. The role of epicardial adipose tissue in cardiac biology: Classic concepts and emerging roles. J. Physiol. 2017, 595, 3907–3917. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G.; Willens, H.J. Echocardiographic epicardial fat: A review of research and clinical applications. J. Am. Soc. Echocardiogr. 2009, 22, 1311–1319, quiz 1417–1318. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G.; Corradi, D.; Sharma, A.M. Epicardial adipose tissue: Anatomic, biomolecular and clinical relationships with the heart. Nat. Clin. Pract. Cardiovasc. Med. 2005, 2, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G.; Pistilli, D.; Gucciardo, M.; Leonetti, F.; Miraldi, F.; Brancaccio, G.; Gallo, P.; di Gioia, C.R. Adiponectin expression in human epicardial adipose tissue in vivo is lower in patients with coronary artery disease. Cytokine 2005, 29, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Sacks, H.S.; Fain, J.N.; Bahouth, S.W.; Ojha, S.; Frontini, A.; Budge, H.; Cinti, S.; Symonds, M.E. Adult epicardial fat exhibits beige features. J. Clin. Endocrinol. Metab. 2013, 98, E1448–E1455. [Google Scholar] [CrossRef] [PubMed]
- Aldiss, P.; Davies, G.; Woods, R.; Budge, H.; Sacks, H.S.; Symonds, M.E. B‘rowning’ the cardiac and peri-vascular adipose tissues to modulate cardiovascular risk. Int. J. Cardiol. 2017, 228, 265–274. [Google Scholar] [CrossRef]
- Chechi, K.; Voisine, P.; Mathieu, P.; Laplante, M.; Bonnet, S.; Picard, F.; Joubert, P.; Richard, D. Functional characterization of the Ucp1-associated oxidative phenotype of human epicardial adipose tissue. Sci. Rep. 2017, 7, 15566. [Google Scholar] [CrossRef]
- Villasante Fricke, A.C.; Iacobellis, G. Epicardial Adipose Tissue: Clinical Biomarker of Cardio-Metabolic Risk. Int. J. Mol. Sci. 2019, 20, 5989. [Google Scholar] [CrossRef] [PubMed]
- Silaghi, A.; Achard, V.; Paulmyer-Lacroix, O.; Scridon, T.; Tassistro, V.; Duncea, I.; Clément, K.; Dutour, A.; Grino, M. Expression of adrenomedullin in human epicardial adipose tissue: Role of coronary status. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1443–E1450. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Tabata, M.; Kurobe, H.; Motoki, T.; Akaike, M.; Nishio, C.; Higashida, M.; Mikasa, H.; Nakaya, Y.; Takanashi, S.; et al. Coronary atherosclerosis is associated with macrophage polarization in epicardial adipose tissue. J. Am. Coll. Cardiol. 2011, 58, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Kurobe, H.; Akaike, M.; Chikugo, F.; Hori, T.; Bando, Y.; Nishio, C.; Higashida, M.; Nakaya, Y.; Kitagawa, T.; et al. Enhanced inflammation in epicardial fat in patients with coronary artery disease. Int. Heart J. 2011, 52, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Bilski, J.; Mazur-Bialy, A.; Wojcik, D.; Surmiak, M.; Magierowski, M.; Sliwowski, Z.; Pajdo, R.; Kwiecien, S.; Danielak, A.; Ptak-Belowska, A.; et al. Role of Obesity, Mesenteric Adipose Tissue, and Adipokines in Inflammatory Bowel Diseases. Biomolecules 2019, 9, 780. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Tan, J.; Chi, Y.; Zhang, F.; Xu, J.; Song, Y.; Cong, X.; Wu, N.; Liu, Y. Mesenteric adipose tissue contributes to intestinal barrier integrity and protects against nonalcoholic fatty liver disease in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G659–G670. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.M.D.; Chacon, P.A.I.; Ramalho, F.N.Z.; Foss, M.C.; Schmidt, A. Cardiac Alterations in Patients with Familial Lipodystrophy. Arq. Bras. Cardiol. 2020, 114, 305–312. [Google Scholar] [CrossRef]
- Herman, M.A.; Kahn, B.B. Glucose transport and sensing in the maintenance of glucose homeostasis and metabolic harmony. J. Clin. Investig. 2006, 116, 1767–1775. [Google Scholar] [CrossRef]
- Pelleymounter, M.A.; Cullen, M.J.; Baker, M.B.; Hecht, R.; Winters, D.; Boone, T.; Collins, F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science 1995, 269, 540–543. [Google Scholar] [CrossRef]
- Sacks, H.S.; Fain, J.N.; Cheema, P.; Bahouth, S.W.; Garrett, E.; Wolf, R.Y.; Wolford, D.; Samaha, J. Inflammatory genes in epicardial fat contiguous with coronary atherosclerosis in the metabolic syndrome and type 2 diabetes: Changes associated with pioglitazone. Diabetes Care 2011, 34, 730–733. [Google Scholar] [CrossRef]
- Distel, E.; Penot, G.; Cadoudal, T.; Balguy, I.; Durant, S.; Benelli, C. Early induction of a brown-like phenotype by rosiglitazone in the epicardial adipose tissue of fatty Zucker rats. Biochimie 2012, 94, 1660–1667. [Google Scholar] [CrossRef] [PubMed]
- Melvin, A.; Adams, C.; Flanagan, C.; Gaff, L.; Gratton, B.; Gribble, F.; Roberts, G.; Semple, R.K.; O’Rahilly, S.; Rubino, F.; et al. Roux-en-Y Gastric Bypass Surgery in the Management of Familial Partial Lipodystrophy Type 1. J. Clin. Endocrinol. Metab. 2017, 102, 3616–3620. [Google Scholar] [CrossRef] [PubMed]
- Banning, F.; Rottenkolber, M.; Freibothe, I.; Seissler, J.; Lechner, A. Insulin secretory defect in familial partial lipodystrophy Type 2 and successful long-term treatment with a glucagon-like peptide 1 receptor agonist. Diabet. Med. 2017, 34, 1792–1794. [Google Scholar] [CrossRef] [PubMed]
- Valerio, C.M.; de Almeida, J.S.; Moreira, R.O.; Aguiar, L.B.S.; Siciliano, P.O.; Carvalho, D.P.; Godoy-Matos, A.F. Dipeptidyl peptidase-4 levels are increased and partially related to body fat distribution in patients with familial partial lipodystrophy type 2. Diabetol. Metab. Syndr. 2017, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Hartinger, R.; Lederer, E.M.; Schena, E.; Lattanzi, G.; Djabali, K. Impact of Combined Baricitinib and FTI Treatment on Adipogenesis in Hutchinson-Gilford Progeria Syndrome and Other Lipodystrophic Laminopathies. Cells 2023, 12, 1350. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Qiao, X.; Fong, L.G.; Young, S.G. Treatment with a farnesyltransferase inhibitor improves survival in mice with a Hutchinson-Gilford progeria syndrome mutation. Biochim. Biophys. Acta 2008, 1781, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Young, S.G.; Meta, M.; Yang, S.H.; Fong, L.G. Prelamin A farnesylation and progeroid syndromes. J. Biol. Chem. 2006, 281, 39741–39745. [Google Scholar] [CrossRef]
- Clements, C.S.; Bikkul, M.U.; Ofosu, W.; Eskiw, C.; Tree, D.; Makarov, E.; Kill, I.R.; Bridger, J.M. Presence and distribution of progerin in HGPS cells is ameliorated by drugs that impact on the mevalonate and mTOR pathways. Biogerontology 2019, 20, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.B.; Massaro, J.; D’Agostino, R.B., Sr.; Campbell, S.E.; Brazier, J.; Brown, W.T.; Kleinman, M.E.; Kieran, M.W. Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation 2014, 130, 27–34. [Google Scholar] [CrossRef]
- Strandberg, T.E. Role of Statin Therapy in Primary Prevention of Cardiovascular Disease in Elderly Patients. Curr. Atheroscler. Rep. 2019, 21, 28. [Google Scholar] [CrossRef]
- Langdahl, B.L.; Hofbauer, L.C.; Forfar, J.C. Cardiovascular Safety and Sclerostin Inhibition. J. Clin. Endocrinol. Metab. 2021, 106, 1845–1853. [Google Scholar] [CrossRef]
- Gordon, L.B.; Kleinman, M.E.; Miller, D.T.; Neuberg, D.S.; Giobbie-Hurder, A.; Gerhard-Herman, M.; Smoot, L.B.; Gordon, C.M.; Cleveland, R.; Snyder, B.D.; et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2012, 109, 16666–16671. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.J.; Mastronardi, C.A.; Paz-Filho, G.J. New advances in the treatment of generalized lipodystrophy: Role of metreleptin. Ther. Clin. Risk Manag. 2015, 11, 1391–1400. [Google Scholar] [CrossRef]
- Chan, J.L.; Koda, J.; Heilig, J.S.; Cochran, E.K.; Gorden, P.; Oral, E.A.; Brown, R.J. Immunogenicity associated with metreleptin treatment in patients with obesity or lipodystrophy. Clin. Endocrinol. 2016, 85, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Püschel, J.; Miehle, K.; Müller, K.; Villringer, A.; Stumvoll, M.; Fasshauer, M.; Schlögl, H. Beneficial effects of leptin substitution on impaired eating behavior in lipodystrophy are sustained beyond 150 weeks of treatment. Cytokine 2019, 113, 400–404. [Google Scholar] [CrossRef]
- Cook, K.; Ali, O.; Akinci, B.; Foss de Freitas, M.C.; Montenegro, R.M.; Fernandes, V.O.; Gupta, D.; Lou, K.J.; Tuttle, E.; Oral, E.A.; et al. Effect of Leptin Therapy on Survival in Generalized and Partial Lipodystrophy: A Matched Cohort Analysis. J. Clin. Endocrinol. Metab. 2021, 106, e2953–e2967. [Google Scholar] [CrossRef] [PubMed]
- Chong, A.Y.; Lupsa, B.C.; Cochran, E.K.; Gorden, P. Efficacy of leptin therapy in the different forms of human lipodystrophy. Diabetologia 2010, 53, 27–35. [Google Scholar] [CrossRef]
- Vasandani, C.; Clark, G.O.; Adams-Huet, B.; Quittner, C.; Garg, A. Efficacy and Safety of Metreleptin Therapy in Patients With Type 1 Diabetes: A Pilot Study. Diabetes Care 2017, 40, 694–697. [Google Scholar] [CrossRef]
- Sekizkardes, H.; Cochran, E.; Malandrino, N.; Garg, A.; Brown, R.J. Efficacy of Metreleptin Treatment in Familial Partial Lipodystrophy Due to PPARG vs. LMNA Pathogenic Variants. J. Clin. Endocrinol. Metab. 2019, 104, 3068–3076. [Google Scholar] [CrossRef]
- Oral, E.A.; Gorden, P.; Cochran, E.; Araújo-Vilar, D.; Savage, D.B.; Long, A.; Fine, G.; Salinardi, T.; Brown, R.J. Long-term effectiveness and safety of metreleptin in the treatment of patients with partial lipodystrophy. Endocrine 2019, 64, 500–511. [Google Scholar] [CrossRef]
- Simsir, I.Y.; Yurekli, B.S.; Polat, I.; Saygili, F.; Akinci, B. Metreleptin replacement treatment improves quality of life and psychological well-being in congenital generalized lipodystrophy. Natl. Med. J. India 2020, 33, 278–280. [Google Scholar] [CrossRef] [PubMed]
- Özalkak, Ş.; Demiral, M.; Ünal, E.; Taş, F.F.; Onay, H.; Demirbilek, H.; Özbek, M.N. Metreleptin Treatment in a Boy with Congenital Generalized Lipodystrophy due to Homozygous c.465_468delGACT (p.T156Rfs*8) Mutation in the BSCL2 Gene: Results From the First-year. J. Clin. Res. Pediatr. Endocrinol. 2023, 15, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Gryn, S.E.; Hegele, R.A. New oral agents for treating dyslipidemia. Curr. Opin. Lipidol. 2016, 27, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.A.K.; Cornicelli, J.A.; Markham, B.; Bisgaier, C.L. Gemcabene, a first-in-class lipid-lowering agent in late-stage development, down-regulates acute-phase C-reactive protein via C/EBP-δ-mediated transcriptional mechanism. Mol. Cell Biochem. 2018, 449, 167–183. [Google Scholar] [CrossRef] [PubMed]
- Akinci, B.; Swaidan, M.; Foss-Freitas, M.C.; Luo, Y.; Neidert, A.H.; Hench, R.P.; Chenevert, T.L.; Longcore, A.; Bakker-Arkema, R.; Bisgaier, C.L.; et al. 2214-PUB: An Open-Label Study of Gemcabene in Adults with Familial Partial Lipodystrophy. Diabetes 2020, 69. [Google Scholar] [CrossRef]
- Raal, F.J.; Rosenson, R.S.; Reeskamp, L.F.; Hovingh, G.K.; Kastelein, J.J.P.; Rubba, P.; Ali, S.; Banerjee, P.; Chan, K.C.; Gipe, D.A.; et al. Evinacumab for Homozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 383, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Reiner, Ž. Triglyceride-Rich Lipoproteins and Novel Targets for Anti-atherosclerotic Therapy. Korean Circ. J. 2018, 48, 1097–1119. [Google Scholar] [CrossRef]
- Paik, J.; Duggan, S. Volanesorsen: First Global Approval. Drugs 2019, 79, 1349–1354. [Google Scholar] [CrossRef]
- Alexander, V.J.; Xia, S.; Hurh, E.; Hughes, S.G.; O’Dea, L.; Geary, R.S.; Witztum, J.L.; Tsimikas, S. N-acetyl galactosamine-conjugated antisense drug to APOC3 mRNA, triglycerides and atherogenic lipoprotein levels. Eur. Heart J. 2019, 40, 2785–2796. [Google Scholar] [CrossRef]
- Sponton, C.H.; Kajimura, S. AAV-mediated gene therapy as a strategy to fight obesity and metabolic diseases. EMBO Mol. Med. 2018, 10, e9431. [Google Scholar] [CrossRef]
- Jimenez, V.; Jambrina, C.; Casana, E.; Sacristan, V.; Muñoz, S.; Darriba, S.; Rodó, J.; Mallol, C.; Garcia, M.; León, X.; et al. FGF21 gene therapy as treatment for obesity and insulin resistance. EMBO Mol. Med. 2018, 10, e8791. [Google Scholar] [CrossRef] [PubMed]
- Degirolamo, C.; Sabbà, C.; Moschetta, A. Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23. Nat. Rev. Drug Discov. 2016, 15, 51–69. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Barkho, B.Z.; Ruiz, S.; Diep, D.; Qu, J.; Yang, S.L.; Panopoulos, A.D.; Suzuki, K.; Kurian, L.; Walsh, C.; et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature 2011, 472, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xiong, Z.M.; Cao, K. Mechanisms controlling the smooth muscle cell death in progeria via down-regulation of poly(ADP-ribose) polymerase 1. Proc. Natl. Acad. Sci. USA 2014, 111, E2261–E2270. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 2005, 11, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.B.; Harten, I.A.; Patti, M.E.; Lichtenstein, A.H. Reduced adiponectin and HDL cholesterol without elevated C-reactive protein: Clues to the biology of premature atherosclerosis in Hutchinson-Gilford Progeria Syndrome. J. Pediatr. 2005, 146, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Hamczyk, M.R.; Villa-Bellosta, R.; Gonzalo, P.; Andrés-Manzano, M.J.; Nogales, P.; Bentzon, J.F.; López-Otín, C.; Andrés, V. Vascular Smooth Muscle-Specific Progerin Expression Accelerates Atherosclerosis and Death in a Mouse Model of Hutchinson-Gilford Progeria Syndrome. Circulation 2018, 138, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Hegele, R.A. Familial partial lipodystrophy: A monogenic form of the insulin resistance syndrome. Mol. Genet. Metab. 2000, 71, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Weinreich, M.; Frishman, W.H. Antihyperlipidemic therapies targeting PCSK9. Cardiol. Rev. 2014, 22, 140–146. [Google Scholar] [CrossRef]
- Levenson, A.E.; Haas, M.E.; Miao, J.; Brown, R.J.; de Ferranti, S.D.; Muniyappa, R.; Biddinger, S.B. Effect of Leptin Replacement on PCSK9 in ob/ob Mice and Female Lipodystrophic Patients. Endocrinology 2016, 157, 1421–1429. [Google Scholar] [CrossRef]
- Cesaro, A.; Gragnano, F.; Fimiani, F.; Moscarella, E.; Diana, V.; Pariggiano, I.; Concilio, C.; Natale, F.; Limongelli, G.; Bossone, E.; et al. Impact of PCSK9 inhibitors on the quality of life of patients at high cardiovascular risk. Eur. J. Prev. Cardiol. 2020, 27, 556–558. [Google Scholar] [CrossRef]
- Lappegård, K.T.; Enebakk, T.; Thunhaug, H.; Hovland, A. Transition from LDL apheresis to evolocumab in heterozygous FH is equally effective in lowering LDL, without lowering HDL cholesterol. Atherosclerosis 2016, 251, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Lagathu, C.; Béréziat, V.; Gorwood, J.; Fellahi, S.; Bastard, J.P.; Vigouroux, C.; Boccara, F.; Capeau, J. Metabolic complications affecting adipose tissue, lipid and glucose metabolism associated with HIV antiretroviral treatment. Expert. Opin. Drug Saf. 2019, 18, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, R.V. Treatment of Dyslipidemia in HIV. Curr. Atheroscler. Rep. 2015, 17, 17. [Google Scholar] [CrossRef] [PubMed]
- Ewald, H.; Santini-Oliveira, M.; Bühler, J.E.; Vuichard, D.; Schandelmaier, S.; Stöckle, M.; Briel, M.; Bucher, H.C.; Hemkens, L.G. Comparative effectiveness of tenofovir in HIV-infected treatment-experienced patients: Systematic review and meta-analysis. HIV Clin. Trials 2017, 18, 17–27. [Google Scholar] [CrossRef]
- Araújo-Vilar, D.; Lattanzi, G.; González-Méndez, B.; Costa-Freitas, A.T.; Prieto, D.; Columbaro, M.; Mattioli, E.; Victoria, B.; Martínez-Sánchez, N.; Ramazanova, A.; et al. Site-dependent differences in both prelamin A and adipogenic genes in subcutaneous adipose tissue of patients with type 2 familial partial lipodystrophy. J. Med. Genet. 2009, 46, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Peinado, J.R.; Quirós, P.M.; Pulido, M.R.; Mariño, G.; Martínez-Chantar, M.L.; Vázquez-Martínez, R.; Freije, J.M.; López-Otín, C.; Malagón, M.M. Proteomic profiling of adipose tissue from Zmpste24-/- mice, a model of lipodystrophy and premature aging, reveals major changes in mitochondrial function and vimentin processing. Mol. Cell Proteom. 2011, 10, M111.008094. [Google Scholar] [CrossRef] [PubMed]
- Caron, M.; Vigouroux, C.; Bastard, J.P.; Capeau, J. Adipocyte dysfunction in response to antiretroviral therapy: Clinical, tissue and in-vitro studies. Curr. Opin. HIV AIDS 2007, 2, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Sawada, D.; Kato, H.; Kaneko, H.; Kinoshita, D.; Funayama, S.; Minamizuka, T.; Takasaki, A.; Igarashi, K.; Koshizaka, M.; Takada-Watanabe, A.; et al. Senescence-associated inflammation and inhibition of adipogenesis in subcutaneous fat in Werner syndrome. Aging 2023, 15, 9948–9964. [Google Scholar] [CrossRef]
- Neves, J.; Sousa-Victor, P. Regulation of inflammation as an anti-aging intervention. FEBS J. 2020, 287, 43–52. [Google Scholar] [CrossRef]
- Bieber, T.; Paller, A.S.; Kabashima, K.; Feely, M.; Rueda, M.J.; Ross Terres, J.A.; Wollenberg, A. Atopic dermatitis: Pathomechanisms and lessons learned from novel systemic therapeutic options. J. Eur. Acad. Dermatol. Venereol. 2022, 36, 1432–1449. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Arnold, R.; Henriques, G.; Djabali, K. Inhibition of JAK-STAT Signaling with Baricitinib Reduces Inflammation and Improves Cellular Homeostasis in Progeria Cells. Cells 2019, 8, 1276. [Google Scholar] [CrossRef] [PubMed]
- Arnold, R.; Vehns, E.; Randl, H.; Djabali, K. Baricitinib, a JAK-STAT Inhibitor, Reduces the Cellular Toxicity of the Farnesyltransferase Inhibitor Lonafarnib in Progeria Cells. Int. J. Mol. Sci. 2021, 22, 7474. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, G.A.M.; Reinhardt, A.; Ramsey, S.; Wittkowski, H.; Hashkes, P.J.; Berkun, Y.; Schalm, S.; Murias, S.; Dare, J.A.; Brown, D.; et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J. Clin. Investig. 2018, 128, 3041–3052. [Google Scholar] [CrossRef] [PubMed]
- Bajetto, A.; Pattarozzi, A.; Sirito, R.; Barbieri, F.; Florio, T. Metformin potentiates immunosuppressant activity and adipogenic differentiation of human umbilical cord-mesenchymal stem cells. Int. Immunopharmacol. 2023, 124, 111078. [Google Scholar] [CrossRef] [PubMed]
- Egesipe, A.L.; Blondel, S.; Lo Cicero, A.; Jaskowiak, A.L.; Navarro, C.; Sandre-Giovannoli, A.; Levy, N.; Peschanski, M.; Nissan, X. Metformin decreases progerin expression and alleviates pathological defects of Hutchinson-Gilford progeria syndrome cells. NPJ Aging Mech. Dis. 2016, 2, 16026. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.Y.; Anderson, S.S.; Chicoine, N.H.; Mayfield, J.R.; Academia, E.C.; Wilson, J.A.; Pongkietisak, C.; Thompson, M.A.; Lagmay, E.P.; Miller, D.M.; et al. Rapamycin Reverses Metabolic Deficits in Lamin A/C-Deficient Mice. Cell Rep. 2016, 17, 2542–2552. [Google Scholar] [CrossRef] [PubMed]
- Cenni, V.; Capanni, C.; Columbaro, M.; Ortolani, M.; D’Apice, M.R.; Novelli, G.; Fini, M.; Marmiroli, S.; Scarano, E.; Maraldi, N.M.; et al. Autophagic degradation of farnesylated prelamin A as a therapeutic approach to lamin-linked progeria. Eur. J. Histochem. 2011, 55, e36. [Google Scholar] [CrossRef]
- Cenni, V.; Capanni, C.; Mattioli, E.; Columbaro, M.; Wehnert, M.; Ortolani, M.; Fini, M.; Novelli, G.; Bertacchini, J.; Maraldi, N.M.; et al. Rapamycin treatment of Mandibuloacral dysplasia cells rescues localization of chromatin-associated proteins and cell cycle dynamics. Aging 2014, 6, 755–770. [Google Scholar] [CrossRef]
- Wipperman, M.F.; Montrose, D.C.; Gotto, A.M., Jr.; Hajjar, D.P. Mammalian Target of Rapamycin: A Metabolic Rheostat for Regulating Adipose Tissue Function and Cardiovascular Health. Am. J. Pathol. 2019, 189, 492–501. [Google Scholar] [CrossRef]
- Osorio, F.G.; Bárcena, C.; Soria-Valles, C.; Ramsay, A.J.; de Carlos, F.; Cobo, J.; Fueyo, A.; Freije, J.M.; López-Otín, C. Nuclear lamina defects cause ATM-dependent NF-κB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012, 26, 2311–2324. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, S.; Onder, G.; Liperoti, R.; Russo, A.; Carter, C.; Capoluongo, E.; Pahor, M.; Bernabei, R.; Landi, F. Interleukin-6, C-reactive protein, and tumor necrosis factor-alpha as predictors of mortality in frail, community-living elderly individuals. J. Am. Geriatr. Soc. 2011, 59, 1679–1685. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Tocilizumab: A Review in Rheumatoid Arthritis. Drugs 2017, 77, 1865–1879. [Google Scholar] [CrossRef] [PubMed]
- Squarzoni, S.; Schena, E.; Sabatelli, P.; Mattioli, E.; Capanni, C.; Cenni, V.; D’Apice, M.R.; Andrenacci, D.; Sarli, G.; Pellegrino, V.; et al. Interleukin-6 neutralization ameliorates symptoms in prematurely aged mice. Aging Cell 2021, 20, e13285. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.J.; Lee, R.G.; Brandt, T.A.; Tai, L.-J.; Fu, W.; Peralta, R.; Yu, R.; Hurh, E.; Paz, E.; McEvoy, B.W.; et al. Cardiovascular and Metabolic Effects of ANGPTL3 Antisense Oligonucleotides. N. Engl. J. Med. 2017, 377, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Dewey, F.E.; Gusarova, V.; Dunbar, R.L.; O’Dushlaine, C.; Schurmann, C.; Gottesman, O.; McCarthy, S.; Van Hout, C.V.; Bruse, S.; Dansky, H.M.; et al. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicki, A.S.; Viljoen, A. Anti-sense oligonucleotide therapies for the treatment of hyperlipidaemia. Expert. Opin. Biol. Ther. 2016, 16, 1125–1134. [Google Scholar] [CrossRef]
- Pelka, K.; Shibata, T.; Miyake, K.; Latz, E. Nucleic acid-sensing TLRs and autoimmunity: Novel insights from structural and cell biology. Immunol. Rev. 2016, 269, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Ruda, V.M.; Chandwani, R.; Sehgal, A.; Bogorad, R.L.; Akinc, A.; Charisse, K.; Tarakhovsky, A.; Novobrantseva, T.I.; Koteliansky, V. The roles of individual mammalian argonautes in RNA interference in vivo. PLoS ONE 2014, 9, e101749. [Google Scholar] [CrossRef]
- Sharma, V.K.; Watts, J.K. Oligonucleotide therapeutics: Chemistry, delivery and clinical progress. Future Med. Chem. 2015, 7, 2221–2242. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Cannon, C.P.; Morrow, D.A.; Ray, K.K.; Pfeffer, M.A.; Braunwald, E. Can low-density lipoprotein be too low? The safety and efficacy of achieving very low low-density lipoprotein with intensive statin therapy: A PROVE IT-TIMI 22 substudy. J. Am. Coll. Cardiol. 2005, 46, 1411–1416. [Google Scholar] [CrossRef] [PubMed]
- Fruchart, J.C.; Sacks, F.M.; Hermans, M.P.; Assmann, G.; Brown, W.V.; Ceska, R.; Chapman, M.J.; Dodson, P.M.; Fioretto, P.; Ginsberg, H.N.; et al. The Residual Risk Reduction Initiative: A call to action to reduce residual vascular risk in dyslipidaemic patient. Diab. Vasc. Dis. Res. 2008, 5, 319–335. [Google Scholar] [CrossRef] [PubMed]
- Macchi, C.; Sirtori, C.R.; Corsini, A.; Santos, R.D.; Watts, G.F.; Ruscica, M. A new dawn for managing dyslipidemias: The era of rna-based therapies. Pharmacol. Res. 2019, 150, 104413. [Google Scholar] [CrossRef] [PubMed]
- Arca, M.; Hsieh, A.; Soran, H.; Rosenblit, P.; O’Dea, L.; Stevenson, M. The effect of volanesorsen treatment on the burden associated with familial chylomicronemia syndrome: The results of the ReFOCUS study. Expert. Rev. Cardiovasc. Ther. 2018, 16, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Nimesh, S.; Gupta, N.; Chandra, R. Cationic polymer based nanocarriers for delivery of therapeutic nucleic acids. J. Biomed. Nanotechnol. 2011, 7, 504–520. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, S.; Gholami, M.H.; Ang, H.L.; Hashemi, F.; Zarrabi, A.; Zabolian, A.; Hushmandi, K.; Delfi, M.; Khan, H.; Ashrafizadeh, M.; et al. Pre-Clinical and Clinical Applications of Small Interfering RNAs (siRNA) and Co-Delivery Systems for Pancreatic Cancer Therapy. Cells 2021, 10, 3348. [Google Scholar] [CrossRef] [PubMed]
- Shim, G.; Jeong, S.; Oh, J.L.; Kang, Y. Lipid-based nanoparticles for photosensitive drug delivery systems. J. Pharm. Investig. 2022, 52, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.E.; Drummond, D.C.; Kirpotin, D.B.; Zheng, W.W.; Noble, C.O.; Park, J.W.; Marks, J.D.; Benz, C.C.; Hong, K. Genospheres: Self-assembling nucleic acid-lipid nanoparticles suitable for targeted gene delivery. Gene Ther. 2006, 13, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Ickenstein, L.M.; Garidel, P. Lipid-based nanoparticle formulations for small molecules and RNA drugs. Expert. Opin. Drug Deliv. 2019, 16, 1205–1226. [Google Scholar] [CrossRef]
- Morales-Becerril, A.; Aranda-Lara, L.; Isaac-Olivé, K.; Ocampo-García, B.E.; Morales-Ávila, E. Nanocarriers for delivery of siRNA as gene silencing mediator. EXCLI J. 2022, 21, 1028–1052. [Google Scholar] [CrossRef]
- Wong, F.M.; MacAdam, S.A.; Kim, A.; Oja, C.; Ramsay, E.C.; Bally, M.B. A lipid-based delivery system for antisense oligonucleotides derived from a hydrophobic complex. J. Drug Target. 2002, 10, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Paunovska, K.; Loughrey, D.; Dahlman, J.E. Drug delivery systems for RNA therapeutics. Nat. Rev. Genet. 2022, 23, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, G.; Kubben, N.; Wickert, U.; Göhler, H.; Hoffmann, K.; Misteli, T. Ageing-related chromatin defects through loss of the NURD complex. Nat. Cell Biol. 2009, 11, 1261–1267. [Google Scholar] [CrossRef] [PubMed]
- Min, H.S.; Kim, H.J.; Naito, M.; Ogura, S.; Toh, K.; Hayashi, K.; Kim, B.S.; Fukushima, S.; Anraku, Y.; Miyata, K.; et al. Systemic Brain Delivery of Antisense Oligonucleotides across the Blood-Brain Barrier with a Glucose-Coated Polymeric Nanocarrier. Angew. Chem. Int. Ed. Engl. 2020, 59, 8173–8180. [Google Scholar] [CrossRef] [PubMed]
- Araújo, D.; Braz, J.; Dencheva, N.V.; Carvalho, I.; Henriques, M.; Denchev, Z.Z.; Malfois, M.; Silva, S. Polyamide Microsized Particulate Polyplex Carriers for the 2’-OMethylRNA EFG1 Antisense Oligonucleotide. ACS Appl. Bio Mater. 2021, 4, 4607–4617. [Google Scholar] [CrossRef] [PubMed]
- Kilicay, E.; Karahaliloglu, Z.; Alpaslan, P.; Hazer, B.; Denkbas, E.B. In vitro evaluation of antisense oligonucleotide functionalized core-shell nanoparticles loaded with α-tocopherol succinate. J. Biomater. Sci. Polym. Ed. 2017, 28, 1762–1785. [Google Scholar] [CrossRef] [PubMed]
- Mainini, F.; Eccles, M.R. Lipid and Polymer-Based Nanoparticle siRNA Delivery Systems for Cancer Therapy. Molecules 2020, 25, 2692. [Google Scholar] [CrossRef]
- Zhao, J.; Weng, G.; Li, J.; Zhu, J.; Zhao, J. Polyester-based nanoparticles for nucleic acid delivery. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 92, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Dunbar, C.E.; High, K.A.; Joung, J.K.; Kohn, D.B.; Ozawa, K.; Sadelain, M. Gene therapy comes of age. Science 2018, 359, eaan4672. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, S.M.; Hinkle, C.; Chen, S.J.; Sandhu, A.; Hovhannisyan, R.; Stephan, S.; Lagor, W.R.; Ahima, R.S.; Johnston, J.C.; Reilly, M.P. Targeting adipose tissue via systemic gene therapy. Gene Ther. 2014, 21, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Uhrig-Schmidt, S.; Geiger, M.; Luippold, G.; Birk, G.; Mennerich, D.; Neubauer, H.; Grimm, D.; Wolfrum, C.; Kreuz, S. Gene delivery to adipose tissue using transcriptionally targeted rAAV8 vectors. PLoS ONE 2014, 9, e116288. [Google Scholar] [CrossRef] [PubMed]
- Sommer, N.; Roumane, A.; Han, W.; Delibegović, M.; Rochford, J.J.; McIlroy, G.D. Gene therapy restores adipose tissue and metabolic health in a pre-clinical mouse model of lipodystrophy. Mol. Ther. Methods Clin. Dev. 2022, 27, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, A.C.; Evitt, N.H.; Lv, W.; Musunuru, K. Reduced Blood Lipid Levels With In Vivo CRISPR-Cas9 Base Editing of ANGPTL3. Circulation 2018, 137, 975–977. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Good news in the nuclear envelope: Loss of lamin A might be a gain. J. Clin. Investig. 2006, 116, 632–634. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.-J.; Coffinier, C.; Choe, Y.; Beigneux, A.P.; Davies, B.S.J.; Yang, S.H.; Barnes, R.H.; Hong, J.; Sun, T.; Pleasure, S.J.; et al. Regulation of prelamin A but not lamin C by miR-9, a brain-specific microRNA. Proc. Natl. Acad. Sci. USA 2012, 109, E423–E431. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Guan, Y.; Chen, Y.; Zhang, C.; Yu, L.; Gao, H.; Du, H.; Liu, B.; Wang, X. miRNA-9 expression is upregulated in the spinal cord of G93A-SOD1 transgenic mice. Int. J. Clin. Exp. Pathol. 2013, 6, 1826–1838. [Google Scholar]
- Liu, G.-H.; Suzuki, K.; Qu, J.; Sancho-Martinez, I.; Yi, F.; Li, M.; Kumar, S.; Nivet, E.; Kim, J.; Soligalla, R.D.; et al. Targeted gene correction of laminopathy-associated LMNA mutations in patient-specific iPSCs. Cell Stem Cell 2011, 8, 688–694. [Google Scholar] [CrossRef]
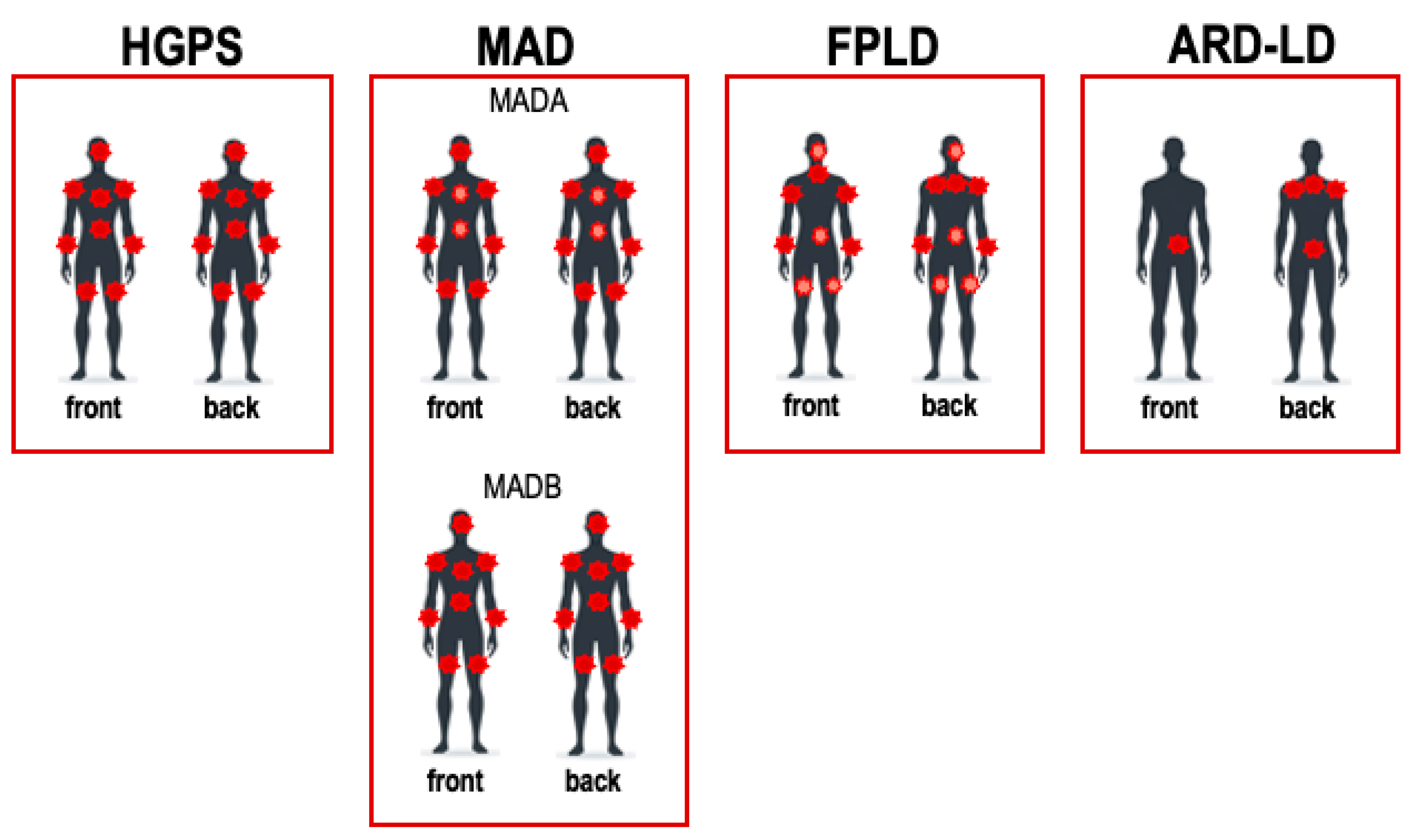
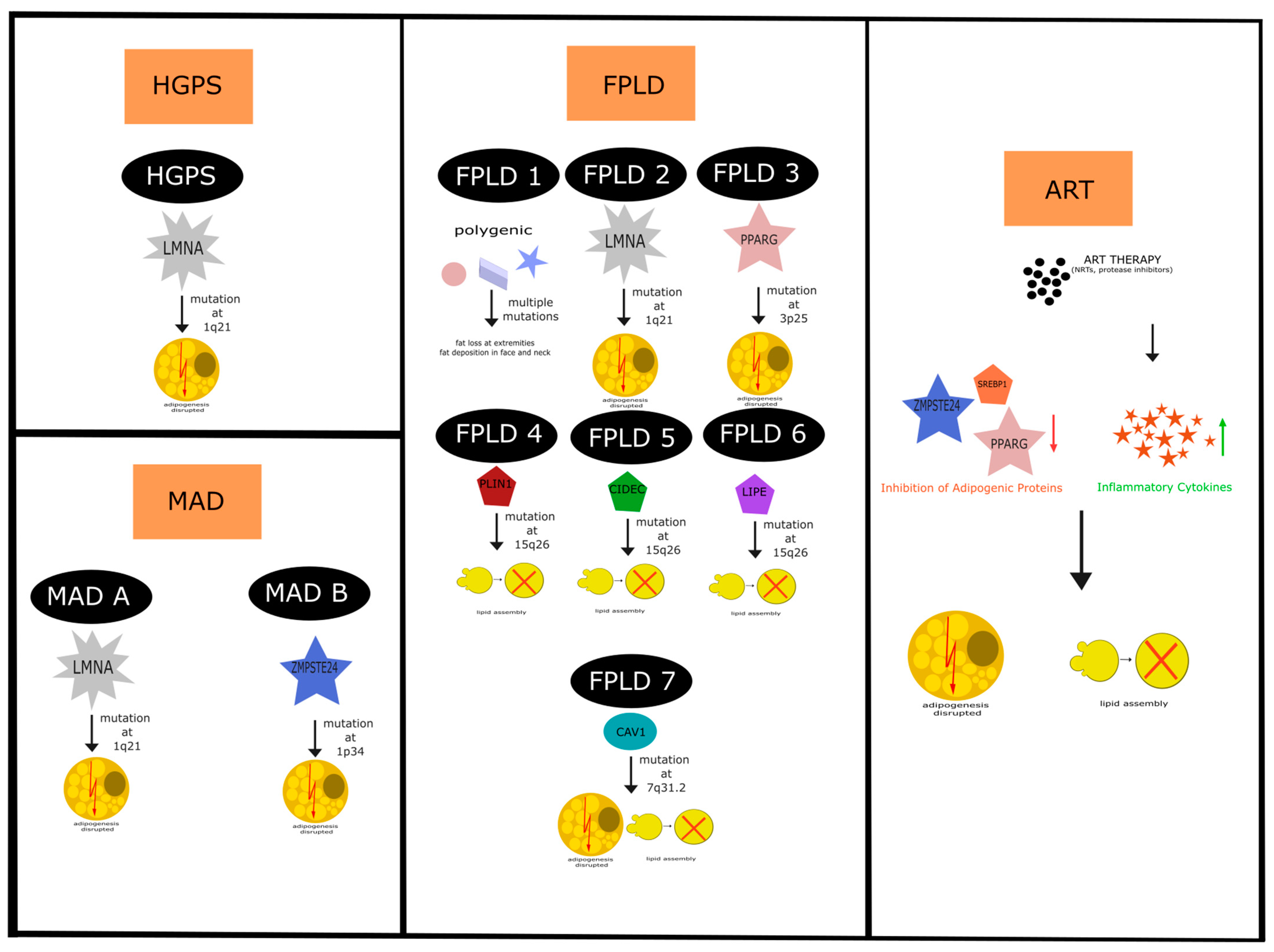
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krüger, P.; Hartinger, R.; Djabali, K. Navigating Lipodystrophy: Insights from Laminopathies and Beyond. Int. J. Mol. Sci. 2024, 25, 8020. https://doi.org/10.3390/ijms25158020
Krüger P, Hartinger R, Djabali K. Navigating Lipodystrophy: Insights from Laminopathies and Beyond. International Journal of Molecular Sciences. 2024; 25(15):8020. https://doi.org/10.3390/ijms25158020
Chicago/Turabian StyleKrüger, Peter, Ramona Hartinger, and Karima Djabali. 2024. "Navigating Lipodystrophy: Insights from Laminopathies and Beyond" International Journal of Molecular Sciences 25, no. 15: 8020. https://doi.org/10.3390/ijms25158020