Abstract
Caffeic acid phenethyl ester (CAPE) is a phenolic natural product with a wide range of biological activities, including anticancer activity; however, the ester group of CAPE is metabolically labile. The corresponding amide, CAPA, has improved metabolic stability but limited anticancer activity relative to CAPE. We report the synthesis using flow and on-water Wittig reaction approaches of five previously reported and five novel CAPA analogues. All of these analogues lack the reactive catechol functionality of CAPA and CAPE. Cytotoxicity studies of CAPE, CAPA, and these CAPA analogues in HeLa and BE(2)-C cells were carried out. Surprisingly, we found that CAPA is cytotoxic against the neuroblastoma BE(2)-C cell line (IC50 = 12 µM), in contrast to the weak activity of CAPA against HeLa cells (IC50 = 112 µM), and the literature reports of the absence of activity for CAPA against a variety of other cancer cell lines. One novel CAPA analogue, 3f, was identified as having cytotoxic activity similar to CAPE in HeLa cells (IC50 = 63 µM for 3f vs. 32 µM for CAPE), albeit with lower activity against BE(2)-C cells (IC50 = 91 µM) than CAPA. A different CAPA analogue, 3g, was found to have similar effects against BE(2)-C cells (IC50 = 92 µM). These results show that CAPA is uniquely active against neuroblastoma cells and that specific CAPA analogues that are predicted to be more metabolically stable than CAPE can reproduce CAPA’s activity against neuroblastoma cells and CAPE’s activity against HeLa cells.
1. Introduction
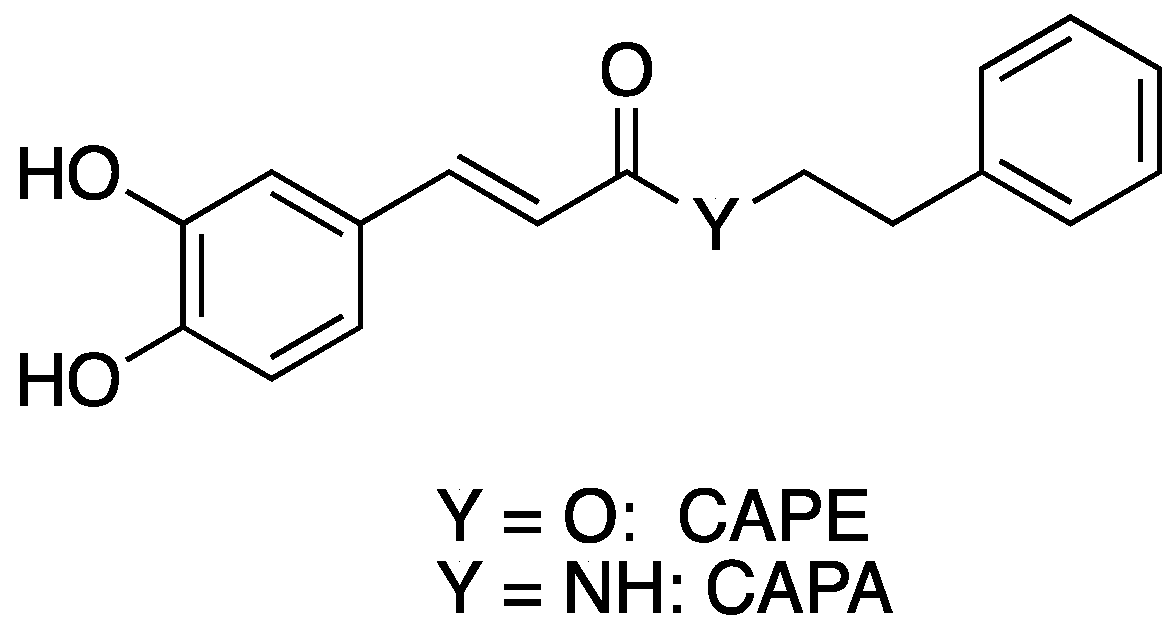
Caffeic acid phenethyl ester (CAPE) (Figure 1) is a phenolic natural product isolated from bee propolis. Like propolis, CAPE has been reported to have a wide range of biological activity, including antimicrobial, anti-inflammatory, antioxidant, and immunostimulatory effects [1,2,3,4,5]. However, the metabolic liability of CAPE has led to the search for more metabolically stable analogues [6]. One promising approach to CAPE analogs with increased stability involves replacing the metabolically unstable ester linkage in CAPE with an amide moiety to give caffeic acid phenethyl amide (CAPA) (Figure 1). This change in going from CAPE to CAPA introduces “amidicity” [7], which has been shown to be a fundamental phenomenon and driving force controlling reactivity or functionality and can be measured and indexed. In some cases, the biological activity of CAPA mirrors that of CAPE. For example, CAPA is similar to CAPE in its reported cytoprotective activity against oxidative insult to human umbilical vein endothelial cells (HUVEC) [8]. CAPE and CAPA both are potent inhibitors of leukotriene biosynthesis in vitro [9], antifungal agents that synergize with fluconazole [10], and inhibitors of biofilm formation and quorum sensing [11]. However, CAPA does not possess all the biological activities associated with CAPE; the anti-cancer activity of CAPA is much lower than that of CAPE [12,13,14,15].
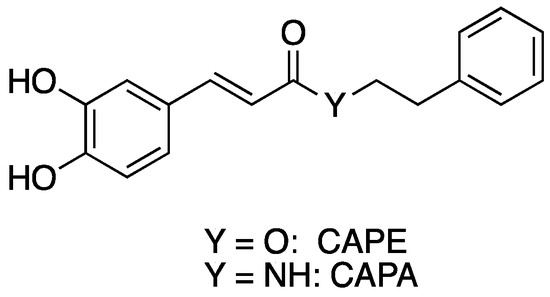
Figure 1.
Structure of CAPE and CAPA.
To address the issue of CAPA’s relative lack of anticancer activity compared to CAPE, we set out to explore analogues of CAPA that may recapture the anticancer properties of CAPE while possessing the increased metabolic stability associated with the ester-to-amide replacement. The mechanism of the anticancer activity of CAPE is not well established and likely varies from one cancer cell type to another [12,13,14,15]. The hydrolysis of CAPE to caffeic acid and 2-phenylethanol does not appear to be involved, as caffeic acid itself does not display cytotoxicity to cancer cells, and other esters of caffeic acid can have superior in vitro activity to CAPE [13,14,15]. Our focus here was on less readily hydrolyzed CAPA analogs with varied substituents on the caffeic acid catechol ring. Substituting the catechol moiety of CAPE or CAPA with alternative substituted phenyl groups has the added benefit of further increasing the metabolic stability of these analogs by avoiding the reactivity and extensive phase II metabolism associated with catechol groups.
There are a variety of synthetic approaches that have been used to prepare CAPE and CAPA analogues, but the most common include the ester or amide coupling of caffeic acid derivatives or the Wittig olefination of appropriate benzaldehydes [16]. Continuous flow methods offer several advantages to traditional batch methods for the synthesis of biologically active compounds in terms of safety, sustainability, and efficiency [17]. Specifically, Wittig olefination carried out in flow reactors has been used to prepare cinnamic ester derivatives [18] and cinnamides [19,20]. However, there have been no reports of flow reaction conditions to prepare CAPE and CAPA analogues. Wittig reactions have been carried out in water employing water-soluble phosphonium salts [21], but the advantages of “on-water” Wittig reactions of typical, water-insoluble partners have only recently been recognized [22,23]. While caffeic esters have been prepared on-water from preformed phosphorylides [24], there are no reports of this green approach to caffeic amides employing precursor phosphonium salts in aqueous base. Here, we report the flow and on-water synthesis of previously reported and novel CAPA analogues. We also report the unique and unexpected cytotoxicity of CAPA against BE(2)-C neuroblastoma cells and describe the cancer cell cytotoxicity of these CAPA analogues against HeLa adenocarcinoma and BE(2)-C cells in comparison to the activity of CAPE and CAPA.
2. Results
Amides are more stable than esters to hydrolysis, and the reactivity of specific amides can be related to the “amidicity” of these compounds, a quantitative measure of the degree to which the amide group is stabilized through conjugation [7]. Amidity is determined computationally by comparison of the enthalpy of hydrogen addition to the amide carbonyl relative to that of model systems defined as 100% (N,N-dimethylacetamide) or 0% (azaadamantane-2-one) amidity. Based on this method, we determined that CAPA has 115.8% amidity, indicating that the amide carbonyl group of CAPA is more resistant to attack than N,N-dimethylacetamide, which is reflective of a highly stable amide moiety.
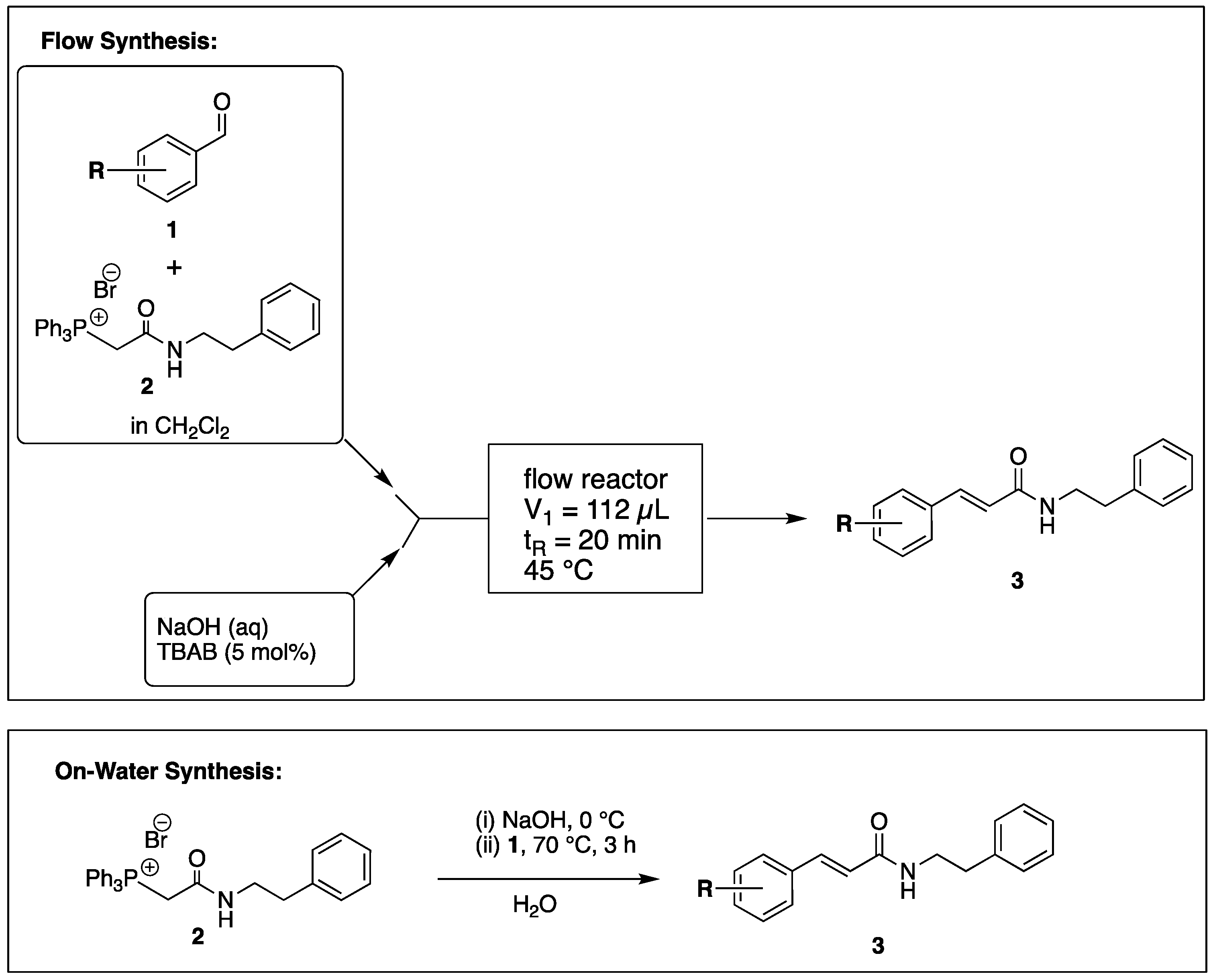
The flow reactor design for the preparation of CAPA analogues is shown in Scheme 1. A glass microreactor with two inlets and channel dimensions of 700 µm width × 500 µm depth containing a mixing region and overall effective reaction volume of 112 µL (Figure S1) was heated to 45 °C. One inlet was connected to a syringe containing a solution of the substituted benzaldehyde (1) and (2-oxo-2-(phenethylamino)ethyl)triphenylphosphonium bromide (2) in CH2Cl2. The other inlet was connected to a syringe containing 160 mM NaOH in water containing 5 mol% of tetrabutylammonium bromide (TBAB). The flow from both syringes was equal and set to provide a residence time of 45 min. The outlet was plumbed to a collection flask, and after the run was complete, the product was isolated after liquid/liquid separation. Purification was carried out by flash chromatography.
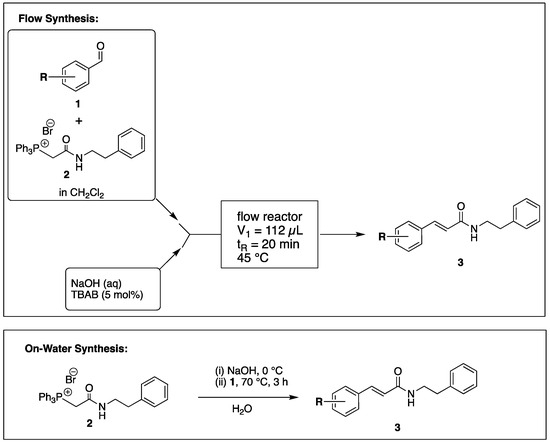
Scheme 1.
Procedure for Flow and On-Water Synthesis of CAPA Analogues.
Wittig reactions of benzaldehydes (1) and phosphonium bromide (2) on-water were carried out in water (0.8 M with respect to both 1 and 2) with the addition of NaOH (4 equiv.) and heating at 70 °C for 3 h, followed by extraction of products into CH2Cl2 and purification by flash chromatography.
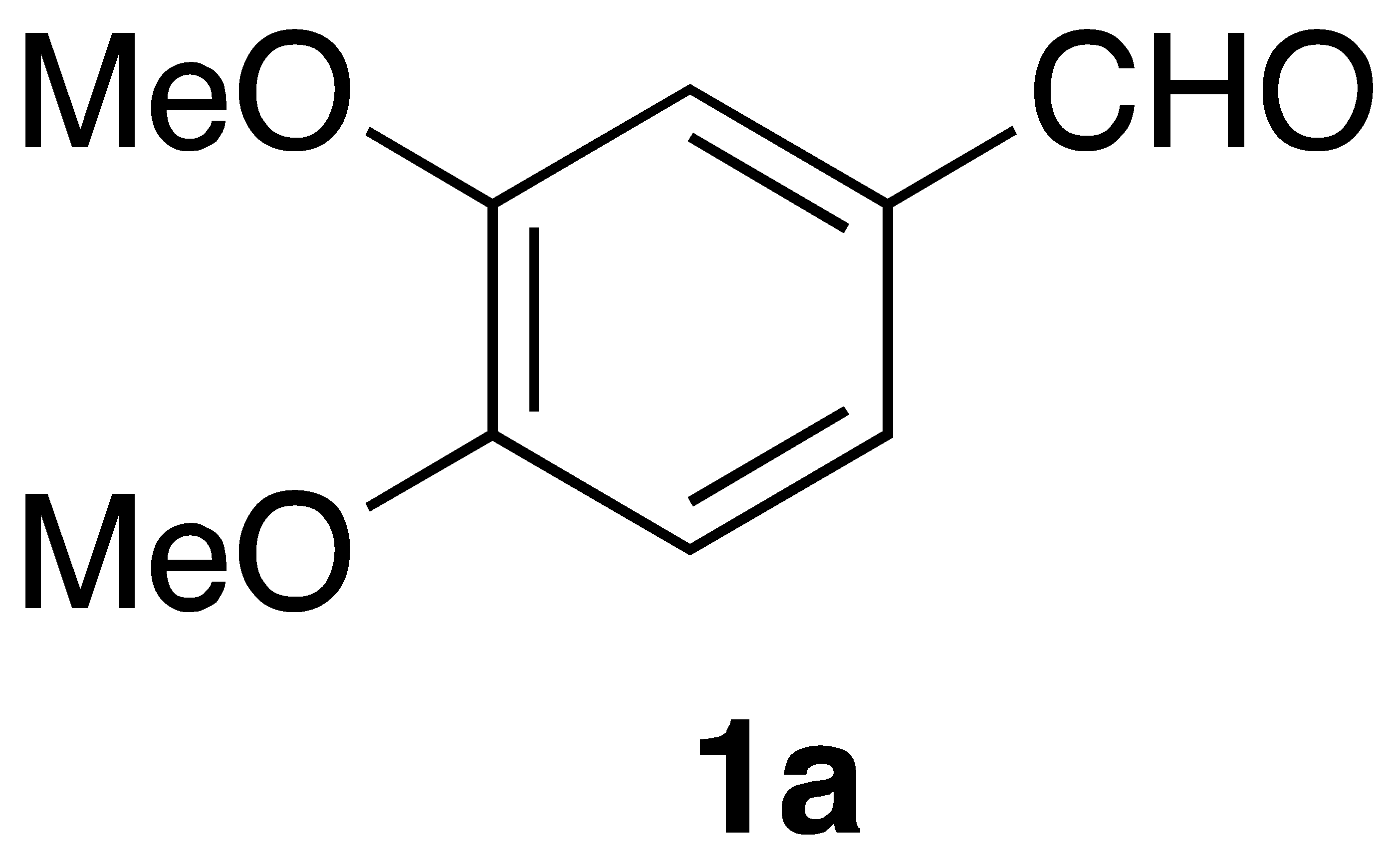
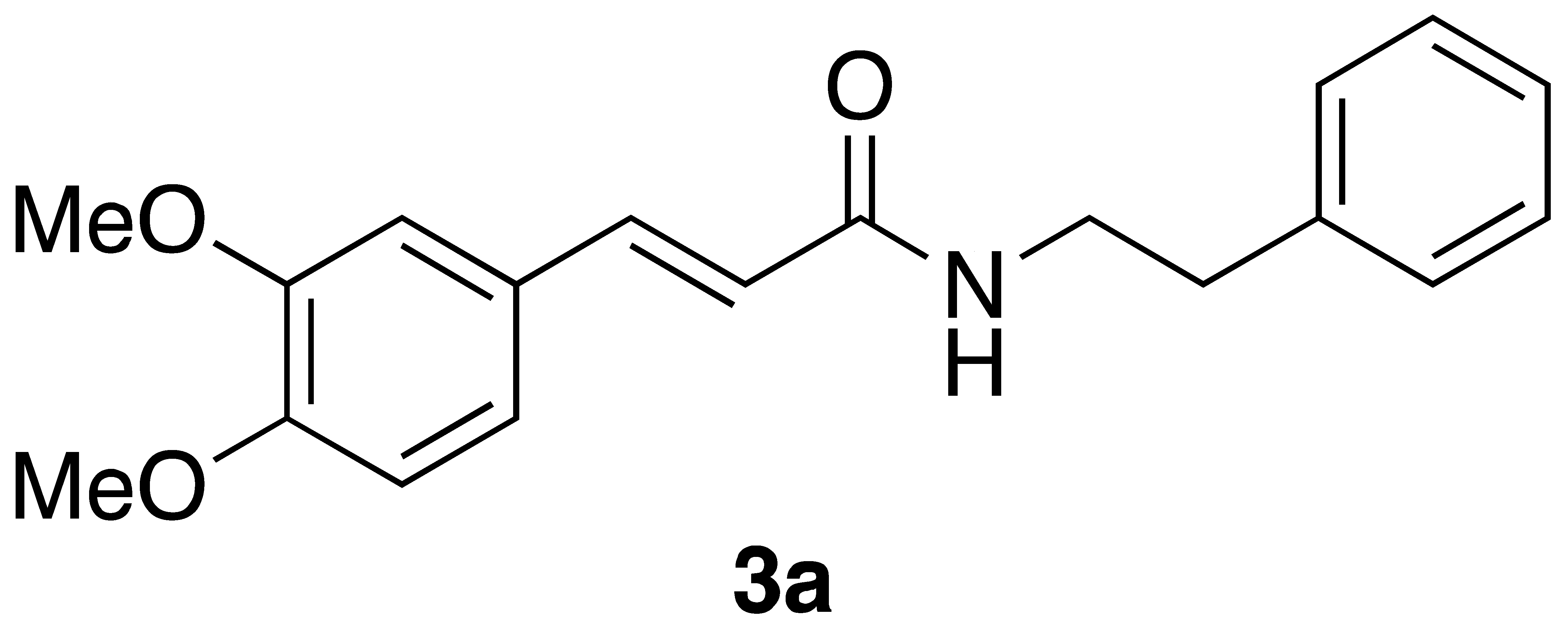
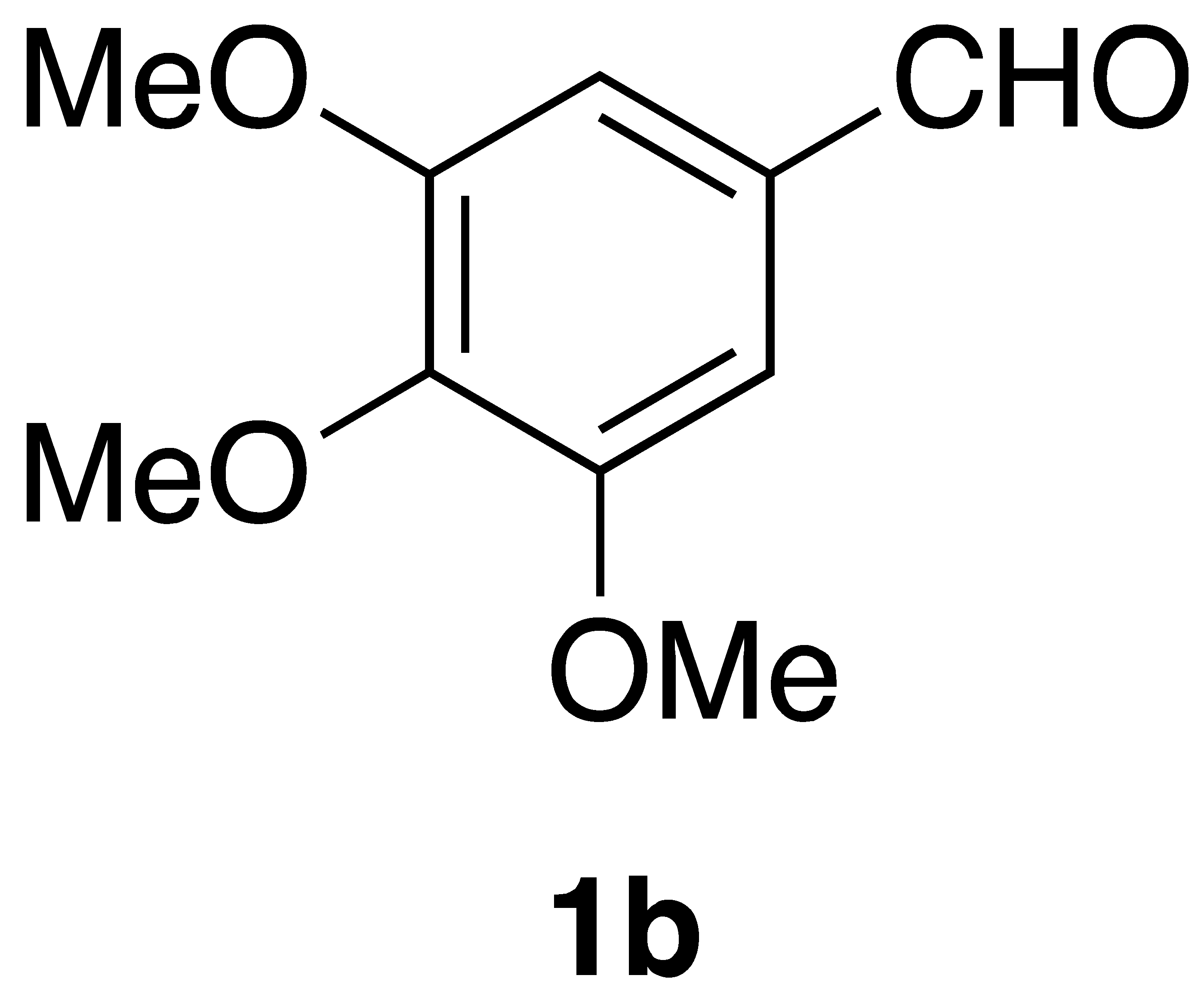
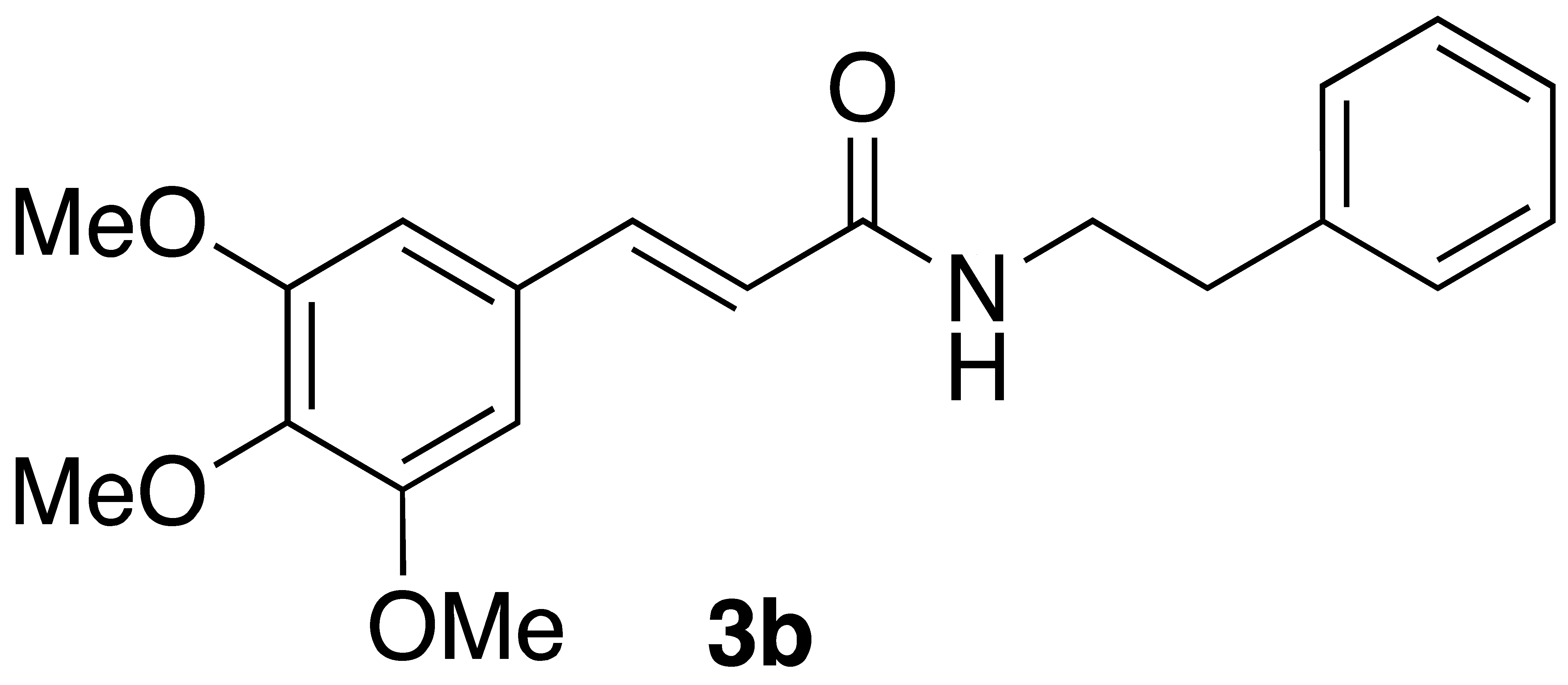
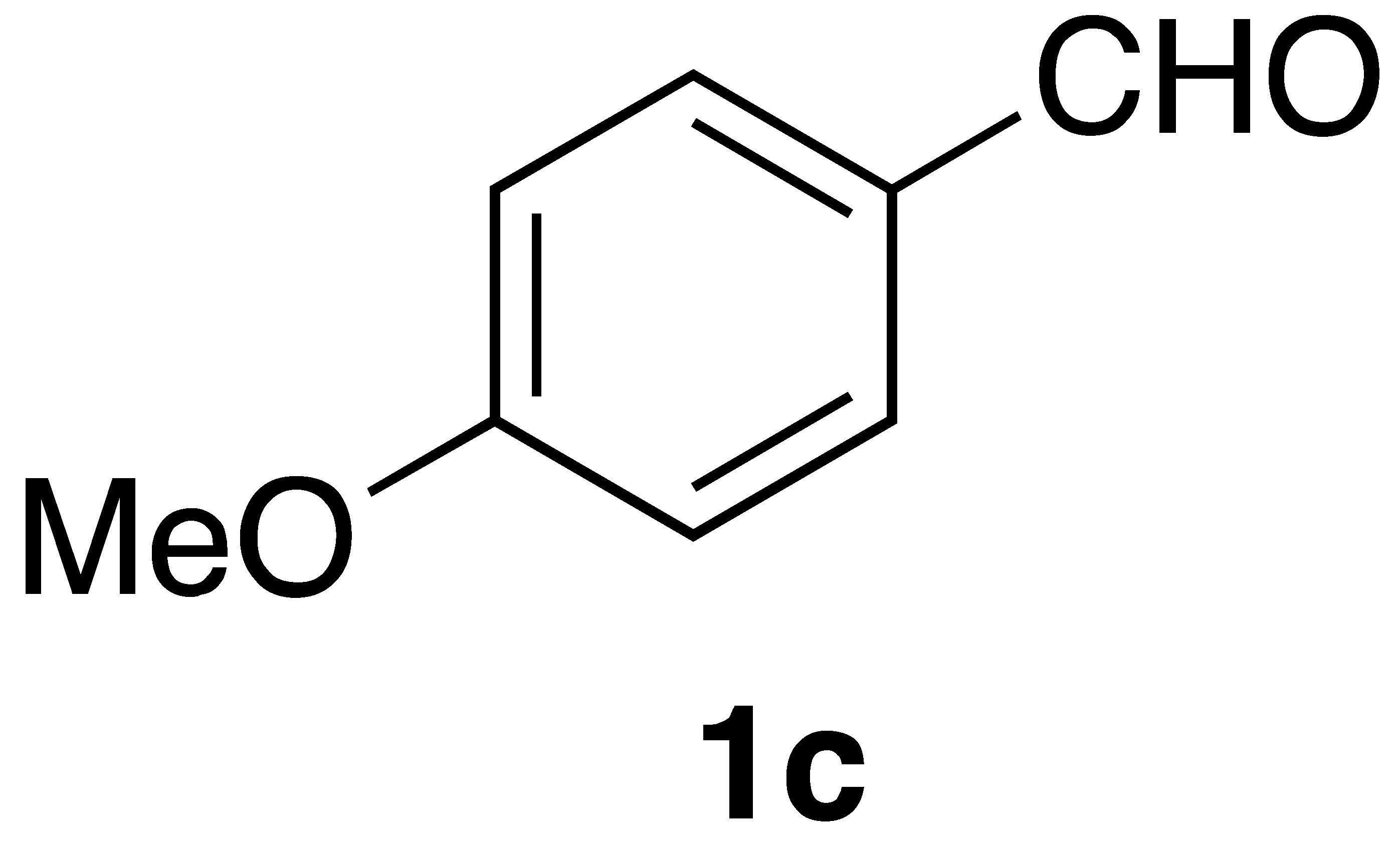
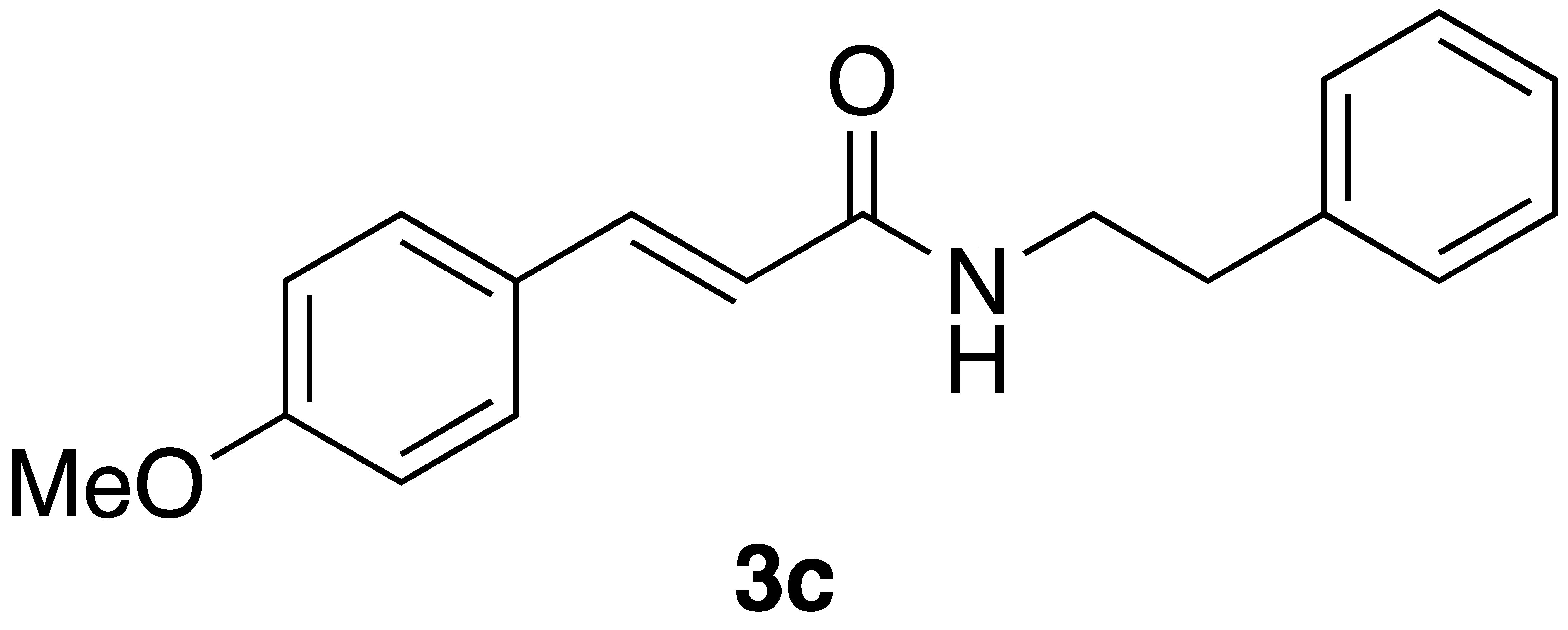
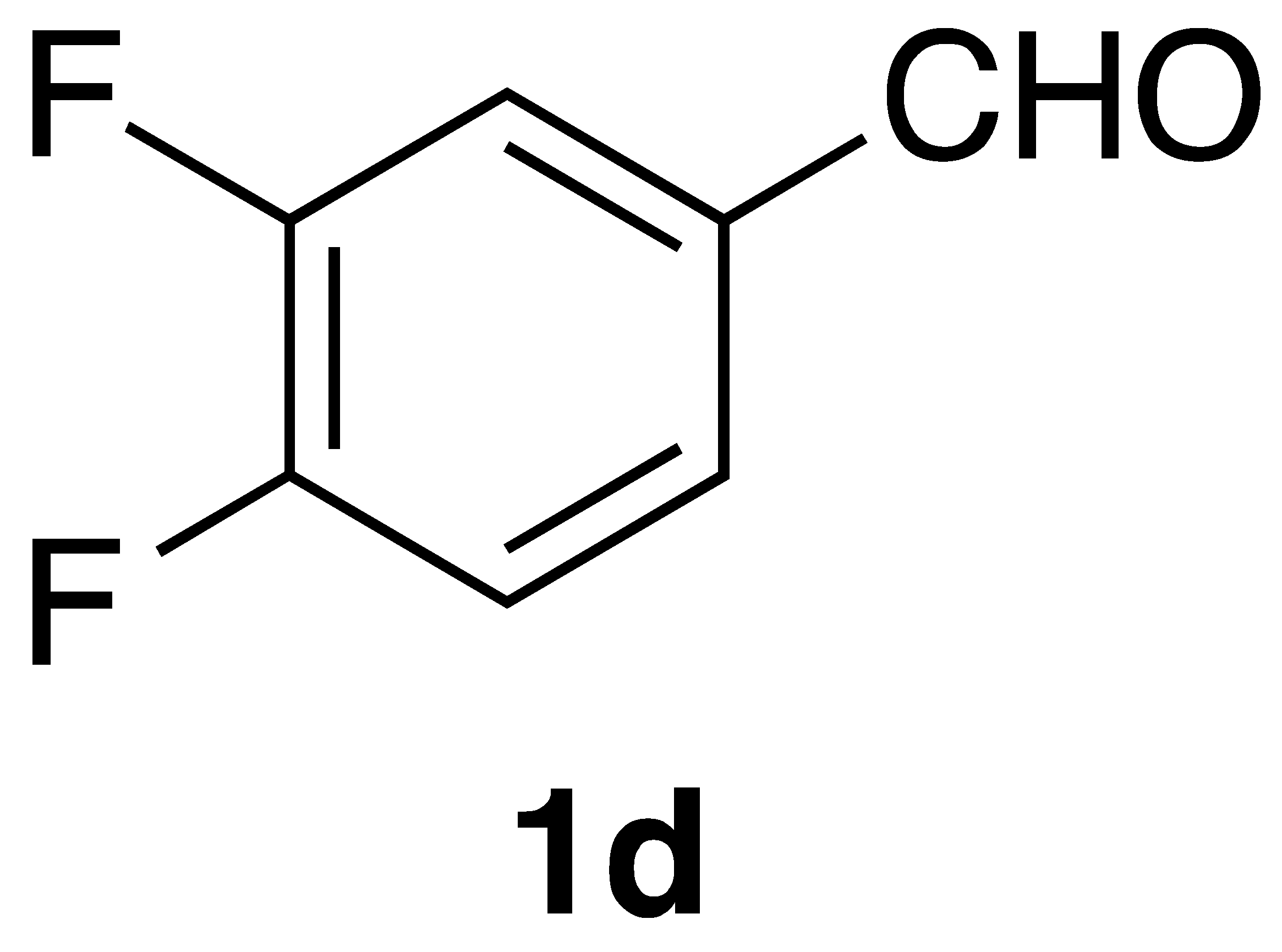
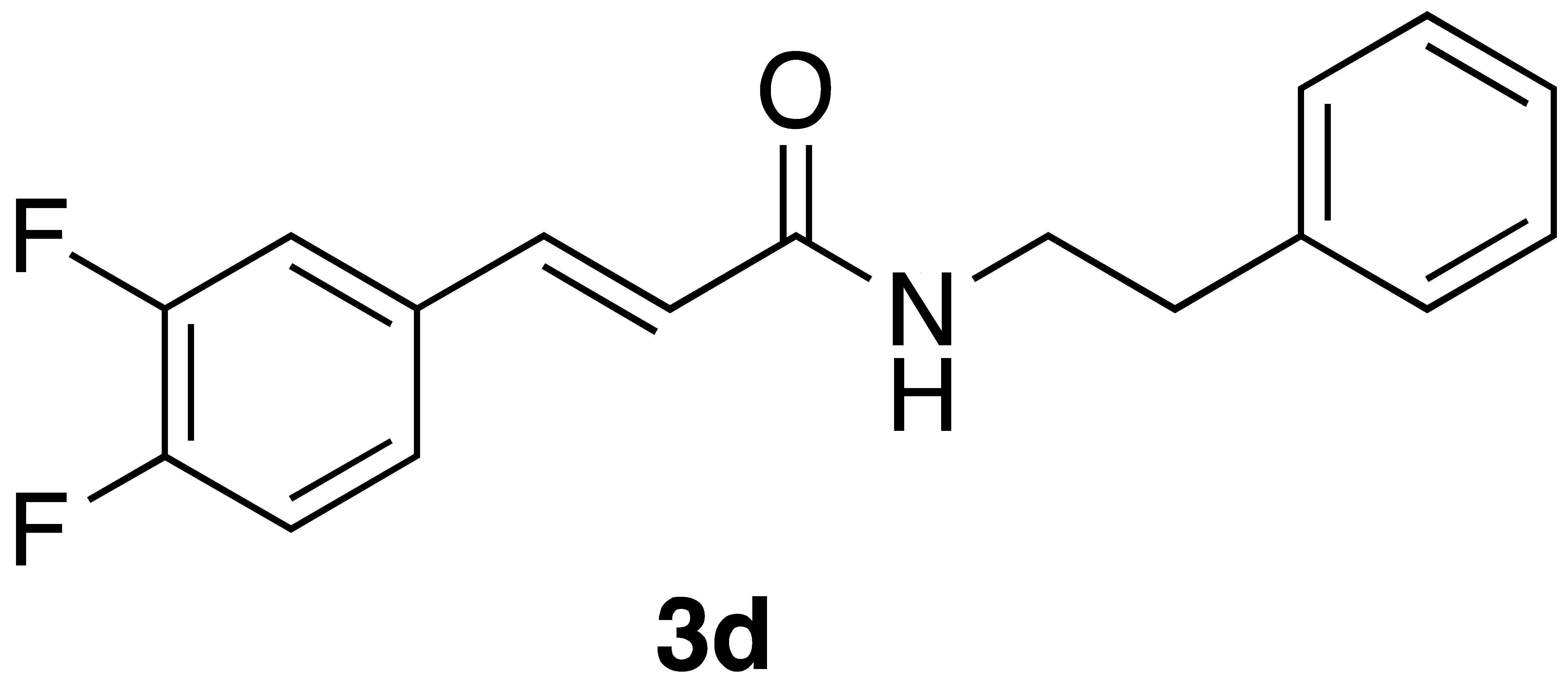
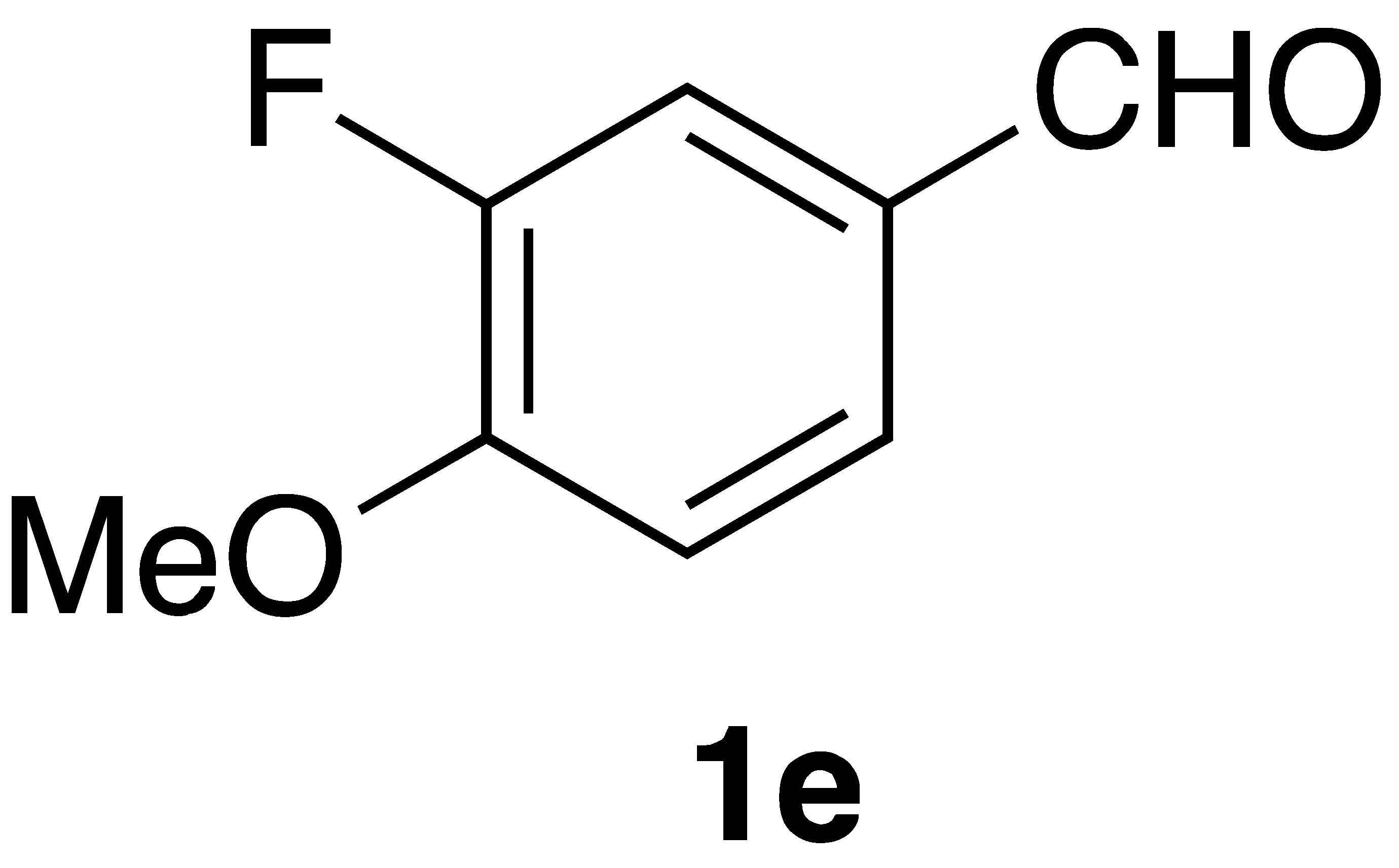
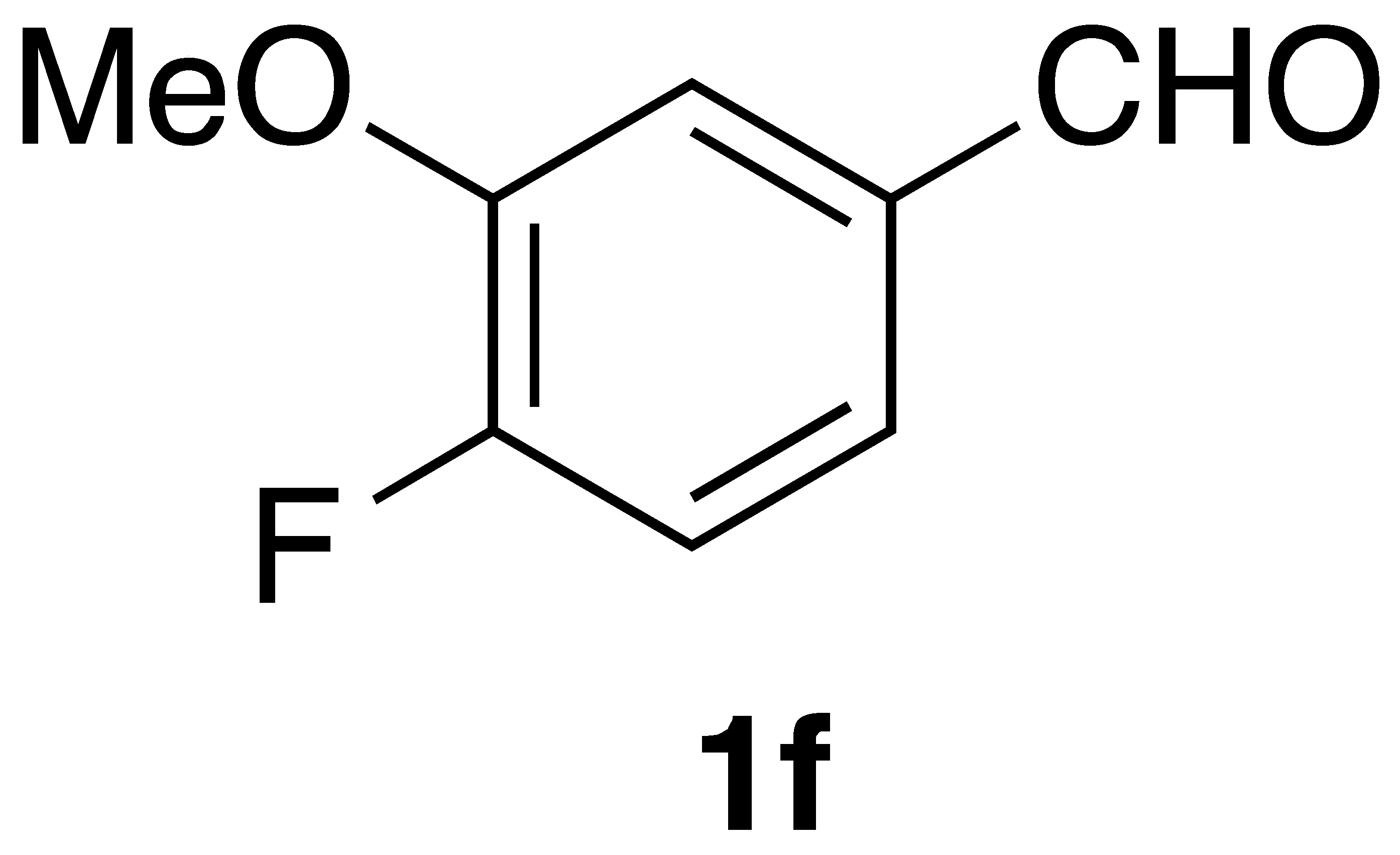
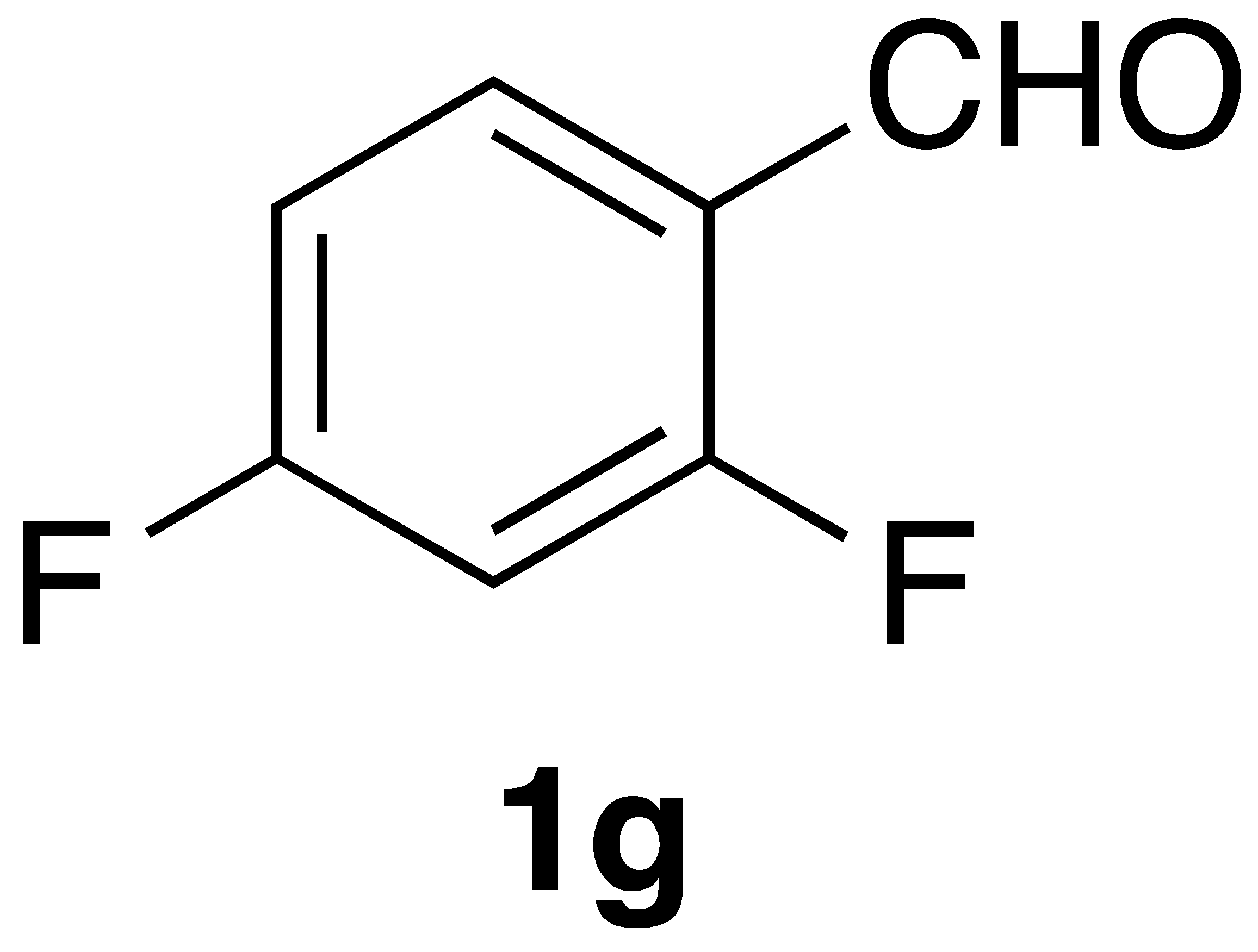
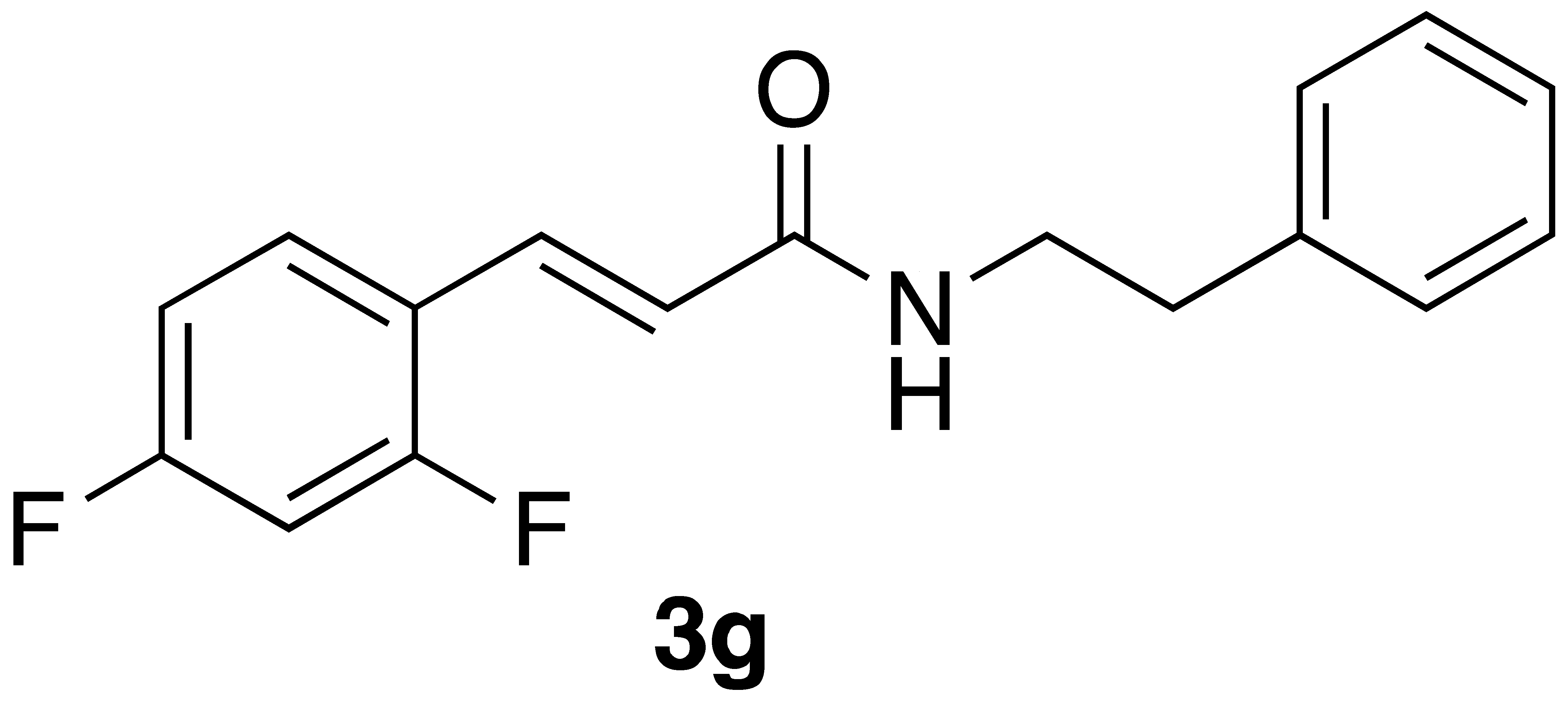
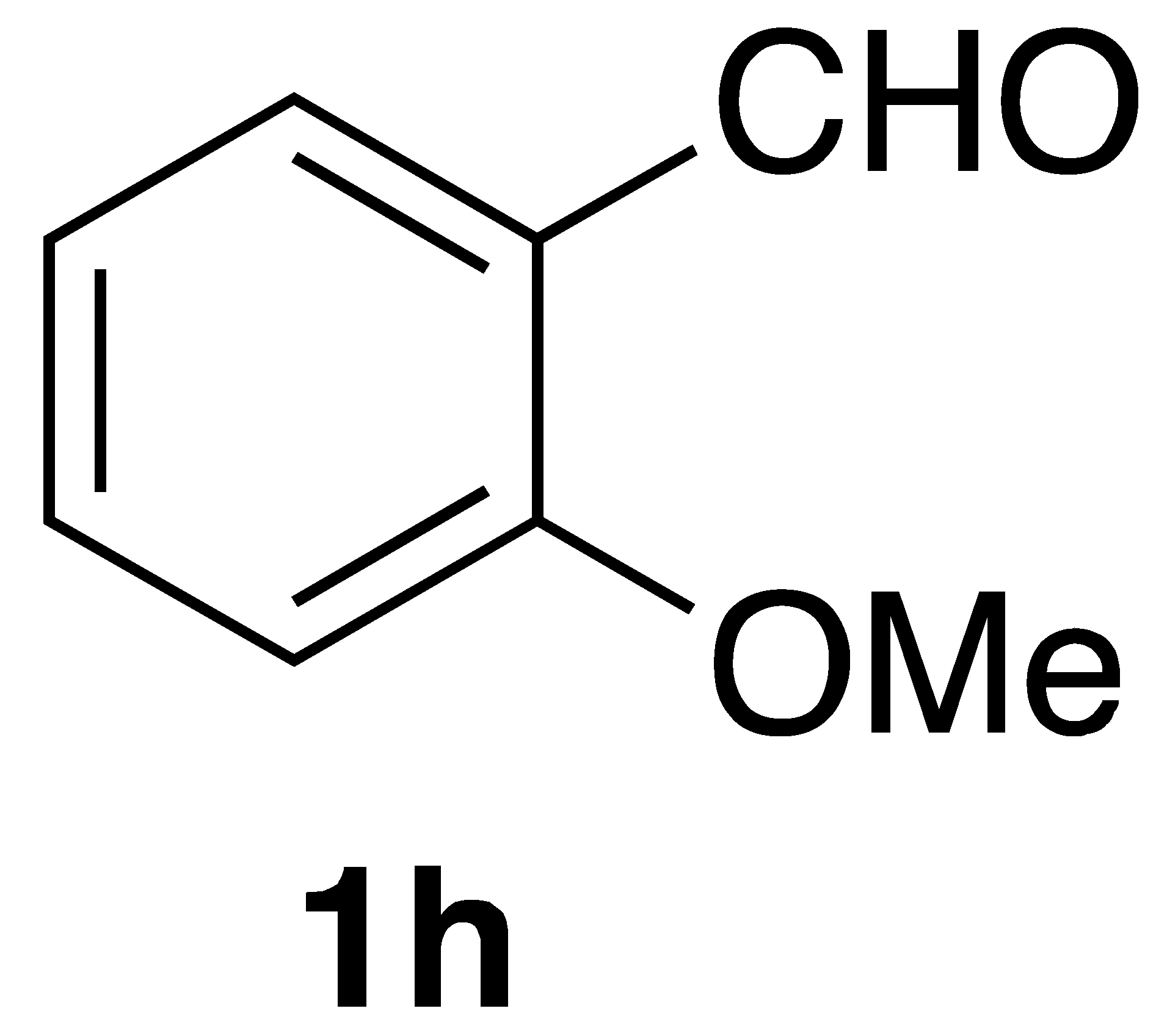
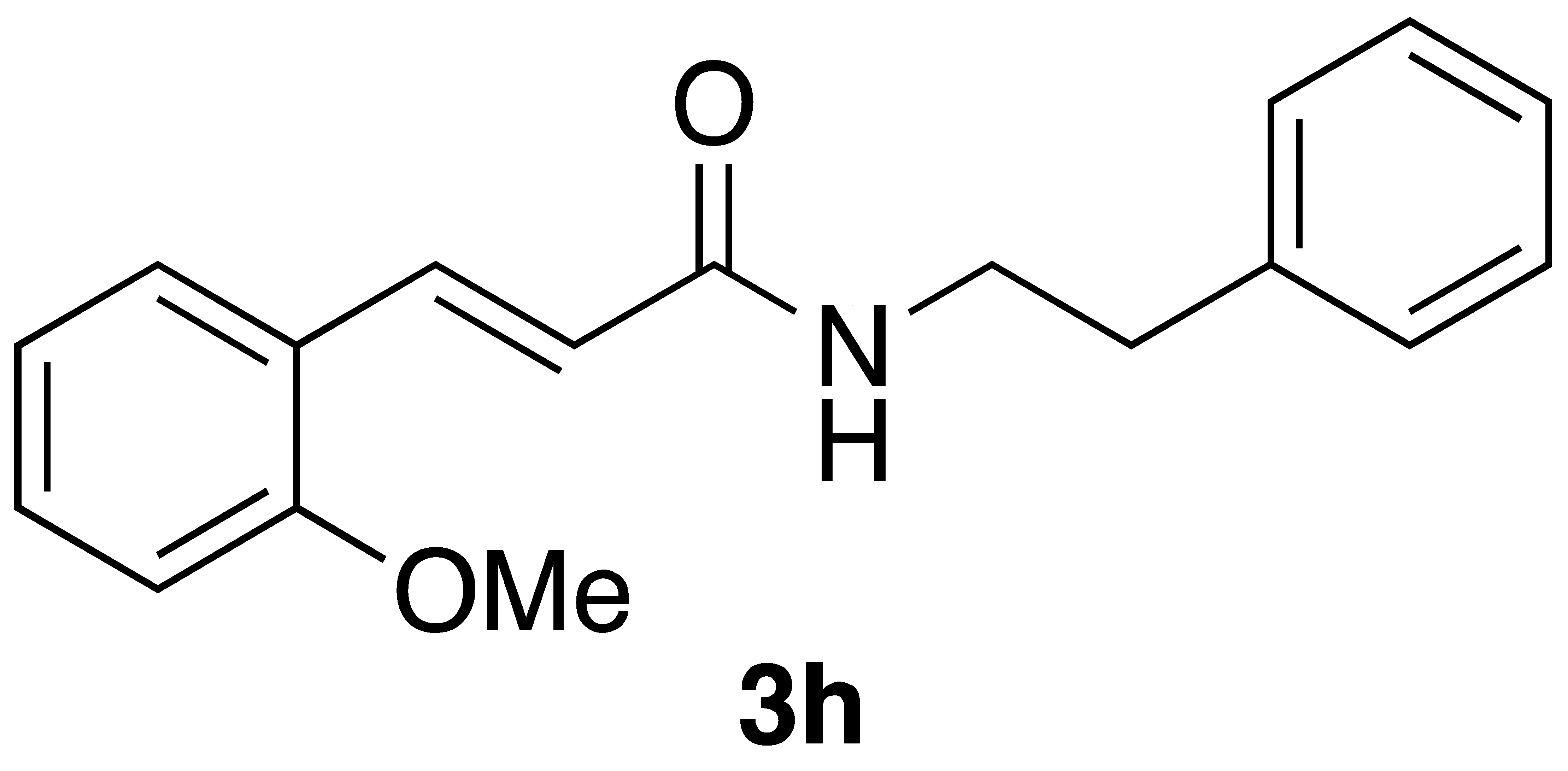
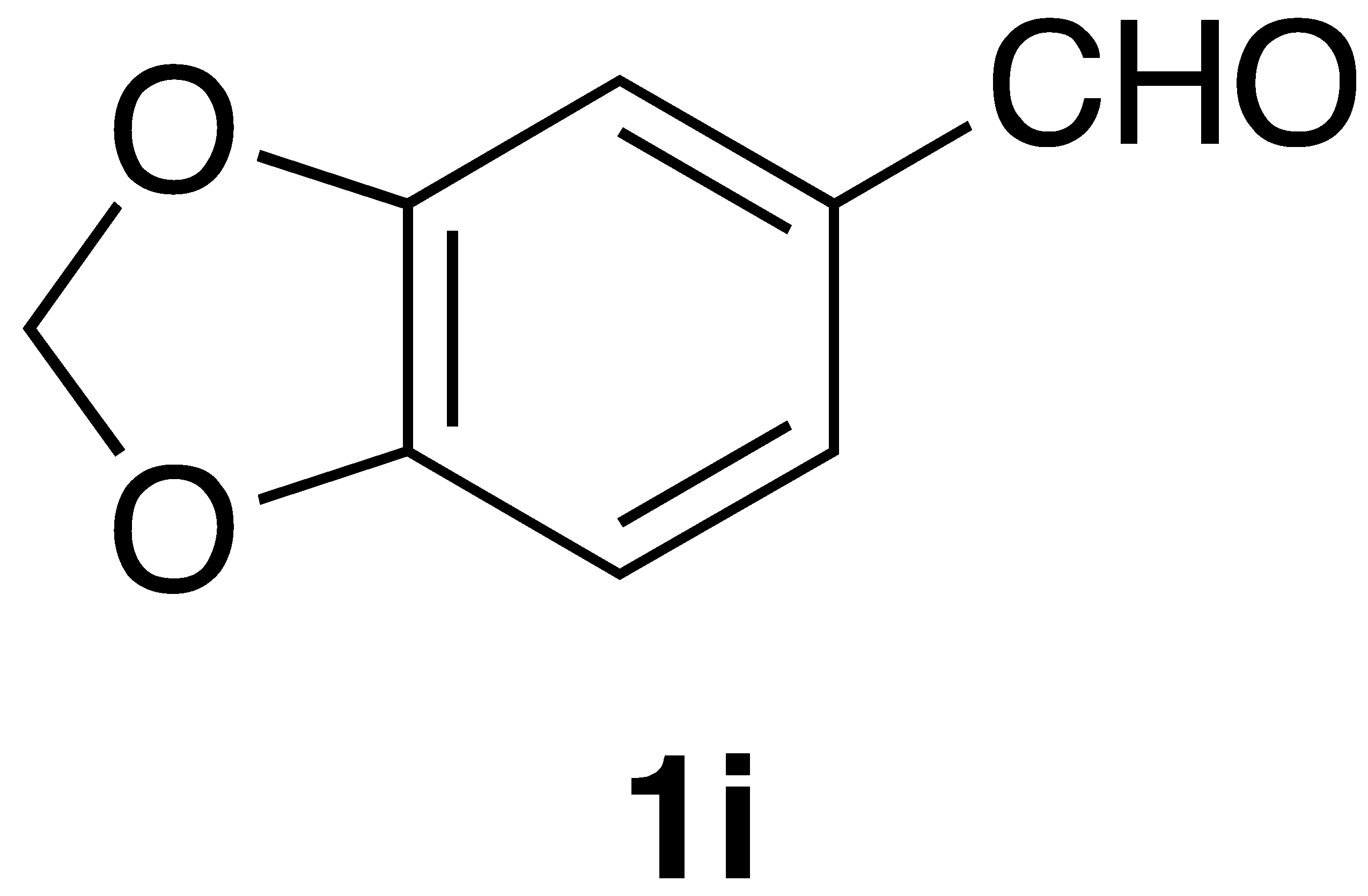
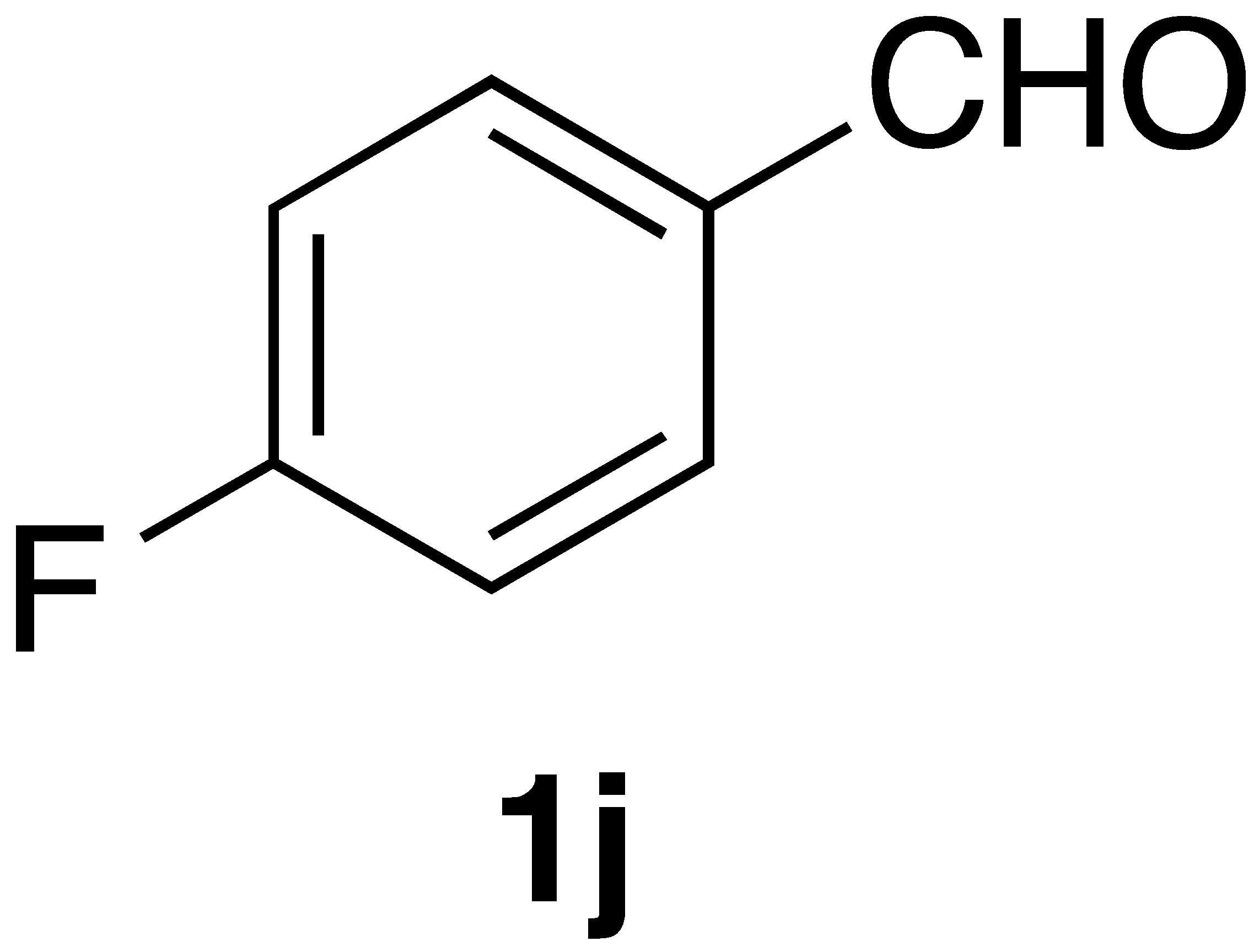
By employing different substituted benzaldehyde inputs (1a–j), ten different CAPA analogs (3a–j) were prepared, as shown in Table 1. The yields for the flow reactions were generally low, as most reactions failed to go to completion. Optimization efforts to improve the yields included increasing the equivalents of the phosphonium bromide reagent 2, increasing the temperature, and increasing the residence time. Control reactions carried out in the absence of phase-transfer catalyst TBAB did not afford product. The yields reported for flow reactions represent the best conditions found and described in Table 1, which were limited by the temperature that could be attained and the maximum residence time attainable in the flow reactor. Reactions carried out on-water generally proceeded with higher, albeit moderate yields. Notably, both methods afforded >90% diastereomeric purity of the desired (E)-isomers, which is superior to some other methods plagued by the formation of mixtures of difficult-to-separate (E)- and (Z)-isomers [8].

Table 1.
Synthesis of CAPA Analogues.
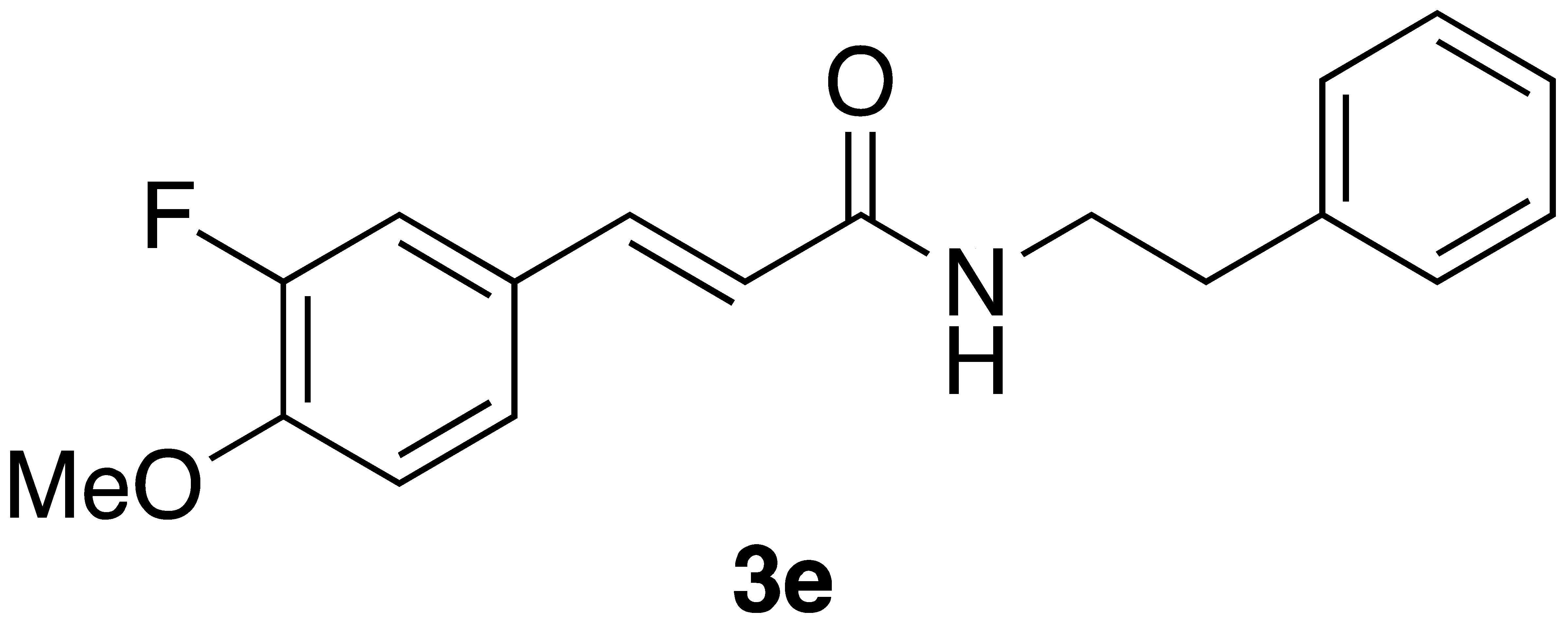
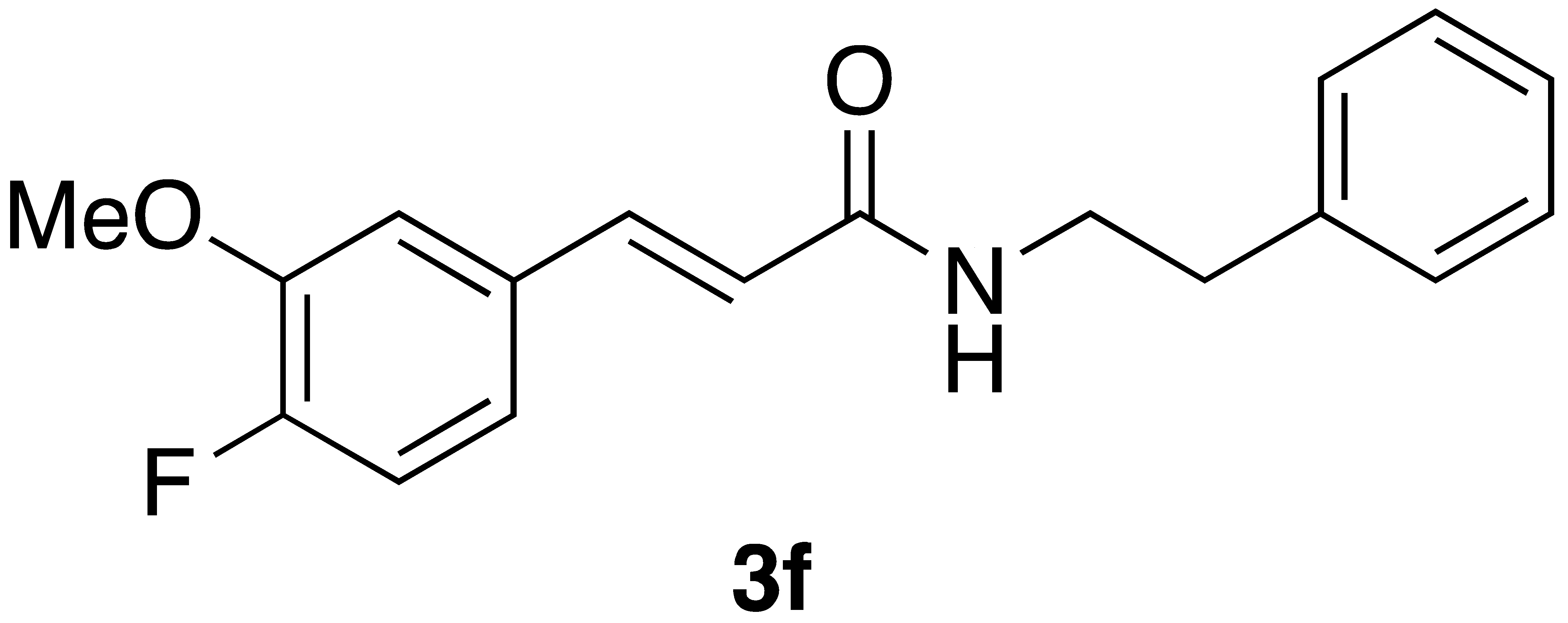
With the CAPA analogues 3a–j in-hand, we set out to determine if any of these display cytotoxicity against cancer cells that is superior to that of CAPA and perhaps even similar to that of CAPE itself. For these studies, we selected HeLa adenocarcinoma cells for our initial cytotoxicity screen, as the activity of CAPE in this cell type is well established [25,26]. Cytotoxicity was determined using the MTT assay [27]. As shown in Table 2, CAPE displays µM activity against HeLa cells, in accord with the literature data [25,26]. In contrast, CAPA has very low activity that is statistically different from that of CAPE (p < 0.05). While the CAPA analogues 3a–e and i–j were inactive against HeLa cells, analogue 3g displayed similar activity to CAPA, and analogue 3f displayed activity that is superior to CAPA and close to that of CAPE. Analogue 3h showed very low activity against HeLa cells.
Table 2.
Cytotoxicity of CAPE, CAPA, and CAPA Analogues.
We next examined the activity of CAPE, CAPA, and selected CAPA analogues against the neuroblastoma cell line BE(2)-C. Interestingly, while CAPE was more active against BE(2)-C than CAPA (p = 0.07), CAPA displayed markedly better activity against BE(2)-C than HeLa cells (p < 0.05) (Table 2). CAPA analogues 3a, 3d, 3i, and 3j, which were inactive against HeLa cells, were also inactive against the neuroblastoma cell line. CAPA analogues 3f and 3g displayed activity against both HeLa and BE(2)-C cells. While analogue 3f was less active against BE(2)-C cells compared to HeLa cells, analogues 3g and 3h were similar to CAPA in being more active against the neuroblastoma cell line (see Table S1 for statistical analysis).
3. Discussion
CAPA has been shown to have no cytotoxicity against LNCaP human androgen-dependent prostate cancer cells [15], U-937 human lymphoma cells [28], or KMM-1 human myeloma cells [14]. Here, we show that CAPA is only weakly active against HeLa human adenocarcinoma cells (IC50 > 100 µM), as would be expected based upon the literature reports in other human cancer cell lines. However, we found that CAPA is active against human neuroblastoma BE(2)-C cells, with an IC50 of 21 µM. Given that CAPA is non-cytotoxic against normal human cells [8,29], the finding of anti-neuroblastoma activity for CAPA is significant, as selectivity for cancer cells is an important requirement for anticancer therapy.
A number of previously unreported CAPA analogs (3d–h) as well as the previously reported compounds 3a–c and 3i,j were prepared using Wittig flow and on-water chemistry. In general, the on-water method was found superior in terms of yields and has the advantage that no special equipment is required; however, both methods are operationally simple and provide a high degree of control of the stereochemistry of the product caffeic amides. CAPE, CAPA, and these analogues were assayed for cancer cell cytotoxicity against HeLa cells. One new compound, 3f, with 4-fluoro-3-methoxy substituents, displayed cytotoxicity against HeLa cells that is superior to CAPA and about two times that of CAPE. This is remarkable in that the regioisomeric 3-fluoro-4-methoxy analogue 3e was inactive against HeLa cells. While the 4-fluoro analogue 3j and 3,4-difluoro analogue 3d were also inactive against HeLa cells, the 2,4-difluoro compound 3g displayed weak activity against HeLa cells, similar to CAPA. The 2-methoxy analogue 3h showed only very weak activity against HeLa cells. Taken together, these results point to a tight geometric requirement for substituents about the ring corresponding to the catechol ring of CAPA for HeLa cell activity, with preference for 4-fluoro substituent in combination with substituents at the 2- or 3-positions.
CAPE, CAPA, and these analogues were also tested against the neuroblastoma cell line BE(2)-C. Surprisingly CAPA displayed activity in this cell line, although CAPE was more active (p < 0.05). This indicates that neuroblastoma may be a good target for CAPA and its analogues. CAPA analogues 3f–h were also active against BE(2)-C cells, although none of these had activity superior to that of CAPA. Interestingly, while 3f was the most active of the analogues examined here against HeLa cells, 3g and 3f had similar activity against BE(2)-C cells. This may indicate a similar but distinct structure–activity relationship for CAPA analogues in these two cancer cell lines.
4. Materials and Methods
Unless otherwise noted, all materials were obtained from commercial suppliers and were used without further purification: CAPE (>98%) from CaymanChemical Co., Ann Arbor, MI, USA; CAPA (98%) from TargetMol, Boston, MA, USA; 4-fluorobenzaldehyde (98%), 3,4-dimethoxybenzaldehyde (99%) from Acros, Fair Lawn, NJ, USA; 3,4-difluorobenzaldehyde (95%) and 4-fluoro-3-methoxybenzaldehyde (97%) from Matrix Scientific, Columbia, SC, USA; 3-fluoro-4-methoxybenzaldehyde (99%), piperonal (99%), p-anisaldehyde (99%), and 3,4,5-trimethoxybenzaldehyde (98%) from Millipore Sigma, St. Louis Missouri, MI, USA; and 2,4-difluorobenzaldehyde (98%) from TCI, Houston, TX, USA. (2-Oxo-2-(phenethylamino)ethyl)triphenylphosphonium bromide (2) was prepared by the literature method [8]. Flash chromatography was performed on a Teledyne CombiFlash with RediSep Rf silica gel (230–400 mesh) using the mobile phase indicated. Melting points (open capillary) were determined on a Electrothermal series IA 9000 digital melting point apparatus and are uncorrected. Unless otherwise noted, 1H and 13C NMR spectra were obtained on a Bruker Advance Spectrometers operating at 400 MHz (100 MHz for 13C) or 500 MHz (126 MHz for 13C) and were determined in CDCl3. Chemical shifts are reported in ppm using solvent as internal standard (7.26 ppm for 1H and 77.0 ppm for 13C in CDCl3). Low-resolution mass spectra were obtained on a Advion ExpressionL compact mass spectrometer with atmospheric pressure chemical ionization (APCI) or electrospray ionization (ESI). High-resolution mass spectra were obtained on a Waters SynaptXS spectrometer with ESI or and Agilent 6530 Q-TOF LC/MS with chemical ionization (CI). Copies of all NMR and MS spectra are available in the Supporting Information. Flow chemistry was carried out on a Future Chemistry FlowStart Evo system with a M-111 microreactor.
General Procedure 1—Flow Wittig Reaction: (E)-phenethyl 3-(3,4,5-trimethoxyphenyl)acrylamide (3b): A syringe was charged with a solution of 15.7 mg (0.08 mmol) of 3,4,5-trimethoxybenzaldehyde and 73.59 mg (0.16 mmol) of (2-oxo-2-(phenethylamino)ethyl)triphenylphosphonium bromide (2) in 1 mL of CH2Cl2. A second syringe was charged with a solution of 1.29 mg (5 mol%) TBAB and 6.4 mg (0.16 mmol) NaOH in 1 mL in H2O. The flow reactor was set to run with equal flow from both syringes and a residence time of 20 min at 45 °C. The resultant mixture was then transferred to a 30 mL sep. funnel, and the aqueous layer was washed with CH2Cl2. (3 × 3 mL), and then, the organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressured to give a slightly yellow oil that was subjected to flash chromatography (55%/45% EtOAc:Hex with 0.1% triethylamine) to afford 14.8 mg (52% yield) of compound 3b as a white solid after chromatography: 1H NMR (400 MHz, CDCl3) δ: 7.52 (1H, d, J = 15.5 Hz, CH=CH), 7.35–7.31 (2H, m, ArH), 7.23 (3H, m, ArH), 6.70 (2H, s, ArH), 6.23 (1H, d, J = 15.5 Hz, CH=CH), 5.66–5.64 (1H, m, NH), 3.86 (6H, s, -OCH3), 3.86 (3H, s, -OCH3), 3.67 (1H, q, J = 6.4 Hz, CH2CH2), 2.89 (2H, t, J = 6.8 Hz, CH2CH2); 13C NMR (100 MHz, CDCl3) δ: 165.8, 153.4, 141.0, 139,6, 138,.8, 130.3, 128.8, 128,7, 126.5, 119.9, 104.9, 60.9, 56.1, 40.7, 35.6; APCI MS m/z: 342 (MH+). Matches lit [30].
(E)-phenethyl 3-(3,4-dimethoxyphenyl)acrylamide (3a): Following the general procedure 1 starting with 3,4-dimethoxybenzaldehyde afforded 27 mg (24% yield) of a white solid after flash chromatography: mp = 131–132 °C (lit. 126–128 °C); 1H NMR (400 MHz, CDCl3) δ: 7.55 (1H, d, J = 15.5 Hz, CH=CH), 7.31–7.27 (2H, m, ArH), 7.23–7.19 (3H, m, ArH), 7.04 (1H, dd, J = 8.3, 1.8 Hz, ArH), 6.98 (1H, d, J = 1.8 Hz, ArH), 6.80 (1H, d, J = 8.3 Hz, ArH), 6.25 (1H, d, J = 15.5 Hz, CH=CH), 6.01 (1H, s, -OCH3), 3.86 (3H, s, -OCH3), 3.84 (3H, s, -OCH3), 3.64 (2H, q, J = 5.9 Hz, CH2CH2), 2.87 (2H, t, J = 6.9 Hz, CH2CH2); 13C NMR (100 MHz, CDCl3) δ: 166.2, 150.5, 149.0, 140.7, 138.8, 128.7, 128.6, 127.7, 126.3, 121.8, 118.4, 111.0, 109.6, 55.8, 55.7, 40.7. 35.6; APCI MS m/z: 312 (MH+). Matches lit [30].
(E)-phenethyl 3-(4-methoxyphenyl)acrylamide (3c): Following the general procedure 1 starting with 4-methoxybenzaldehyde afforded 13.6 mg (12% yield) of a white solid after flash chromatography (60% EtOAc/hex): 1H NMR (400 MHz, CDCl3) δ: 7.57 (1H, d, J = 15.6 Hz, CH=CH), 7.43–7.42 (2H, m, ArH), 7.34–7.31 (2H, m, ArH), 7.24–7.22 (3H, m, ArH), 6.88–6.87 (2H, m, ArH), 6.19 (1H, d, J = 15.6 Hz, CH2CH2), 5.59 (1H, s(br), NH), 3.82 (3H, s, -OCH3), 3.66 (2H, q, J = 6.4 Hz, CH2CH2), 2.89 (2H, t, J = 6.9 Hz, CH2CH2). 13C NMR (100 MHz) δ: 166.2, 160.9, 140.7, 138.9, 129.3, 128.8, 128.7, 127.5, 126.5, 118.2, 114.3, 55.3, 40.7, 35.7; APCI MS m/z: 282 (MH+). Matches lit [30].
(E)-phenethyl 3-(3,4-difluorophenyl)acrylamide (3d): Following the general procedure 1 starting with 3,4-difluorobenzaldehyde afforded 13.2 mg (14% yield) of a white solid after flash chromatography (15–40% EtOAc/hex): mp 143–145 °C; IR 3303, 11,656, 1622, 1550, 1518, 1275, 1197, 966 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.51 (1H, d, J = 15.5 Hz, CH=CH), 7.34–7.29 (2H, m, ArH), 7.28–7.11 (6H, m, ArH), 6.25 (1H, d, J = 15.5 Hz, CH=CH), 5.78 (1H, s(br), NH), 3.66 (2H, q, J = 6.4 Hz, CH2CH2), 2.89 (2H, t, J = 6.4 Hz, CH2CH2); 13C NMR (100 MHz, CDCl3) δ: 165.2, 150.9 (dd, JC-F = 252.4, 13.1 Hz), 150.5 (dd, JC-F = 249.1, 12.9 Hz), 138.8, 138.7, 132.0 (dd, JC-F = 6.1, 4.1 Hz), 128.7 (d, JC-F = 4.7 Hz), 126.6, 124.5 (dd, JC-F = 6.4, 3.4 Hz), 121.7, 121.6, 117.6 (d, JC-F = 17.7 Hz), 115.8 (d, JC-F = 17.6 Hz), 40.8, 35.6; CI MS m/z: 288.1 (MH+); HR CIMS calcd. for C17H16NOF2 288.1200 (MH+), found 288.1200.
(E)-phenethyl 3-(3-fluoro-4-methoxyphenyl)acrylamide (3e): Following the general procedure 1 starting with 3-fluoro-4-methoxybenzaldehyde afforded 44 mg (37% yield) of a white solid after flash chromatography (40% EtOAc/hex): mp 152–152.5 °C; IR 3297, 3078, 3029, 29,665, 1657, 1613, 1577, 1547, 1519, 1270, 1119 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.51 (1H, d, J = 15.5 Hz, CH=CH), 7.32–7.28 (1H, m, ArH), 7.24–7.14 (5H, m, ArH), 6.88 (1H, t, J = 8.5 Hz, ArH), 6.23 (1H, d, J = 15.5 Hz, CH=CH), 6.03 (1H, s(br), NH), 3.88 (3H, s, -OCH3), 3.64 (2H, q, J = 6.3 Hz, CH2CH2), 2.88 (2H, t, J = 7.0 Hz, CH2CH2). 13C NMR (100 MHz) δ: 165.8, 152.2 (d, JC-F = 246.7 Hz), 148.8 (d, JC-F = 11.0 Hz), 139.5 (d, JC-F = 2.4 Hz), 138.7, 128.7, 128.5, 128.1 (d, JC-F 6.6 Hz), 126.5, 125.0 (d, JC-F = 3.1 Hz), 119.6, 114.1 (d, JC-F = 18.9 Hz), 113.1 (d, JC-F = 2.1 Hz), 56.1, 40.8, 35.6. CIMS m/z 300 (MH+); HR CIMS calcd. for C18H20FNO2 (MH+) 300.1400, found 300.1398.
(E)-phenethyl 3-(4-fluoro-3-methoxyphenyl) acrylamide (3f). Following the general procedure 1 starting with 4-fluoro-3-methoxybenzaldehyde afforded 40 mg (33% yield) of a white needles after flash chromatography (40% EtOAc/hex). Follow the general procedure 2 starting with 62 mg of aldehyde afforded 59 mg (49% yield) after flash chromatography: mp = 119–120 °C; IR (ATR, neat): 3297, 3071, 3029, 2946, 1739, 1654, 1621, 1603, 1546, 1519, 1467, 1410, 1374, 1319, 1299, 1283, 1261, 1209, 1191, 1162, 1118, 1045, 1029 cm−1; 1H NMR (400 MHz, CD3COCD3) δ: 7.49 (1H, d, J = 15.6 Hz, CH=CH), 7.39 (1H, s(br), NH), 7.33–7.30 (1H, m, ArH), 7.29–7.23 (5H, m, ArH), 7.21–7.18 (2H, m, ArH), 6.63 (1H, dd, J = 15.6, 0.5 Hz, CH=CH), 3.91 (3H, s, -OCH3), 2.86 (2H, t, J = 7.4 Hz, CH2CH2); 13C NMR (100 MHz, CD3COCD3) δ: 165.9, 153.7 (d, JC-F = 248.0 Hz), 148.8 (d, JC-F = 11.0 Hz), 140.5, 139.2 133.2 (d, JC-F = 3.9 Hz), 129.2, 127.0, 122.9 (d, JC-F = 2.3 Hz), 121.4, 121.3, 116.9 (d, JC-F = 18.5 Hz), 113.3 (d, JC-F = 2.1 Hz), 56.4, 41.6, 36.5. CIMS m/z: 300 (MH+); HR CIMS calcd. for C18H20FNO2 (MH+) 300.1400, found 300.1402.
General Procedure 2—On-Water Wittig Reaction: (E)-3-(2,4-difluorophenyl)-N-phenethylacrylamide (3g): To a stirred solution of phosphonium salt (201 mg, 0.4 mmol) in 2 mL of water at 0 °C, sodium hydroxide (64 mg, 1.6 mmol) portion wise added and followed by addition of 2,4-difluorobenzaldehyde (56.8 mg, 0.4 mmol). The resulting mixture was stirred at 70 °C for 3 h. After completion of the reaction (monitored by TLC), the reaction mixture was diluted with water (10 mL) and the aqueous layer was extracted with CH2Cl2 (2 × 25 mL). The combined organic layers were washed with brine solution (20 mL), dried over sodium sulphate, and concentrated under reduced pressure. Purification by flash chromatography (40% EtOAc/hex) followed by recrystallisation from CH2Cl2 and hexane afforded 59 mg (52% yield) of compound 3g as a white solid: mp = 131–133 °C; IR 3393, 1670, 1616, 1550, 1521, 1472, 1167 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.66 (1H, d, J = 15.7 Hz, CH=CH), 7.47 (1H, q, J = 8.4 Hz, ArH), 7.38–7.31 (2H, m, ArH), 7.29–7.25 (3H, m, J = 6.7 Hz, ArH), 6.92–6.84 (2H, m, ArH), 6.42 (1H, d, J = 15.6 Hz, CH=CH), 5.68 (1H, s(br), NH), 3.69 (2H, q, J = 6.7 Hz, CH2CH2), 2.92 (2H, t, J = 6.9 Hz, CH2CH2); 13C NMR (126 MHz, CDCl3) δ: 165.8, 163.4 (dd, JC-F = 252.7, 12.4 Hz), 161.5 (dd, JC-F = 256.0, 12.1 Hz), 138.8, 133.0, 130.6 (dd, JC-F = 9.5, 4.5 Hz), 128.8, 128.7, 126.6, 123.2 (d, JC-F = 6.4 Hz), 119.4 (dd, JC-F = 11.2, 3.2 Hz), 111.9 (dd, JC-F = 21.7, 2.5 Hz), 104.6 (t, JC-F = 25.7 Hz), 40.9, 35.7; 19F NMR (470 MHz, CDCl3) δ −107.25, −109.59; APCI MS m/z: 288.1 (MH+); HR CIMS calcd. for C17H16F2NO (MH+) 288.1194, found 288.1201.
(E)-3-(2-methoxyphenyl)-N-phenethylacrylamide (3h): Following the general procedure 2 starting with 2-methoxy benzaldehyde (54 mg) afforded 46 mg (41% yield) of white needles after flash chromatography (EtOAc/hex): mp = 131–133 °C; IR 3248, 1670, 1600, 1568, 1548, 1472, 1167 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.61 (1H, d, J = 15.6 Hz, CH=CH), 7.40–7.32 (2H, m, ArH), 7.30 (1H, dd, J = 6.3, 4.7 Hz, ArH), 7.28–7.23 (3H, m, ArH), 7.10 (1H, d, J = 7.7 Hz, ArH), 7.05–6.99 (1H, m, ArH), 6.92 (1H, dd, J = 8.2, 1.9 Hz, ArH), 6.33 (1H, d, J = 15.6 Hz, CH=CH), 5.68 (1H, s(br), NH), 3.84 (3H, s, -OCH3), 3.69 (2H, q, J = 6.7 Hz, 2H), 2.92 (2H, t, J = 6.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 165.8, 159.8, 141.0, 138.9, 136.2, 129.8, 128.8, 128.7, 126.6, 120.9, 120.4, 115.4, 112.9, 55.3, 40.8, 35.7; APCI MS m/z: 282.1 (MH+); HR CIMS calcd. for C18H20NO2 (MH+) 282.1489, found 282.1505.
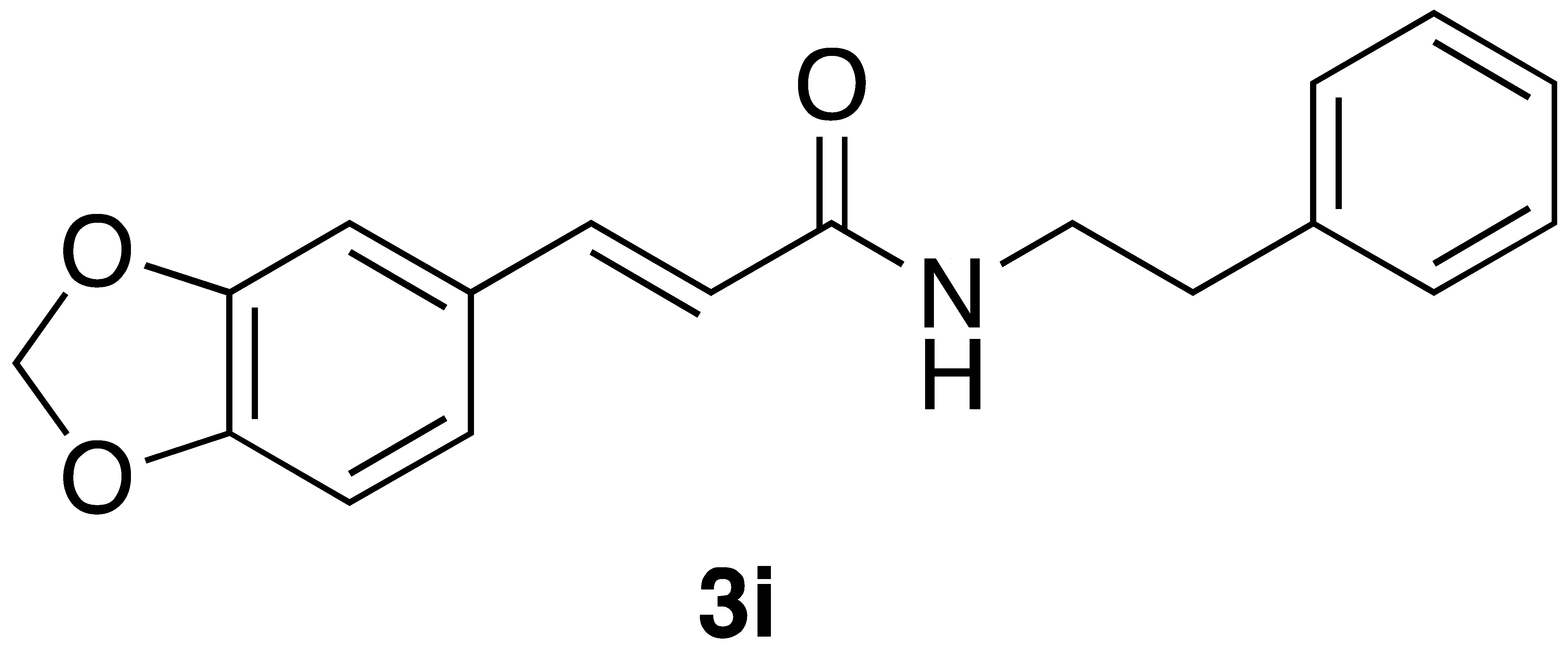
(E)-3-(benzo[d][1,3]dioxol-5-yl)-N-phenethylacrylamide (3i): Following the general procedure 2 starting with benzo[d][1,3]dioxole-5-carbaldehyde (60 mg) afforded 78 mg (66% yield) of a white solid after flash chromatography (EtOAc/hex): mp = 138–140 °C (lit. 133–136 °C); 1H NMR (400 MHz, CDCl3) δ: 7.56 (1H, d, J = 15.4 Hz, CH=CH), 7.35 (2H, t, J = 7.4 Hz, ArH), 7.26 (3H, dd, J = 13.8, 7.2 Hz, ArH), 6.99 (2H, d, J = 8.4 Hz, ArH), 6.80 (1H, d, J = 7.8 Hz, ArH), 6.19 (1H, d, J = 15.4 Hz, CH=CH), 5.99 (2H, s, OCH2O-), 5.74 (1H, s(br), NH), 3.68 (2H, q, J = 4.6 Hz, CH2CH2), 2.91 (2H, t, J = 6.8 Hz, CH2CH2); 13C NMR (126 MHz, CDCl3) δ: 166.1, 149.1, 148.2, 140.9, 138.9, 129.2, 128.8, 128.7, 126.6, 123.9, 118.6, 108.5, 106.4, 101.4, 40.8, 35.7; APCI MS m/z: 296.1 (MH+). Matches lit [30].
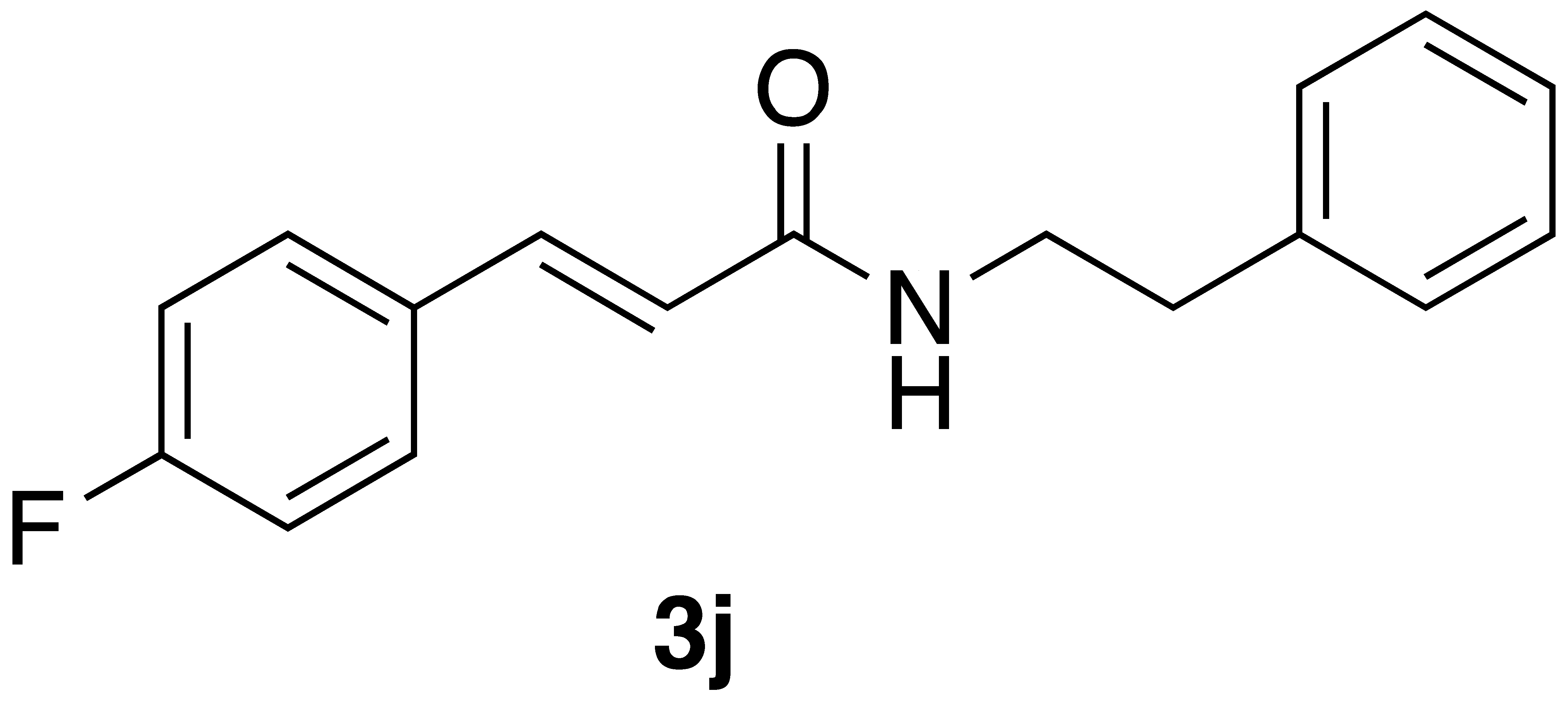
(E)-3-(4-fluorophenyl)-N-phenethylacrylamide (3j): Following the general procedure 2 starting with 4-fluorobenzaldehyde (50 mg) afforded 59 mg (55% yield) of a white solid after flash chromatography (EtOAc/hex): mp = 151–153 °C; 1H NMR (500 MHz, CDCl3) δ: 7.61 (1H, d, J = 15.4 Hz, CH=CH), 7.49 (2H, m, ArH), 7.36 (2H, m, ArH), 7.27 (3H, m, ArH), 7.07 (2H, t, J = 8.4 Hz, ArH), 6.27 (1H, d, J = 15.4 Hz, CH=CH), 5.64 (1H, s(br), NH), 3.70 (2H, q, J = 4.8 Hz, CH2CH2), 2.92 (2H, t, J = 6.5 Hz, CH2CH2); 13C NMR (126 MHz, CDCl3) δ: 165.8, 163.5 (d, JC-F = 250.3 Hz), 139.8, 138.9, 131.1 (d, JC-F = 2.5 Hz), 129.6 (d, JC-F = 8.3 Hz), 128.8, 128.7, 126.6, 120.4, 115.9 (d, JC-F = 21.9), 40.8, 35.7; 19F NMR (470 MHz, CDCl3) δ: −110.72; APCI MS m/z: 270.1 (MH+). Matches lit [31].
Computational Studies: All computational studies were carried out with the Gaussian16 [32] package. Geometry optimization and subsequent frequency calculations were carried out at the B3LYP/6-31G(d,p) level. The enthalpy of reaction for the addition of hydrogen to CAPA was determined from thermodynamic values calculated at 298 K. The resulting enthalpy of reaction was fit to Equation (1) in order to determine the amidity of CAPA.
Percent Amidity = [1.258 × ΔH (kJ/mol)] + 56.126
Cytotoxicity Assays: HeLa cells were maintained in RPMI culture medium supplemented with 10% Equafetal bovine serum (EFBS) and 1% penicillin-streptomycin (P/S). Neuroblastoma BE(2)-C cells were maintained in DMEM/F12 culture medium supplemented with 10% EFBS and 1% P/S. For measuring the dose-dependent effect of a compound on cell viability, cells were plated into a 96-well plate and treated with a series of dilutions of individual compounds in four replicates for 4 days. After 4 days, cell viability was determined by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay [27]. Dose-dependent graphs were generated, and IC50 values were determined using the GraphPad Prism 9 software. The IC50 values correspond to the concentrations of the compounds that reduced the cell viability by 50% in comparison to the carrier (DMSO)-treated control condition. The mean and standard deviation (SD) of IC50 for each compound in each cell line were calculated from IC50’s obtained from at least two independent experiments. Statistical analysis was carried out by two-tailed t-test (p < 0.050) to determine non-equivalence of IC50 values.
5. Conclusions
The results reported here are significant in several ways. First, the discovery of CAPA’s activity against neuroblastoma cells is remarkable given the lack of activity against other human cancer cell types. The lack of cytotoxicity of CAPA in normal human cells indicates that CAPA may be a highly selective agent for neuroblastoma. The amidity of CAPA indicates that it is more metabolically stable to hydrolysis as compared to CAPE, which is a positive attribute for further exploration of CAPA and its analogues as a potential therapy for neuroblastoma.
Several CAPA analogues were prepared using both flow and on-water Wittig couplings. In general, the on-water method was found to be superior, as it does not require special equipment, provides higher yields, and is environmentally friendly. While CAPA is only weakly active against HeLa cells, it is significant that one of these analogues, 3f, displays cytotoxicity against HeLa cells that is similar to that of CAPE. This shows that replacement of the catechol ring of CAPA can afford compounds that recapture the broad anticancer activity that is lost going from CAPE to CAPA while having the additional benefit of avoiding the reactivity of this catechol group. The lack of activity of the regioisomeric CAPA analogue 3e indicates that there is a significant structure–activity relationship (SAR) for cytotoxicity against HeLa cells in these CAPA analogues. It is also notable that 3g and 3f are equipotent against BE(2)-C neuroblastoma cells, albeit less active than CAPA itself. This indicates a distinct SAR for BE(2)-C cell cytotoxicity for CAPA analogues compared to HeLa cell activity.
There is an interest in metabolically stable CAPE analogues that incorporate an amide isostere, as in CAPA, that can overcome the limited cancer cell cytotoxicity of CAPA. The anti-neuroblastoma activity of CAPA and analogues 3f and 3g reported here provides a starting point for future studies on the mechanism of this selective anti-cancer activity. In addition, the identification of the HeLa cell-active CAPA analogue 3f demonstrates that judicious modification of the CAPA structure can lead to enhanced activity even in cancer cell lines in which CAPA itself is inactive. The work reported here focused on improvements of the catechol group of CAPA; future work that builds from the identification of 3f and 3g as anticancer CAPA analogues will focus on further optimization of other structural features.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms25158051/s1.
Author Contributions
Conceptualization, S.M.K.; methodology, A.S. and M.S.; investigation, A.S., M.S., M.J., N.L.M.-D., J.L.S. and A.V.; writing—original draft preparation, S.M.K.; writing—review and editing, M.S. and L.D.; supervision, L.D. and S.M.K. All authors have read and agreed to the published version of the manuscript.
Funding
Funding was provided by grants from the National Science Foundation (1955432, to S.M.K.) and the National Institutes of Health (CA213199, to L.D. and the R25 Bridges to the Doctorate Program GM102783, to N.M.D.). We also acknowledge support from the Texas State University Postdoctoral Researcher Catalyst Program (M.S.).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the Supplementary Materials.
Acknowledgments
We are grateful to Ian Riddington in the Department of Chemistry at the University of Texas at Austin and David Schilter in the Department of Chemistry and Biochemistry at Texas State University for the H.R.M.S data.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Olgierd, B.; Kamila, Z.; Anna, B.; Emilia, M. The Pluripotent Activities of Caffeic Acid Phenethyl Ester. Molecules 2021, 26, 1335. [Google Scholar] [CrossRef]
- Balaha, M.; De Filippis, B.; Cataldi, A.; di Giacomo, V. CAPE and neuroprotection: A review. Biomolecules 2021, 11, 176. [Google Scholar] [CrossRef] [PubMed]
- Erdemli, H.K.; Akyol, S.; Armutcu, F.; Akyol, O. Antiviral properties of caffeic acid phenethyl ester and its potential application. J. Intercult. Ethnopharmacol. 2015, 4, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Pittala, V.; Salerno, L.; Romeo, G.; Acquaviva, R.; Di Giacomo, C.; Sorrenti, V. Therapeutic potential of caffeic acid phenethyl ester (cape) in diabetes. Curr. Med. Chem. 2018, 25, 4827–4836. [Google Scholar] [CrossRef] [PubMed]
- Tolba, M.F.; Omar, H.A.; Azab, S.S.; Khalifa, A.E.; Abdel-Naim, A.B.; Abdel-Rahman, S.Z. Caffeic Acid Phenethyl Ester: A Review of Its Antioxidant Activity, Protective Effects against Ischemia-reperfusion Injury and Drug Adverse Reactions. Crit. Rev. Food Sci. Nutr. 2016, 56, 2183–2190. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pang, J.; Maffucci, J.A.; Pade, D.S.; Newman, R.A.; Kerwin, S.M.; Bowman, P.D.; Stavchansky, S. Pharmacokinetics of caffeic acid phenethyl ester and its catechol-ring fluorinated derivative following intravenous administration to rats. Biopharm. Drug Dispos. 2009, 30, 221–228. [Google Scholar] [CrossRef]
- Mucsi, Z.; Chass, G.A.; Csizmadia, I.G. Amidicity Change as a Significant Driving Force and Thermodynamic Selection Rule of Transamidation Reactions. A Synergy between Experiment and Theory. J. Phys. Chem. B 2008, 112, 7885–7893. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Marriner, G.A.; Wang, X.; Bowman, P.D.; Kerwin, S.M.; Stavchansky, S. Synthesis of a Series of Caffeic Acid Phenethyl Amide (CAPA) Fluorinated Derivatives: Comparison of Cytoprotective Effects to Caffeic Acid Phenethyl Ester (CAPE). Bioorg. Med. Chem. 2010, 18, 5032–5038. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, L.H.; Maillet, J.; LeBlanc, L.M.; Jean-François, J.; Touaibia, M.; Flamand, N.; Surette, M.E. Caffeic Acid Phenethyl Ester and Its Amide Analogue Are Potent Inhibitors of Leukotriene Biosynthesis in Human Polymorphonuclear Leukocytes. PLoS ONE 2012, 7, e31833. [Google Scholar] [CrossRef]
- Dai, L.; Zang, C.; Tian, S.; Liu, W.; Tan, S.; Cai, Z.; Ni, T.; An, M.; Li, R.; Gao, Y.; et al. Design, Synthesis, and Evaluation of Caffeic Acid Amides as Synergists to Sensitize Fluconazole-Resistant Candida Albicans to Fluconazole. Bioorg. Med. Chem. Lett. 2015, 25, 34–37. [Google Scholar] [CrossRef]
- David, S.; Mandabi, A.; Uzi, S.; Aharoni, A.; Meijler, M.M. Mining Plants for Bacterial Quorum Sensing Modulators. ACS Chem. Biol. 2018, 13, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Firdaus; Soekamto, N.H.; Seniwati; Islam, M.F.; Sultan. Phenethyl Ester and Amide of Ferulic Acids: Synthesis and Bioactivity against P388 Leukemia Murine Cells. J. Phys. Conf. Ser. 2018, 979, 012016. [Google Scholar] [CrossRef]
- Beauregard, A.-P.; Harquail, J.; Lassalle-Claux, G.; Belbraouet, M.; Jean-Francois, J.; Touaibia, M.; Robichaud, G.A. CAPE Analogs Indue Growth Arrest and Apoptosis in Breast Cancer Cells. Molecules 2015, 20, 12576–12589. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, A.; Lassalle-Claux, G.; Hogan, L.; Vaillancourt, E.; Selka, A.; Luiker, K.; Kim, M.J.; Touaibia, M.; Reiman, T. Antimyeloma Potential of Caffeic Acid Phenethyl Ester and Its Analogues through Sp1 Mediated Downregulation of IKZF1-IRF4-MYC Axis. J. Nat. Prod. 2020, 83, 3526–3535. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, J.T.; Clabault, H.; Patton, C.; Lassalle-Claux, G.; Jean-Francois, J.; Pare, A.F.; Herbert, M.J.G.; Surette, M.E.; Touaibia, M. Antiproliferative, Antiandrogenic and Cytotoxic Effects of Novel Caffeic Acid Derivatives in LNCaP Human Androgen-dependent Prostate Cancer Cells. Bioorg. Med. Chem. 2013, 23, 7192–7193. [Google Scholar] [CrossRef] [PubMed]
- De Armas-Ricard, M.; Ruiz-Reyes, E.; Ramirez-Rodriguez, O. Caffeates and Caffeamides: Synthetic Methodologies and Their Antioxidant Properties. Int. J. Med. Chem. 2019, 2019, 2592609. [Google Scholar] [CrossRef] [PubMed]
- Yoo, W.J.; Ishitani, H.; Saito, Y.; Laroche, B.; Kobayashi, S. Reworking Organic Synthesis for the Modern Age: Synthetic Strategies Based on Continuous-Flow Addition and Condensation Reactions with Heterogeneous Catalysts. J. Org. Chem. 2020, 85, 5132–5145. [Google Scholar] [CrossRef] [PubMed]
- Riccaboni, M.; La Porta, E.; Martorana, A.; Attanasio, R. Effect of Phase Transfer Chemistry, Segmented Fluid Flow, and Sonication on the Synthesis of Cinnamic Esters. Tetrahedron 2010, 66, 4032–4039. [Google Scholar] [CrossRef]
- Baxendale, I.R.; Griffiths-Jones, C.M.; Ley, S.V.; Tranmer, G.K. Preparation of the Neolignan Natural Product Grossamide by a Continuous-Flow Process. Synlett 2006, 2006, 427–430. [Google Scholar] [CrossRef]
- Achanta, S.; Liautard, V.; Paugh, R.; Organ, M.G. The Development of a General Strategy for the Synthesis of Tyramine-Based Natural Products by Using Continuous Flow Techniques. Chem. A Eur. J. 2010, 16, 12797–12800. [Google Scholar] [CrossRef]
- Russell, M.G.; Warren, S. Synthesis of New Water-soluble Phosphonium Salts and Their Wittig Reactions in Water. Chem. Soc. Perkin Trans. 1 2000, 4, 505–513. [Google Scholar] [CrossRef]
- Dambacher, J.; Zhao, W.; El-Batta, A.; Arness, R.; Jiang, C.; Berdgahl, M. Water is an Efficient Medium for Wittig Reactions Employing Stabilized Ylides and Aldehydes. Tetrahedron Lett. 2005, 46, 4473–4477. [Google Scholar] [CrossRef]
- Javaherian, M.; Movaheditabar, P. On-water Biphasic Organic Synthesis. J. Iran. Chem. Soc. 2023, 20, 2103–2125. [Google Scholar] [CrossRef]
- Sidoryk, K.; Jaromin, A.; Filipczak, N.; Cmoch, P.; Cybulski, M. Synthesis and Antioxidant Activity of Caffeic Acid Derivatives. Molecules 2018, 23, 2199. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.-T.; Ma, W.; Yen, P.; Xie, J.-G.; Han, J.; Frenkel, K.; Grunberger, D.; Conney, A.H. Inhibitory effects of caffeic acid phenethyl ester (CAPE) on 12-O-tetradecanoylphorbol-13-acetate-induced tumor promotion in mouse skin and the synthesis of DNA, RNA, and protein in HeLa cells. Carcinogenesis 1996, 17, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.H.; Chu, C.C.; Hung, M.W.; Lee, H.J.; Hsu, H.J.; Chang, T.C. Caffeic acid phenethyl ester induces E2F-1-mediated growth inhibition and cell-cycle arrest in human cervical cancer cells. FEBS J. 2013, 280, 2581–2593. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Nesterenko, V.; Putt, K.S.; Hergenrother, P.J. Identification from a Combinatorial Library of a Small Molecule that Selectively Induces Apoptosis in Cancer Cells. J. Am. Chem. Soc. 2003, 125, 14672–14673. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Wu, P.-Y.W.; Chen, C.-W.; Lyu, J.-L.; Liu, Y.-J.; Wen, K.-C.; Lin, C.-Y.; Kuo, Y.-H.; Chiang, H.-M. Protective Effects and Mechanisms of N-Phenethyl Caffeamide from UVA-Induced Skin Damage in Human Epidermal Keratinocytes through Nrf2/HO-1 Regulation. Int. J. Mol. Sci. 2019, 20, 164. [Google Scholar] [CrossRef]
- Chen, L.; Jin, Y.; Chen, H.; Sun, C.; Fu, W.; Zheng, L.; Lu, M.; Chen, P.; Chen, G.; Zhang, Y.; et al. Discovery of caffeic acid phenethyl ester derivatives as novel myeloid differentiation protein 2 inhibitors for treatment of acute lung injury. Eur. J. Med. Chem. 2018, 143, 361–375. [Google Scholar] [CrossRef]
- Khaldoun, K.; Safer, A.; Saidi-Besbes, A.; Carboni, B.; Le Gueverl, R.; Car-reaux, F. An Efficient Solvent-Free Microwave-Assisted Synthesis of Cinnamamides by Amidation Reaction Using Phenylboronic Acid/Lewis Base Co-catalytic Systems. Synthesis 2019, 51, 3891–3900. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).