

Deciphering the Effects of the PYCR Family on Cell Function, Prognostic Value, Immune Infiltration in ccRCC and Pan-Cancer
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
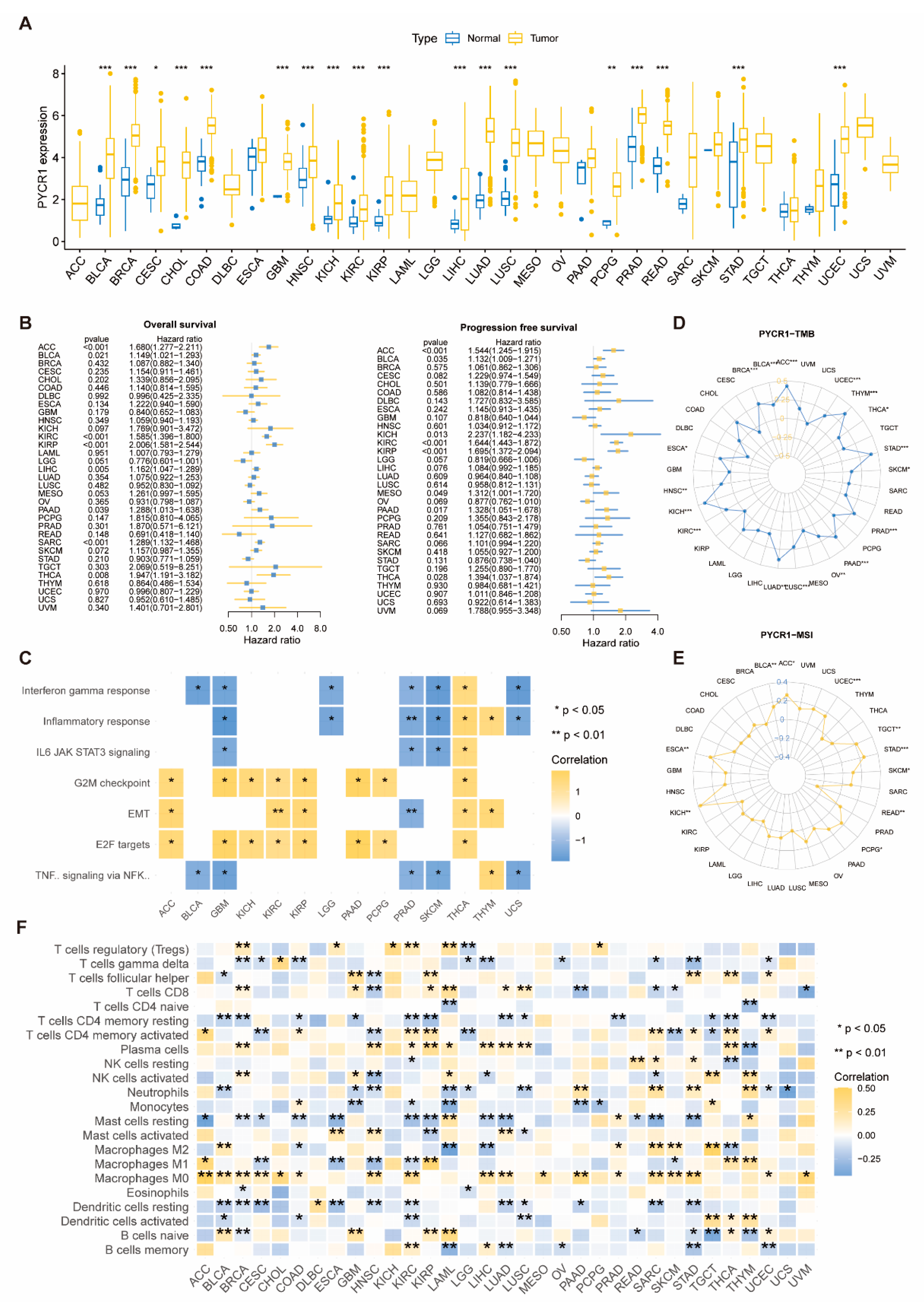
2.1. Analysis of PYCRs in Pan-Cancer
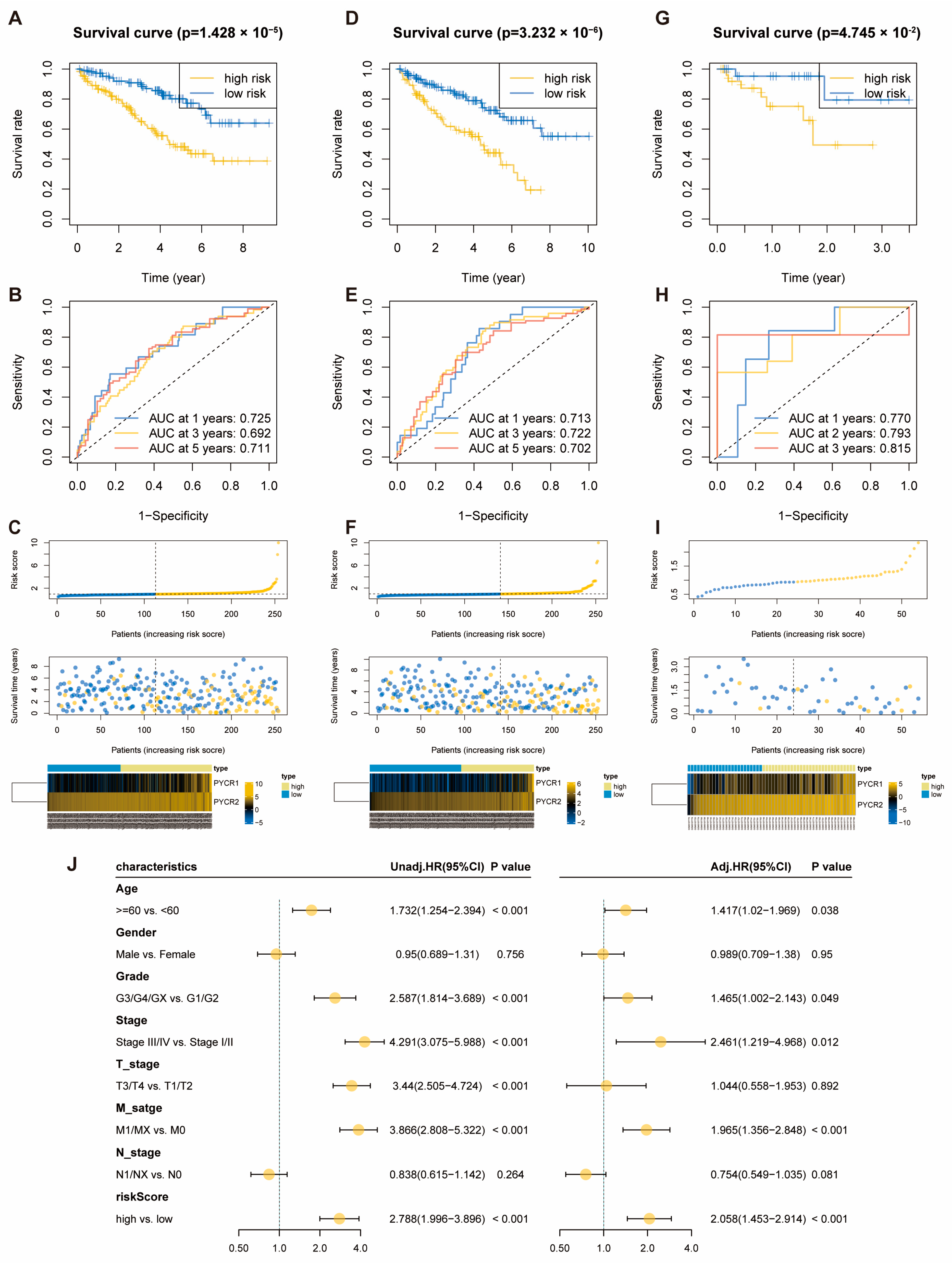
2.2. Construction of OS-Related Prognostic Risk Score Model for PYCRs in KIRC
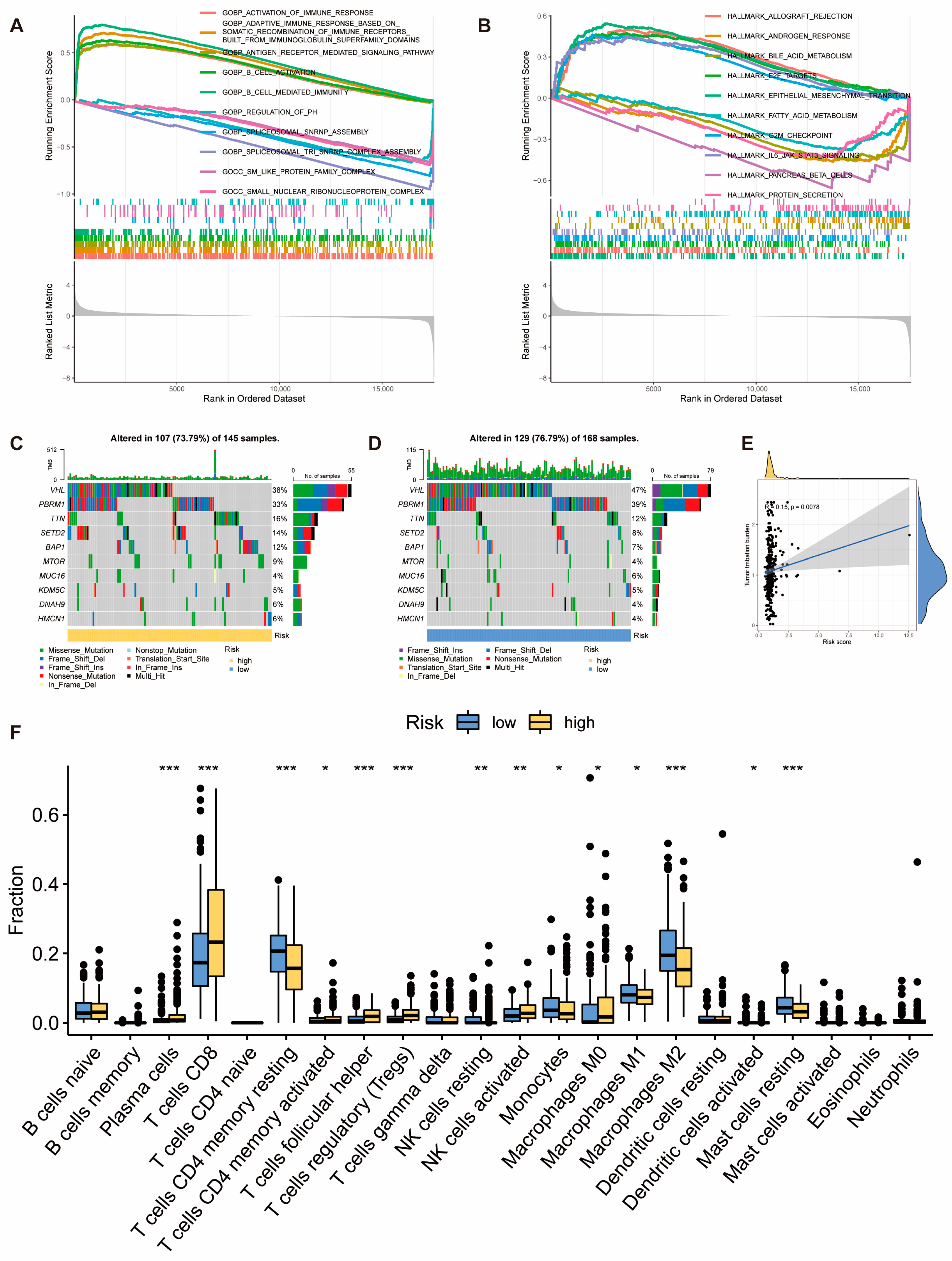
2.3. Molecular and Immune Features of Risk Score Model Subgroups
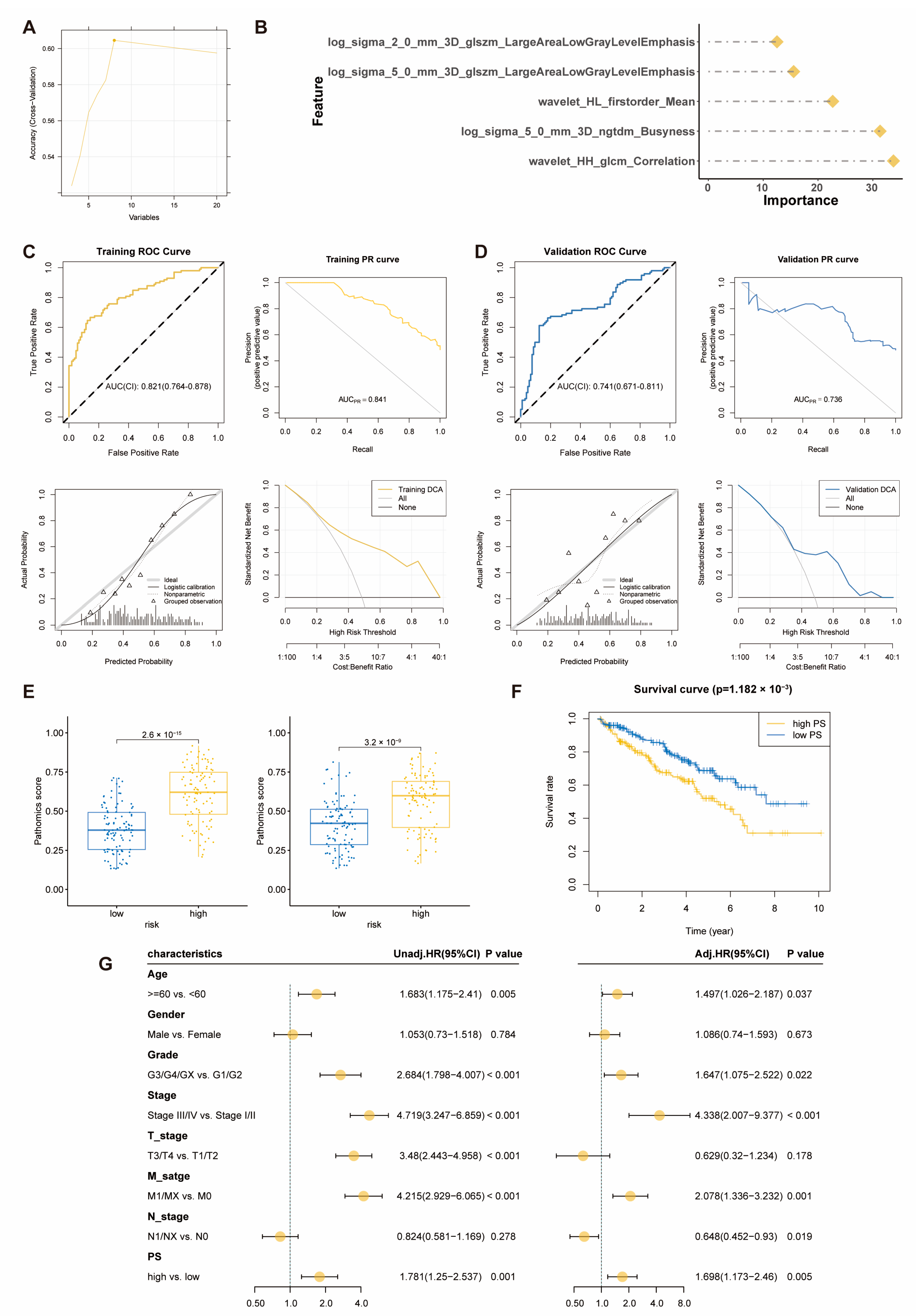
2.4. Pathomics Feature Model Predicts Prognostic Risk Score Model
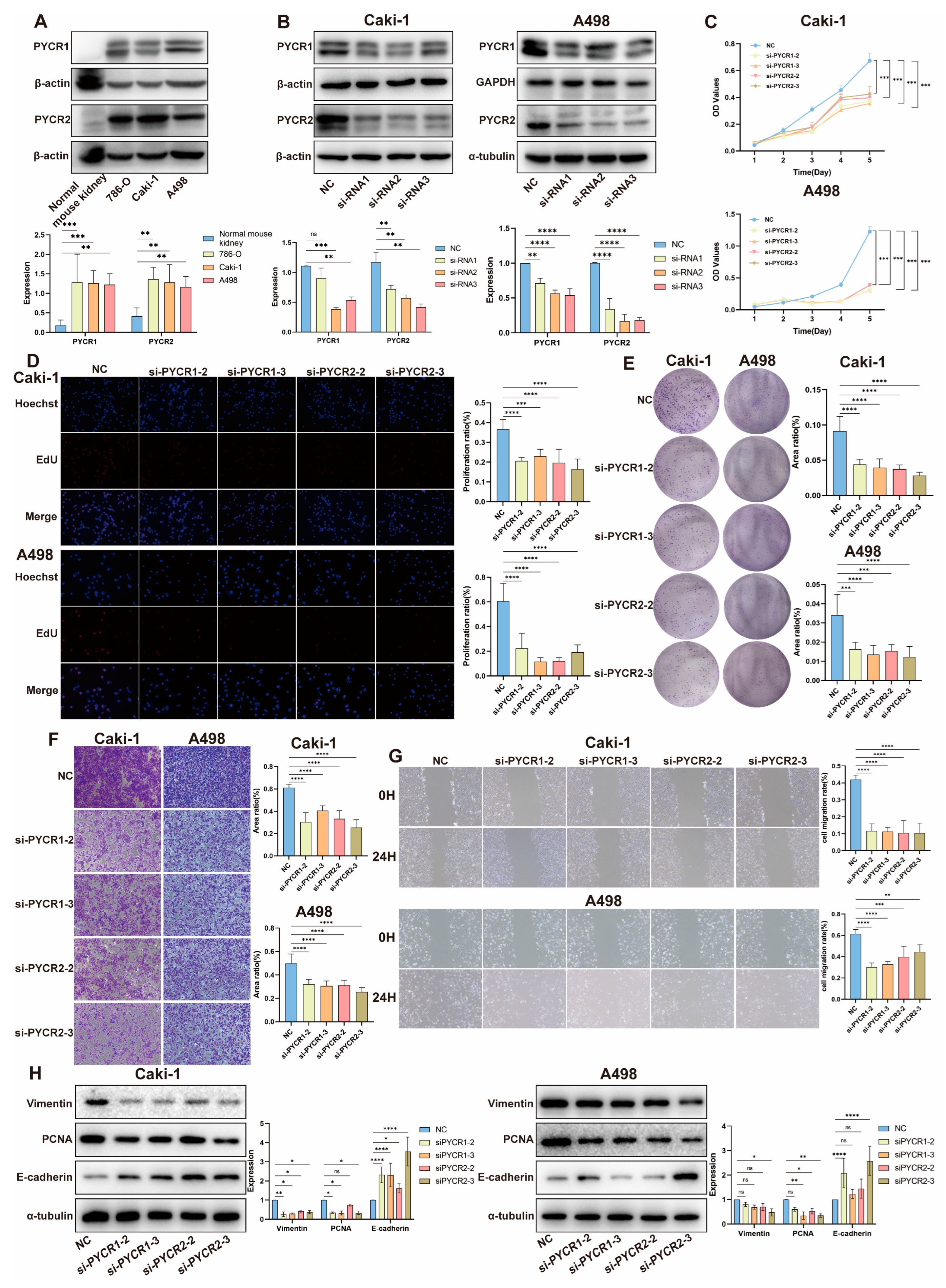
2.5. Impact of PYCRs Knockdown on In Vitro Cell Proliferation and Migration in Human Renal Cell Carcinoma Cell Line Caki-1 and A498 Cells
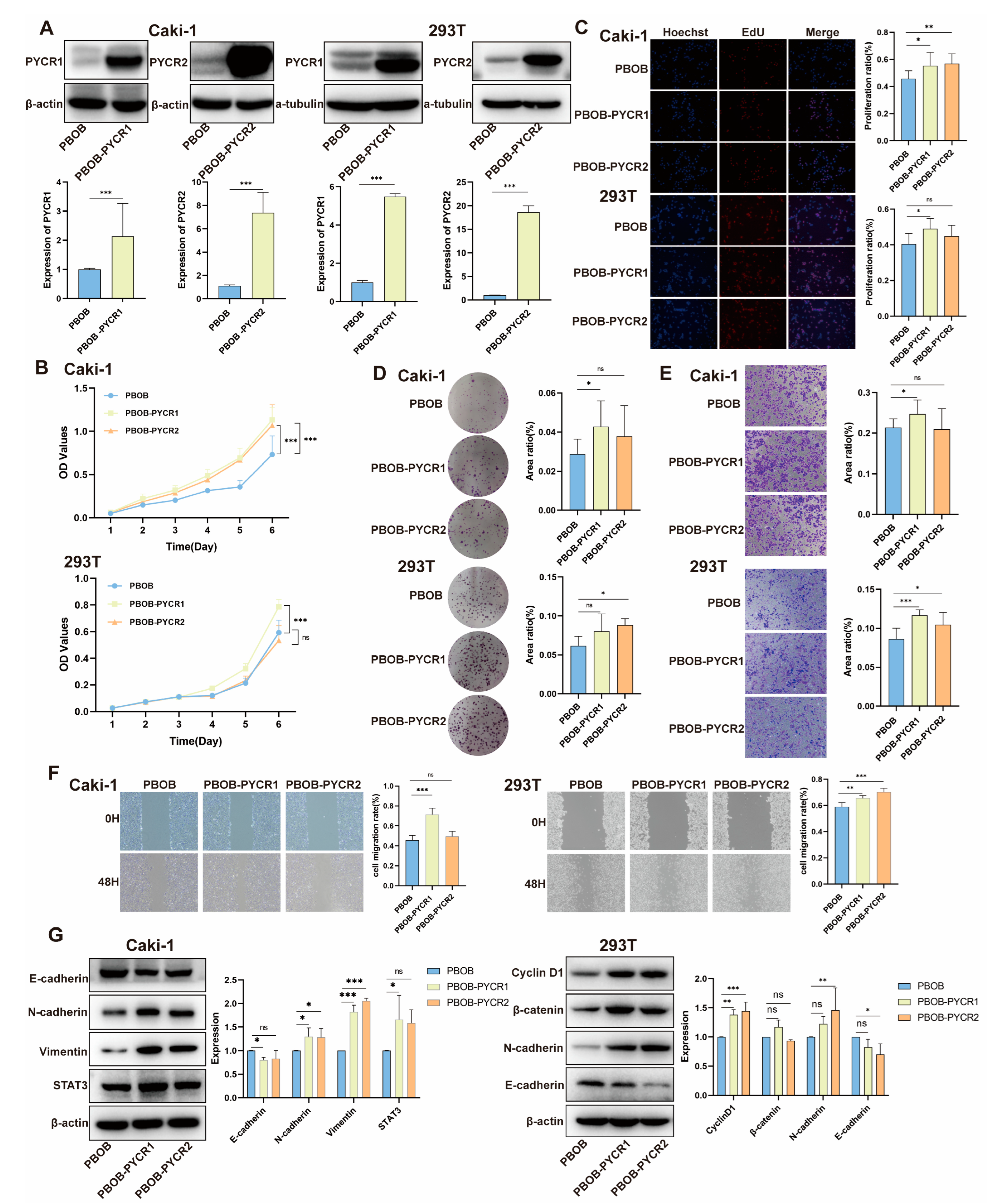
2.6. Impact of PYCRs Overexpression on In Vitro Cell Proliferation and Migration in Human Renal Cell Carcinoma Cell Line Caki-1 and 293T Cells
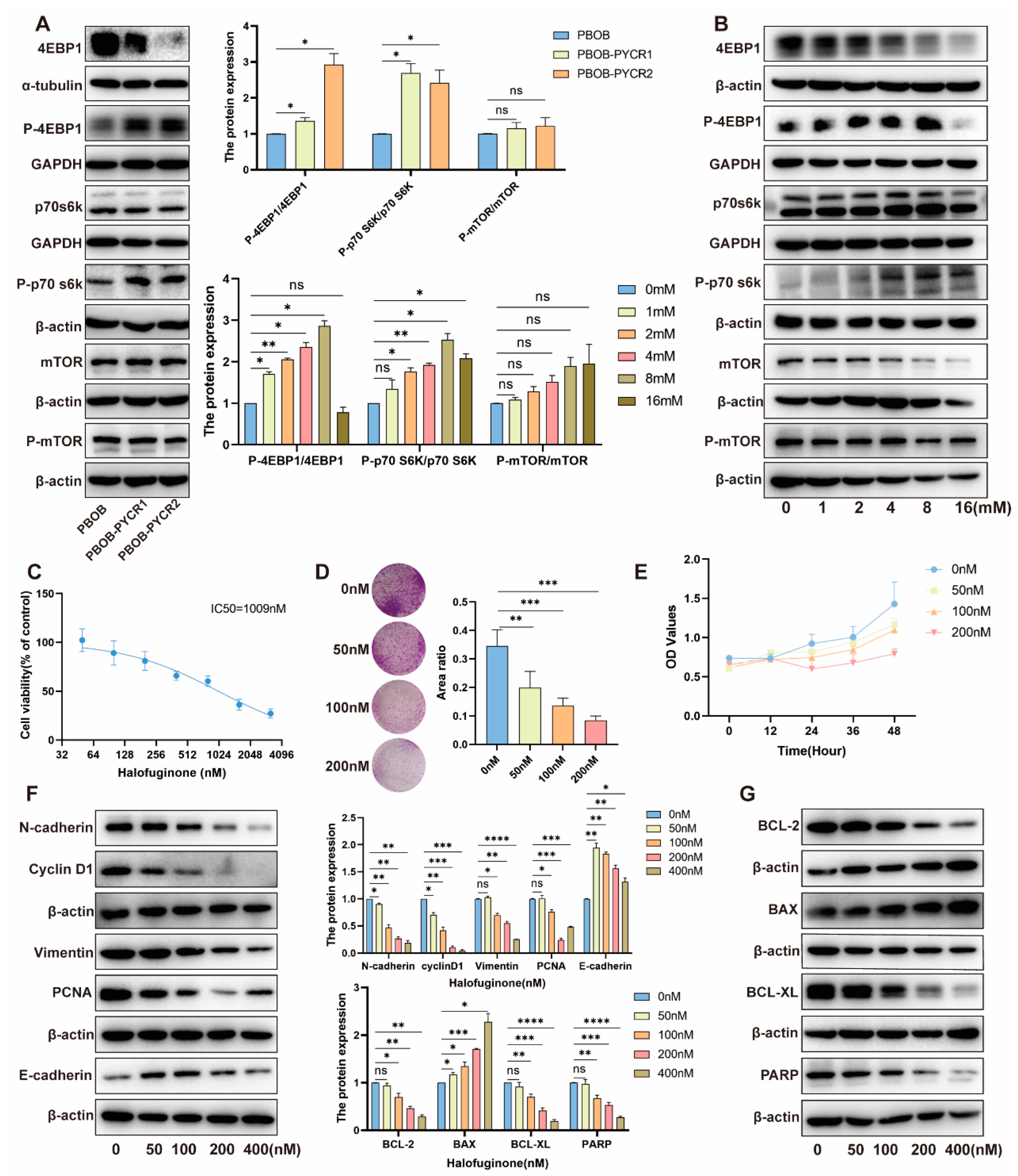
2.7. Regulation of the mTOR Signaling Pathway by PYCRs and Proline in Renal Cell Carcinoma
2.8. Impact and Mechanism Study of Downstream Pathways of PYCRs on the Survival of Renal Cancer Cells
3. Discussion
4. Materials and Methods
4.1. TCGA Pan-Cancer Data and Acquisition/Processing of H&E Pathology Images
4.2. Prognostic Analysis
4.3. Immune Cell Infiltration Analysis and Gene Set Enrichment Analysis (GSEA)
4.4. Tumor Mutation Burden (TMB) and Microsatellite Instability (MSI) Analysis
4.5. Construction of the KIRC Prognostic Risk Score Model
4.6. Pathological Image Segmentation and Feature Extraction
4.7. Pathomics Feature Model Construction
4.8. Cell Culture and Transfection
4.9. Protein Blotting
4.10. Cell Proliferation
4.11. Cell Migration
4.12. Drug Treatment
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Bailey, M.H.; Porta-Pardo, E.; Thorsson, V.; Colaprico, A.; Bertrand, D.; Gibbs, D.L.; Weerasinghe, A.; Huang, K.-L.; Tokheim, C.; et al. Perspective on Oncogenic Processes at the End of the Beginning of Cancer Genomics. Cell 2018, 173, 305–320. [Google Scholar] [CrossRef] [PubMed]
- Chalecka, M.; Kazberuk, A.; Palka, J.; Surazynski, A. P5C as an Interface of Proline Interconvertible Amino Acids and Its Role in Regulation of Cell Survival and Apoptosis. Int. J. Mol. Sci. 2021, 22, 11763. [Google Scholar] [CrossRef]
- Bogner, A.N.; Stiers, K.M.; Tanner, J.J. Structure, biochemistry, and gene expression patterns of the proline biosynthetic enzyme pyrroline-5-carboxylate reductase (PYCR), an emerging cancer therapy target. Amino Acids 2021, 53, 1817–1834. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.M.; Seravalli, J.; Liang, X.; Tanner, J.J.; Becker, D.F. Disease variants of human Delta(1)-pyrroline-5-carboxylate reductase 2 (PYCR2). Arch. Biochem. Biophys. 2021, 703, 108852. [Google Scholar] [CrossRef] [PubMed]
- Pietzner, M.; Wheeler, E.; Carrasco-Zanini, J.; Cortes, A.; Koprulu, M.; Wörheide, M.A.; Oerton, E.; Cook, J.; Stewart, I.D.; Kerrison, N.D.; et al. Mapping the proteo-genomic convergence of human diseases. Science 2021, 374, eabj1541. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Luo, L.; Xie, Y.; Zhao, Y.; Yao, J.; Liu, X. PYCR1 knockdown inhibits the proliferation, migration, and invasion by affecting JAK/STAT signaling pathway in lung adenocarcinoma. Mol. Carcinog. 2020, 59, 503–511. [Google Scholar] [CrossRef]
- Xu, Y.; Zuo, W.; Wang, X.; Zhang, Q.; Gan, X.; Tan, N.; Jia, W.; Liu, J.; Li, Z.; Zhou, B.; et al. Deciphering the effects of PYCR1 on cell function and its associated mechanism in hepatocellular carcinoma. Int. J. Biol. Sci. 2021, 17, 2223–2239. [Google Scholar] [CrossRef]
- Yan, K.; Xu, X.; Wu, T.; Li, J.; Cao, G.; Li, Y.; Ji, Z. Knockdown of PYCR1 inhibits proliferation, drug resistance and EMT in colorectal cancer cells by regulating STAT3-Mediated p38 MAPK and NF-κB signalling pathway. Biochem. Biophys. Res. Commun. 2019, 520, 486–491. [Google Scholar] [CrossRef]
- Wang, S.; Gu, L.; Huang, L.; Fang, J.; Liu, Z.; Xu, Q. The upregulation of PYCR2 is associated with aggressive colon cancer progression and a poor prognosis. Biochem. Biophys. Res. Commun. 2021, 572, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Yang, K.; Luo, J.; Gao, Z.; Gao, Y. Dysregulation of USP18/FTO/PYCR1 signaling network promotes bladder cancer development and progression. Aging 2021, 13, 3909–3925. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhou, X.; Huang, J.; Xu, Z.; Xing, C.; Yang, J.; Zhou, X. MicroRNA hsa-miR-150-5p inhibits nasopharyngeal carcinogenesis by suppressing PYCR1 (pyrroline-5-carboxylate reductase 1). Bioengineered 2021, 12, 9766–9778. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Li, S.; Yuan, Z.; Zhou, L. Pyrroline-5-carboxylate reductase 1 (PYCR1) upregulation contributes to gastric cancer progression and indicates poor survival outcome. Ann. Transl. Med. 2020, 8, 937. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Wu, Y.; Wang, J. Pyrroline-5-carboxylate reductase 1 promotes cell proliferation via inhibiting apoptosis in human malignant melanoma. Cancer Manag. Res. 2018, 10, 6399–6407. [Google Scholar] [CrossRef] [PubMed]
- Bhinder, B.; Gilvary, C.; Madhukar, N.S.; Elemento, O. Artificial Intelligence in Cancer Research and Precision Medicine. Cancer Discov. 2021, 11, 900–915. [Google Scholar] [CrossRef] [PubMed]
- Haug, C.J.; Drazen, J.M. Artificial Intelligence and Machine Learning in Clinical Medicine, 2023. N. Engl. J. Med. 2023, 388, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Kickingereder, P.; Isensee, F.; Tursunova, I.; Petersen, J.; Neuberger, U.; Bonekamp, D.; Brugnara, G.; Schell, M.; Kessler, T.; Foltyn, M.; et al. Automated quantitative tumour response assessment of MRI in neuro-oncology with artificial neural networks: A multicentre, retrospective study. Lancet Oncol. 2019, 20, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Hamet, P.; Tremblay, J. Artificial intelligence in medicine. Metabolism 2017, 69, S36–S40. [Google Scholar] [CrossRef]
- Yu, K.-H.; Zhang, C.; Berry, G.J.; Altman, R.B.; Ré, C.; Rubin, D.L.; Snyder, M. Predicting non-small cell lung cancer prognosis by fully automated microscopic pathology image features. Nat. Commun. 2016, 7, 12474. [Google Scholar] [CrossRef]
- Chen, D.; Fu, M.; Chi, L.; Lin, L.; Cheng, J.; Xue, W.; Long, C.; Jiang, W.; Dong, X.; Sui, J.; et al. Prognostic and predictive value of a pathomics signature in gastric cancer. Nat. Commun. 2022, 13, 6903. [Google Scholar] [CrossRef] [PubMed]
- Keller, T.L.; Zocco, D.; Sundrud, M.S.; Hendrick, M.; Edenius, M.; Yum, J.; Kim, Y.-J.; Lee, H.-K.; Cortese, J.F.; Wirth, D.F.; et al. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat. Chem. Biol. 2012, 8, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Chu, T.L.H.; Guan, Q.; Nguan, C.Y.C.; Du, C. Halofuginone suppresses T cell proliferation by blocking proline uptake and inducing cell apoptosis. Int. Immunopharmacol. 2013, 16, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, A.J.; Hellmann, M.D. Acquired Resistance to Immune Checkpoint Inhibitors. Cancer Cell 2020, 37, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Zhu, H.; Dong, L.; Shi, W.; Chen, R.; Song, Z.; Huang, C.; Li, J.; Dong, X.; Zhou, Y.; et al. Integrated Proteogenomic Characterization of HBV-Related Hepatocellular Carcinoma. Cell 2019, 179, 561–577.e22. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; You, Y.; Zhang, X.; Song, Y.; Xiang, H.; Peng, X.; Qin, J.; Tan, G. Amplification of chromosome 8q21-qter associated with the acquired paclitaxel resistance of nasopharyngeal carcinoma cells. Int. J. Clin. Exp. Pathol. 2015, 8, 12346–12356. [Google Scholar] [PubMed]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Goodman, A.M.; Sokol, E.S.; Frampton, G.M.; Lippman, S.M.; Kurzrock, R. Microsatellite-Stable Tumors with High Mutational Burden Benefit from Immunotherapy. Cancer Immunol. Res. 2019, 7, 1570–1573. [Google Scholar] [CrossRef]
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer-the stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.-Y.; Wang, N.; Lam, W.; Guo, W.; Feng, Y.; Cheng, Y.-C. Targeting tumour microenvironment by tyrosine kinase inhibitor. Mol. Cancer 2018, 17, 43. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Hu, X.; Ye, Y.; Jian, Z.; Zhong, Y.; Gu, L.; Xiong, X. Pan-Cancer Analysis of PIMREG as a Biomarker for the Prognostic and Immunological Role. Front. Genet. 2021, 12, 687778. [Google Scholar] [CrossRef] [PubMed]
- Barnaba, N.; LaRocque, J.R. Targeting cell cycle regulation via the G2-M checkpoint for synthetic lethality in melanoma. Cell Cycle 2021, 20, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Kent, L.N.; Leone, G. The broken cycle: E2F dysfunction in cancer. Nat. Rev. Cancer 2019, 19, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Young, T.M.; Reyes, C.; Pasnikowski, E.; Castanaro, C.; Wong, C.; Decker, C.E.; Chiu, J.; Song, H.; Wei, Y.; Bai, Y.; et al. Autophagy protects tumors from T cell-mediated cytotoxicity via inhibition of TNFα-induced apoptosis. Sci. Immunol. 2020, 5, eabb9561. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, L.; Zhang, Y.; Yan, Z.; Liu, L.; Chen, G. PYCR1 promotes the progression of non-small-cell lung cancer under the negative regulation of miR-488. Biomed. Pharmacother. 2019, 111, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Huang, X.; Xuan, Y. Pyrroline-5-Carboxylate Reductase-2 Promotes Colorectal Cancer Progression via Activating PI3K/AKT/mTOR Pathway. Dis. Markers 2021, 2021, 9950663. [Google Scholar] [CrossRef]
- Zhu, S.; Ding, W.; Chen, Y.; Wang, W.; Xu, R.; Liu, C.; Liu, X.; Deng, H. High VHL Expression Reverses Warburg Phenotype and Enhances Immunogenicity in Kidney Tumor Cells. Genom. Proteom. Bioinform. 2021, 20, 657–669. [Google Scholar] [CrossRef]
- Borst, J.; Ahrends, T.; Babala, N.; Melief, C.J.M.; Kastenmuller, W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef]
- Quazi, S. Artificial intelligence and machine learning in precision and genomic medicine. Med. Oncol. 2022, 39, 120. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Mai, Z.; Ye, Y.; Song, Y.; Zhang, M.; Yang, X.; Xia, W.; Qiu, X. The role of PYCR1 in inhibiting 5-fluorouracil-induced ferroptosis and apoptosis through SLC25A10 in colorectal cancer. Hum. Cell 2022, 35, 1900–1911. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Yao, X.; Ye, J.; Tian, X.; Yin, Z.; Zhou, L. Epigenetic modification facilitates proline synthase PYCR1 aberrant expression in gastric cancer. Biochim. Biophys. Acta Gene Regul. Mech. 2022, 1865, 194829. [Google Scholar] [CrossRef]
- Du, S.; Sui, Y.; Ren, W.; Zhou, J.; Du, C. PYCR1 promotes bladder cancer by affecting the Akt/Wnt/β-catenin signaling. J. Bioenerg. Biomembr. 2021, 53, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yi, W.; Xu, Z.; Shi, M. PYCR2 promotes growth and aerobic glycolysis in human liver cancer by regulating the AKT signaling pathway. Biochem. Biophys. Res. Commun. 2023, 680, 15–24. [Google Scholar] [CrossRef]
- Ou, R.; Zhang, X.; Cai, J.; Shao, X.; Lv, M.; Qiu, W.; Xuan, X.; Liu, J.; Li, Z.; Xu, Y. Downregulation of pyrroline-5-carboxylate reductase-2 induces the autophagy of melanoma cells via AMPK/mTOR pathway. Tumor Biol. 2016, 37, 6485–6491. [Google Scholar] [CrossRef]
- Geng, P.; Qin, W.; Xu, G. Proline metabolism in cancer. Amino Acids 2021, 53, 1769–1777. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shang, L.; Jiang, W.; Wu, W. Shikonin induces apoptosis and autophagy via downregulation of pyrroline-5-carboxylate reductase1 in hepatocellular carcinoma cells. Bioengineered 2022, 13, 7904–7918. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. Available online: https://pubmed.ncbi.nlm.nih.gov/16199517 (accessed on 30 September 2005). [CrossRef] [PubMed]
- Sha, D.; Jin, Z.; Budczies, J.; Kluck, K.; Stenzinger, A.; Sinicrope, F.A. Tumor Mutational Burden as a Predictive Biomarker in Solid Tumors. Cancer Discov. 2020, 10, 1808–1825. [Google Scholar] [CrossRef] [PubMed]
- Hause, R.J.; Pritchard, C.C.; Shendure, J.; Salipante, S.J. Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 2016, 22, 1342–1350. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Chen, Q.; Chen, J.; Mao, Y.; Duan, L.; Ye, D.; Cheng, W.; Chen, J.; Gao, X.; Lin, R.; et al. Deciphering the Effects of the PYCR Family on Cell Function, Prognostic Value, Immune Infiltration in ccRCC and Pan-Cancer. Int. J. Mol. Sci. 2024, 25, 8096. https://doi.org/10.3390/ijms25158096
Chen H, Chen Q, Chen J, Mao Y, Duan L, Ye D, Cheng W, Chen J, Gao X, Lin R, et al. Deciphering the Effects of the PYCR Family on Cell Function, Prognostic Value, Immune Infiltration in ccRCC and Pan-Cancer. International Journal of Molecular Sciences. 2024; 25(15):8096. https://doi.org/10.3390/ijms25158096
Chicago/Turabian StyleChen, Hongquan, Qing Chen, Jinyang Chen, Yazhen Mao, Lidi Duan, Dongjie Ye, Wenxiu Cheng, Jiaxi Chen, Xinrong Gao, Renxi Lin, and et al. 2024. "Deciphering the Effects of the PYCR Family on Cell Function, Prognostic Value, Immune Infiltration in ccRCC and Pan-Cancer" International Journal of Molecular Sciences 25, no. 15: 8096. https://doi.org/10.3390/ijms25158096